The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”?

 ,
, 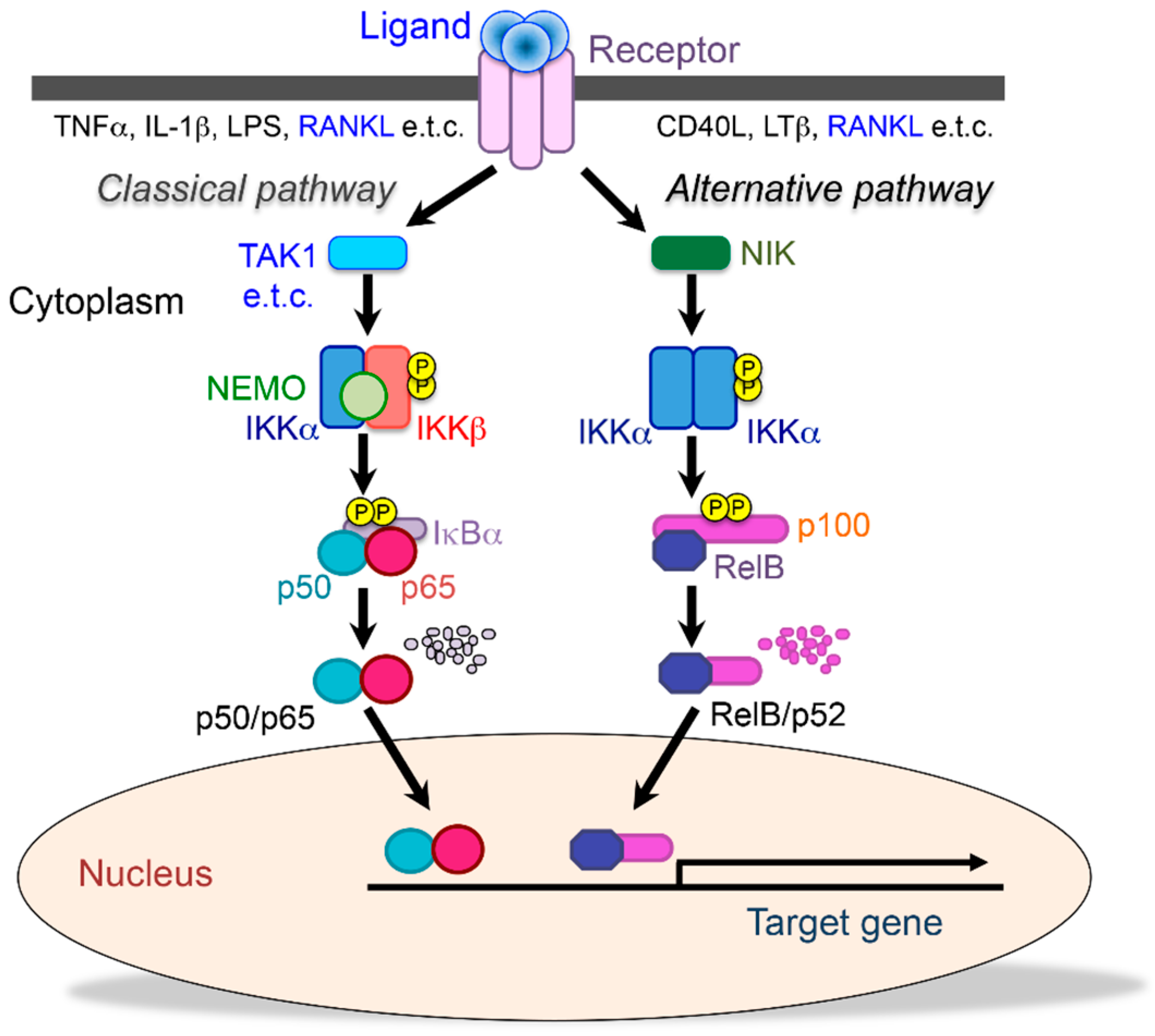{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of NF-κB on Bone Metabolism
2.1. The Functions of NF-κB Signaling in Physiological Osteoclastic Bone Resorption
2.2. NF-κB Inhibition Suppresses Inflammatory Bone Diseases
2.2.1. Rheumatoid Arthritis (RA)
2.2.2. Ankylosing Spondylitis (AS)
2.2.3. Periodontal Disease
2.3. The Activation of NF-κB Suppresses Bone Formation
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NF-κB | Nuclear factor κB |
| IKK | IκB kinase |
| NIK | NF-κB-inducing kinase |
| RA | Rheumatoid arthritis |
| NEMO | NF-κB essential modulator |
| MMPs | Matrix metalloproteases |
References
- Aubin, J.E.; Triffitt, J.T. Mesenchymal stem cells and osteoblast differentiation. In Principles of Bone Biology, 2nd ed.; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 59–81. [Google Scholar]
- De Paula, F.J.; Rosen, C.J. Bone remodeling and energy metabolism: New perspectives. Bone Res. 2013, 1, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Uehara, S.; Udagawa, N.; Takahashi, N. Regulation of bone metabolism by Wnt signals. J. Biochem. 2016, 159, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual framework unifying the immune and skeletal systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Selmi, C. Autoimmunity in 2018. Clin. Rev. Allergy Immunol. 2019, 56, 375–384. [Google Scholar] [CrossRef]
- Favero, M.; Giusti, A.; Geusens, P.; Goldring, S.R.; Lems, W.; Schett, G.; Bianchi, G. OsteoRheumatology: A new discipline? RMD Open 2015, 15, e000083. [Google Scholar] [CrossRef]
- Ceccarelli, F.; Saccucci, M.; Di Carlo, G.; Lucchetti, R.; Pilloni, A.; Pranno, N.; Luzzi, V.; Valesini, G.; Polimeni, A. Periodontitis and rheumatoid arthritis: The same inflammatory mediators? Mediat. Inflamm. 2019, 2019, 6034546. [Google Scholar] [CrossRef]
- Jimi, E.; Ghosh, S. Role of nuclear factor-κB in the immune system and bone. Immunol. Rev. 2005, 208, 80–87. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Britanova, L.; Makeev, V.J.; Kuprash, D.V. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem. Biophys. Res. Commun 2008, 365, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Iotsova, V.; Caamaño, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Z.; Tang, E.; Fan, Z.; McCauley, L.; Franceschi, R.; Guan, K.; Krebsbach, P.H.; Wang, C.Y. Inhibition of osteoblastic boneformation by nuclear factor-κB. Nat. Med. 2009, 15, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Alles, N.; Soysa, N.S.; Hayashi, J.; Khan, M.; Shimoda, A.; Shimokawa, H.; Ritzeler, O.; Akiyoshi, K.; Aoki, K.; Ohya, K. Suppression of NF-κB increases bone formation and ameliorates osteopenia in ovariectomized mice. Endocrinology 2010, 151, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Mizuno, A.; Amizuka, N.; Irie, K.; Murakami, A.; Fujise, N.; Kanno, T.; Sato, Y.; Nakagawa, N.; Yasuda, H.; Mochizuki, S.; et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem. Biophys. Res. Commun. 1998, 247, 610–615. [Google Scholar] [CrossRef]
- Min, H.; Morony, S.; Sarosi, I.; Dunstan, C.R.; Capparelli, C.; Scully, S.; Van, G.; Kaufman, S.; Kostenuik, P.J.; Lacey, D.L.; et al. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J. Exp. Med. 2000, 192, 463–474. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Darnay, B.G.; Haridas, V.; Ni, J.; Moore, P.A.; Aggarwal, B.B. Characterization of the intracellular domain of receptor activator of NF-κB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-κB and c-Jun N-terminal kinase. J. Biol. Chem. 1998, 273, 20551–20555. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.; Azuma, S.; Tanaka, S.; Miyazaki, T.; Takaki, S.; Takatsu, K.; Nakao, K.; Nakamura, K.; Katsuki, M.; Yamamoto, T.; et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 1999, 4, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor kB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef]
- Makris, C.; Godfrey, V.L.; Krähn-Senftleben, G.; Takahashi, T.; Roberts, J.L.; Schwarz, T.; Feng, L.; Johnson, R.S.; Karin, M. Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 2000, 5, 969–979. [Google Scholar] [CrossRef]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israël, A.; Rajewsky, K.; Pasparakis, M. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 2000, 5, 981–992. [Google Scholar] [CrossRef]
- Ruocco, M.G.; Maeda, S.; Park, J.M.; Lawrence, T.; Hsu, L.C.; Cao, Y.; Schett, G.; Wagner, E.F.; Karin, M. IkB kinase (IKK)β, but not IKKα, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J. Exp. Med. 2005, 201, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.E.; Dai, S.; Foglia, D.; Alhawagri, M.; Vacher, J.; Pasparakis, M.; Abu-Amer, Y. Defective osteoclastogenesis by IKKβ-null precursors is a result of receptor activator of NF-κB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J. Biol. Chem. 2008, 283, 24546–24553. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Aoki, K.; Saito, H.; D’Acquisto, F.; May, M.J.; Nakamura, I.; Sudo, T.; Kojima, T.; Okamoto, F.; Fukushima, H.; et al. Selective inhibition of NF-κB blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 2004, 10, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Hirayama, T.; Abbas, S.; Abu-Amer, Y. The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J. Biol. Chem. 2004, 279, 37219–37222. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Gutierrez, A.M.; Lotan, R.; Aggarwal, B.B. N-(4-hydroxyphenyl) retinamide inhibits invasion, suppresses osteoclastogenesis, and potentiates apoptosis through down-regulation of IκBα kinase and nuclear factor-κB-regulated gene products. Cancer Res. 2005, 65, 9555–9565. [Google Scholar] [CrossRef]
- Takatsuna, H.; Asagiri, M.; Kubota, T.; Oka, K.; Osada, T.; Sugiyama, C.; Saito, H.; Aoki, K.; Ohya, K.; Takayanagi, H.; et al. Inhibition of RANKL-induced osteoclastogenesis by (-)-DHMEQ, a novel NF-κB inhibitor, through downregulation of NFATc1. J. Bone Miner. Res. 2005, 20, 653–662. [Google Scholar] [CrossRef]
- Vaira, S.; Alhawagri, M.; Anwisye, I.; Kitaura, H.; Faccio, R.; Novack, D.V. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J. Clin. Investig. 2008, 118, 2088–2097. [Google Scholar] [CrossRef]
- Novack, D.V.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S.L. The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef]
- Aya, K.; Alhawagri, M.; Hagen-Stapleton, A.; Kitaura, H.; Kanagawa, O.; Novack, D.V. NF-κB-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J. Clin. Investig. 2005, 115, 1848–1854. [Google Scholar] [CrossRef]
- Chaisson, M.L.; Branstetter, D.G.; Derry, J.M.; Armstrong, A.P.; Tometsko, M.E.; Takeda, K.; Akira, S.; Dougall, W.C. Osteoclast differentiation is impaired in the absence of inhibitor of κB kinase α. J. Biol. Chem. 2004, 279, 54841–54848. [Google Scholar] [CrossRef]
- Vaira, S.; Johnson, T.; Hirbe, A.C.; Alhawagri, M.; Anwisye, I.; Sammut, B.; O’Neal, J.; Zou, W.; Weilbaecher, K.N.; Faccio, R.; et al. RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Zarei, A.; Yang, C.; Gibbs, J.; Davis, J.L.; Ballard, A.; Zeng, R.; Cox, L.; Veis, D.J. Manipulation of the alternative NF-κB pathway in mice has sexually dimorphic effects on bone. JBMR Plus 2018, 3, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Soysa, N.S.; Alles, N.; Weih, D.; Lovas, A.; Mian, A.H.; Shimokawa, H.; Yasuda, H.; Weih, F.; Jimi, E.; Ohya, K.; et al. The pivotal role of the alternative NF-κB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J. Bone Miner. Res. 2010, 25, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Fukushima, H.; Nakao, K.; Shin, M.; Yasuda, H.; Weih, F.; Doi, T.; Aoki, K.; Alles, N.; Ohya, K.; et al. Processing of the NF-κB2 precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. J. Bone Miner. Res. 2010, 25, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, R.; Fukushima, H.; Osawa, K.; Maruyama, T.; Yasuda, H.; Weih, F.; Doi, T.; Maki, K.; Jim, E. RelB-induced expression of Cot, an MAP3K family member, rescues RANKL-induced osteoclastogenesis in alymphoplasia mice by promoting NF-κB2 processing by IKKα. J. Biol. Chem. 2014, 289, 7349–7361. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Zaka, M.; Abbasi, B.H.; Durdagi, S. Novel tumor necrosis factor-α (TNF-α) inhibitors from small molecule library screening for their therapeutic activity profiles against rheumatoid arthritis using target-driven approaches and binary QSAR models. Biomol. Struct. Dyn. 2019, 37, 2464–2476. [Google Scholar] [CrossRef]
- Tomita, T.; Takeuchi, E.; Tomita, N.; Morishita, R.; Kaneko, M.; Yamamoto, K.; Nakase, T.; Seki, H.; Kato, K.; Kaneda, Y.; et al. Suppressed severity of collagen-induced arthritis by in vivo transfection of nuclear factor κB decoy oligodeoxynucleotides as a gene therapy. Arthritis Rheum. 1999, 42, 2532–2542. [Google Scholar] [CrossRef]
- Tak, P.P.; Gerlag, D.M.; Aupperle, K.R.; van de Geest, D.A.; Overbeek, M.; Bennett, B.L.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. Inhibitor of nuclear factor κB kinase β is a key regulator of synovial inflammation. Arthritis Rheum. 2001, 44, 1897–1907. [Google Scholar] [CrossRef]
- Wen, D.; Nong, Y.; Morgan, J.G.; Gangurde, P.; Bielecki, A.; Dasilva, J.; Keaveney, M.; Cheng, H.; Fraser, C.; Schopf, L.; et al. A selective small molecule IκB kinase β inhibitor blocks nuclear factor κB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. J. Pharmacol. Exp. Ther. 2006, 317, 989–1001. [Google Scholar] [CrossRef]
- Schopf, L.; Savinainen, A.; Anderson, K.; Kujawa, J.; DuPont, M.; Silva, M.; Siebert, E.; Chandra, S.; Morgan, J.; Gangurde, P.; et al. IKKβ inhibition protects against bone and cartilage destruction in a rat model of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 3163–3173. [Google Scholar] [CrossRef]
- Gillooly, K.M.; Pattoli, M.A.; Taylor, T.L.; Chen, L.; Cheng, L.; Gregor, K.R.; Whitney, G.S.; Susulic, V.; Watterson, S.H.; Kempson, J.; et al. Periodic, partial inhibition of IκB Kinase β-mediated signaling yields therapeutic benefit in preclinical models of rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2009, 331, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Imai, K.; Asamitsu, K.; Waguri-Nagaya, Y.; Otsuka, T.; Okamoto, T. Inhibition of inflammatory cytokine production from rheumatoid synovial fibroblasts by a novel IκB kinase inhibitor. J. Pharmacol. Exp. Ther. 2010, 333, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bao, J.; Zeng, J.; Yan, A.; Zhao, C.; Shu, Q. Iguratimod: A valuable remedy from the Asia Pacific region for ameliorating autoimmune diseases and protecting bone physiology. Bone Res. 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Mimori, T.; Harigai, M.; Atsumi, T.; Fujii, T.; Kuwana, M.; Matsuno, H.; Momohara, S.; Takei, S.; Tamura, N.; Takasaki, Y.; et al. Safety and effectiveness of iguratimod in patients with rheumatoid arthritis: Final report of a 52-week, multicenter postmarketing surveillance study. Mod. Rheumatol. 2019, 29, 314–323. [Google Scholar] [CrossRef]
- Xiao, W.; Guo, J.P.; Li, C.; Ye, H.; Wei, W.; Zou, Y.; Dai, L.; Li, Z.; Zhang, M.; Li, X.; et al. Genetic predictors of efficacy and toxicity of iguratimod in patients with rheumatoid arthritis. Pharmacogenomics 2018, 19, 383–392. [Google Scholar] [CrossRef]
- Funk, J.L.; Frye, J.B.; Oyarzo, J.N.; Kuscuoglu, N.; Wilson, J.; McCaffrey, G.; Stafford, G.; Chen, G.; Lantz, R.C.; Jolad, S.D.; et al. Efficacy and mechanism of action of turmeric supplements in the treatment of experimental arthritis. Arthritis Rheum. 2006, 54, 3452–3464. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, A.R.; Lee, J.M.; Kim, S.N.; Choi, J.H.; Kim, D.K.; Kim, J.H.; Kim, B.; Her, E.; Yang, Y.M.; et al. A mixture of Trachelospermi caulis and Moutan cortex radicis extracts suppresses collagen-induced arthritis in mice by inhibiting NF-κB and AP-1. J. Pharm. Pharmacol. 2012, 64, 420–429. [Google Scholar] [CrossRef]
- Kong, X.; Liu, C.; Zhang, C.; Zhao, J.; Wang, J.; Wan, H.; Zhu, H.; Zhang, P.; Chen, W.; Xiao, Y.; et al. The suppressive effects of Saposhnikovia divaricata (Fangfeng) chromone extract on rheumatoid arthritis via inhibition of nuclear factor-κB and mitogen activated proteinkinases activation on collagen-induced arthritis model. J. Ethnopharmacol. 2013, 148, 842–850. [Google Scholar] [CrossRef]
- Min, H.K.; Kim, S.M.; Baek, S.Y.; Woo, J.W.; Park, J.S.; Cho, M.L.; Lee, J.; Kwok, S.K.; Kim, S.W.; Park, S.H. Anthocyanin Extracted from Black Soybean Seed Coats Prevents Autoimmune Arthritis by Suppressing the Development of Th17 Cells and Synthesis of Proinflammatory Cytokines by Such Cells, via Inhibition of NF-κB. PLoS ONE 2015, 10, e0138201. [Google Scholar] [CrossRef]
- Xu, W.; Huang, M.; Zhang, Y.; Li, H.; Zheng, H.; Yu, L.; Chu, K. Extracts of Bauhinia championii (Benth.) Benth. inhibit NF-κB-signaling in a rat model of collagen-induced arthritis and primary synovial cells. J. Ethnopharmacol. 2016, 185, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.X.; Liu, Y.; Zhou, W.; Li, H.W.; Yang, J.; Chen, Z.B. Shikonin inhibits TNF-α production through suppressing PKC-NF-κB-dependent decrease of IL-10 in rheumatoid arthritis-like cell model. J. Nat. Med. 2017, 71, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.B.; Yuan, Y.J.; Zhang, Q.H.; Li, H.; Dai, J.L.; Min, J.K. Salvianolic acid B suppresses inflammatory mediator levels by downregulating NF-κB in a rat modelof rheumatoid arthritis. Med. Sci. Monit. 2018, 24, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Uttra, A.M.; Shahzad, M.; Shabbir, A.; Jahan, S. Ephedra gerardiana aqueous ethanolic extract and fractions attenuate freund complete adjuvant induced arthritis in Sprague Dawley rats by downregulating PGE2, COX2, IL-1β, IL-6, TNF-α, NF-κB and upregulating IL-4 and IL-10. J. Ethnopharmacol. 2018, 224, 482–496. [Google Scholar] [CrossRef]
- Morin, C.; Blier, P.U.; Fortin, S. Eicosapentaenoic acid and docosapentaenoic acid monoglycerides are more potent than docosahexaenoic acid monoglyceride to resolve inflammation in a rheumatoid arthritis model. Arthritis Res. Ther. 2015, 17, 142. [Google Scholar] [CrossRef]
- Proudman, S.M.; Cleland, L.G.; Metcalf, R.G.; Sullivan, T.R.; Spargo, L.D.; James, M.J. Plasma n-3 fatty acids and clinical outcomes in recent-onset rheumatoid arthritis. Br. J. Nutr. 2015, 114, 885–890. [Google Scholar] [CrossRef]
- Rosillo, M.A.; Sánchez-Hidalgo, M.; Sánchez-Fidalgo, S.; Aparicio-Soto, M.; Villegas, I.; Alarcón-de-la-Lastra, C. Dietary extra-virgin olive oil prevents inflammatory response and cartilage matrix degradation in murine collagen-induced arthritis. Eur. J. Nutr. 2016, 55, 315–325. [Google Scholar] [CrossRef]
- Maijer, K.I.; Noort, A.R.; de Hair, M.J.; van der Leij, C.; van Zoest, K.P.; Choi, I.Y.; Gerlag, D.M.; Maas, M.; Tak, P.P.; Tas, S.W. Nuclear Factor-κB-inducing Kinase Is Expressed in Synovial Endothelial Cells in Patients with Early Arthritis and Correlates with Markers of Inflammation: A Prospective Cohort Study. J. Rheumatol. 2015, 42, 1573–1581. [Google Scholar] [CrossRef]
- Noort, A.R.; van Zoest, K.P.; Weijers, E.M.; Koolwijk, P.; Maracle, C.X.; Novack, D.V.; Siemerink, M.J.; Schlingemann, R.O.; Tak, P.P.; Tas, S.W. NF-κB-inducing kinase is a key regulator of inflammation-induced and tumour-associated angiogenesis. J. Pathol. 2014, 234, 375–385. [Google Scholar] [CrossRef]
- Yang, C.; McCoy, K.; Davis, J.L.; Schmidt-Supprian, M.; Sasaki, Y.; Faccio, R.; Novack, D.V. NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS ONE 2010, 5, e15383. [Google Scholar] [CrossRef]
- Wang, H.; Marsters, S.A.; Baker, T.; Chan, B.; Lee, W.P.; Fu, L.; Tumas, D.; Yan, M.; Dixit, V.M.; Ashkenazi, A.; et al. TACI-ligand interactions are required for T cell activation and collagen-induced arthritis in mice. Nat. Immunol. 2001, 2, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Jiang, T.; Chaurasiya, B.; Zhou, Y.; Yu, J.; Wen, J.; Shen, Y.; Ye, X.; Webster, T.J. Advances in nanomedicine for the treatment of ankylosing spondylitis. Int. J. Nanomed. 2019, 14, 8521–8542. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.J.; Maksymowych, W.P. The Pathogenesis of Ankylosing Spondylitis: An Update. Curr. Rheumatol. Rep. 2019, 21, 58. [Google Scholar] [CrossRef] [PubMed]
- Callhoff, J.; Sieper, J.; Weiß, A.; Zink, A.; Listing, J. Efficacy of TNFα blockers in patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis: A meta-analysis. Ann. Rheum. Dis. 2015, 74, 1241–1248. [Google Scholar] [CrossRef]
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Gigante, I.; Colucci, S.; Grano, M. Periodontal disease: Linking the primary inflammation to bone loss. Clin. Dev. Immunol. 2013, 2013, 503754. [Google Scholar] [CrossRef]
- Li, C.H.; Amar, S. Morphometric, histomorphometric, and microcomputed tomographic analysis of periodontal inflammatory lesions in a murine model. J. Periodontol. 2007, 78, 1120–1128. [Google Scholar] [CrossRef]
- Kure, K.; Sato, H.; Suzuki, J.I.; Itai, A.; Aoyama, N.; Izumi, Y. A novel IκB kinase inhibitor attenuates ligature-induced periodontal disease in mice. J. Periodontal. Res. 2019, 54, 164–173. [Google Scholar] [CrossRef]
- Katagiri, T.; Takahashi, N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis. 2002, 8, 147–159. [Google Scholar] [CrossRef]
- Canalis, E. Effects of tumor necrosis factor on bone formation in vitro. Endocrinology 1987, 121, 1596–1604. [Google Scholar] [CrossRef]
- Nakase, T.; Takaoka, K.; Masuhara, K.; Shimizu, K.; Yoshikawa, H.; Ochi, T. Interleukin-1β enhances and tumor necrosis factor-α inhibits bone morphogenetic protein-2-induced alkaline phosphatase activity in MC3T3-E1 osteoblastic cells. Bone 1997, 21, 17–21. [Google Scholar] [CrossRef]
- Gilbert, L.; He, X.; Farmer, P.; Boden, S.; Kozlowski, M.; Rubin, J.; Nanes, M.S. Inhibition of osteoblast differentiation by tumor necrosis factor-α. Endocrinology 2000, 141, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Nanes, M.S. Tumor necrosis factor-α: Molecular and cellular mechanisms in skeletal pathology. Gene 2003, 321, 1–15. [Google Scholar] [CrossRef]
- Yamazaki, M.; Fukushima, H.; Shin, M.; Katagiri, T.; Doi, T.; Takahashi, T.; Jimi, E. Tumor necrosis factor α represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-κB. J. Biol. Chem. 2009, 284, 35987–35995. [Google Scholar] [CrossRef]
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Srivastava, K.; Mansoori, M.N.; Trivedi, R.; Chattopadhyay, N.; Singh, D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: A new candidate in the pathogenesis of osteoporosis. PLoS ONE 2012, 7, e44552. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Jimi, E. The Role of BMP Signaling and NF-κB Signaling on osteoblastic differentiation, cancer development, and vascular diseases—Is the activation of NF-κB a friend or foe of BMP function? Vitam. Horm. 2015, 99, 145–170. [Google Scholar] [CrossRef]
- Li, Y.; Li, A.; Strait, K.; Zhang, H.; Nanes, M.S.; Weitzmann, M.N. Endogenous TNFα lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-κB. J. Bone Miner. Res. 2007, 22, 646–655. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Weitzmann, M.N. Zinc stimulates osteoblastogenesis and suppresses osteoclastogenesis by antagonizing NF-κB activation. Mol. Cell Biochem. 2011, 355, 179–186. [Google Scholar] [CrossRef]
- Tang, Y.; Xie, H.; Chen, J.; Geng, L.; Chen, H.; Li, X.; Hou, Y.; Lu, L.; Shi, S.; Zeng, X.; et al. Activated NF-κB in bone marrow mesenchymal stem cells from systemic lupus erythematosus patients inhibits osteogenic differentiation through downregulating Smad signaling. Stem. Cells Dev. 2013, 22, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Hirata-Tsuchiya, S.; Fukushima, H.; Katagiri, T.; Ohte, S.; Shin, M.; Nagano, K.; Aoki, K.; Morotomi, T.; Sugiyama, G.; Nakatomi, C.; et al. Inhibition of BMP2-induced bone formation by the p65 subunit of NF-κB via an interaction with Smad4. Mol. Endocrinol. 2014, 28, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Urata, M.; Kokabu, S.; Matsubara, T.; Sugiyama, G.; Nakatomi, C.; Takeuchi, H.; Hirata-Tsuchiya, S.; Aoki, K.; Tamura, Y.; Moriyama, Y.; et al. A peptide that blocks the interaction of NF-κB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J. Cell Physiol. 2018, 233, 7356–7366. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, A.L.; Larsen, M.J.; Brusgaard, K.; Novack, D.V.; Knudsen, P.J.; Schrøder, H.D.; Qiu, W.; Eckhardt, C.; McAlister, W.H.; Kassem, M.; et al. Neonatal high bone mass with first mutation of the NF-κB complex: Heterozygous de novo missense (p.Asp512Ser) RELA (Rela/p65). J. Bone Miner. Res. 2016, 31, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Fukushima, H.; Maruyama, T.; Kuroishi, K.N.; Osawa, K.; Nagano, K.; Aoki, K.; Weih, F.; Doi, T.; Zhang, M.; et al. Accumulation of p100, a precursor of NF-κB2, enhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol. Endocrinol. 2012, 26, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Li, Y.; Yin, X.; Dong, Y.; Xing, L.; Boyce, B.F. NF-κB RelB negatively regulates osteoblast differentiation and bone formation. J. Bone Miner. Res. 2014, 29, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Canalis, E. Notch and the skeleton. Mol. Cell Biol. 2010, 30, 886–896. [Google Scholar] [CrossRef]
- Zhang, H.; Hilton, M.J.; Anolik, J.H.; Welle, S.L.; Zhao, C.; Yao, Z.; Li, X.; Wang, Z.; Boyce, B.F.; Xing, L. NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-κB. J. Clin. Investig. 2014, 124, 3200–3214. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimi, E.; Takakura, N.; Hiura, F.; Nakamura, I.; Hirata-Tsuchiya, S. The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”? Cells 2019, 8, 1636. https://doi.org/10.3390/cells8121636
Jimi E, Takakura N, Hiura F, Nakamura I, Hirata-Tsuchiya S. The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”? Cells. 2019; 8(12):1636. https://doi.org/10.3390/cells8121636
Chicago/Turabian StyleJimi, Eijiro, Nana Takakura, Fumitaka Hiura, Ichiro Nakamura, and Shizu Hirata-Tsuchiya. 2019. "The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”?" Cells 8, no. 12: 1636. https://doi.org/10.3390/cells8121636
APA StyleJimi, E., Takakura, N., Hiura, F., Nakamura, I., & Hirata-Tsuchiya, S. (2019). The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”? Cells, 8(12), 1636. https://doi.org/10.3390/cells8121636