The Butterfly Effect of RNA Alterations on Transcriptomic Equilibrium



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. MicroRNAs: Small, But Powerful Post-Transcriptional Regulators
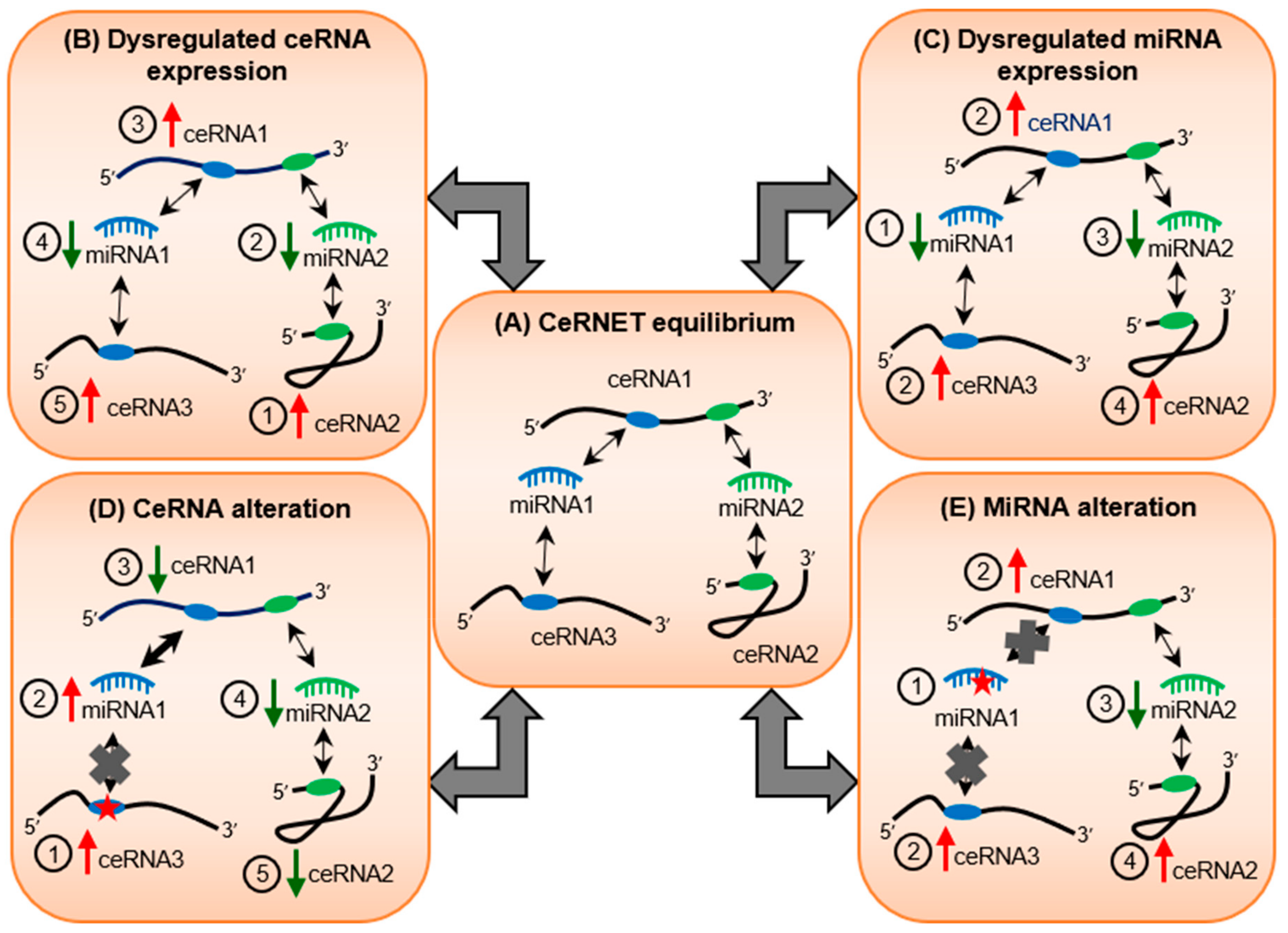
3. Webs of Co-Regulatory miRNA/Target Interactions May Cause Ripple Effects on Transcriptomic Equilibrium
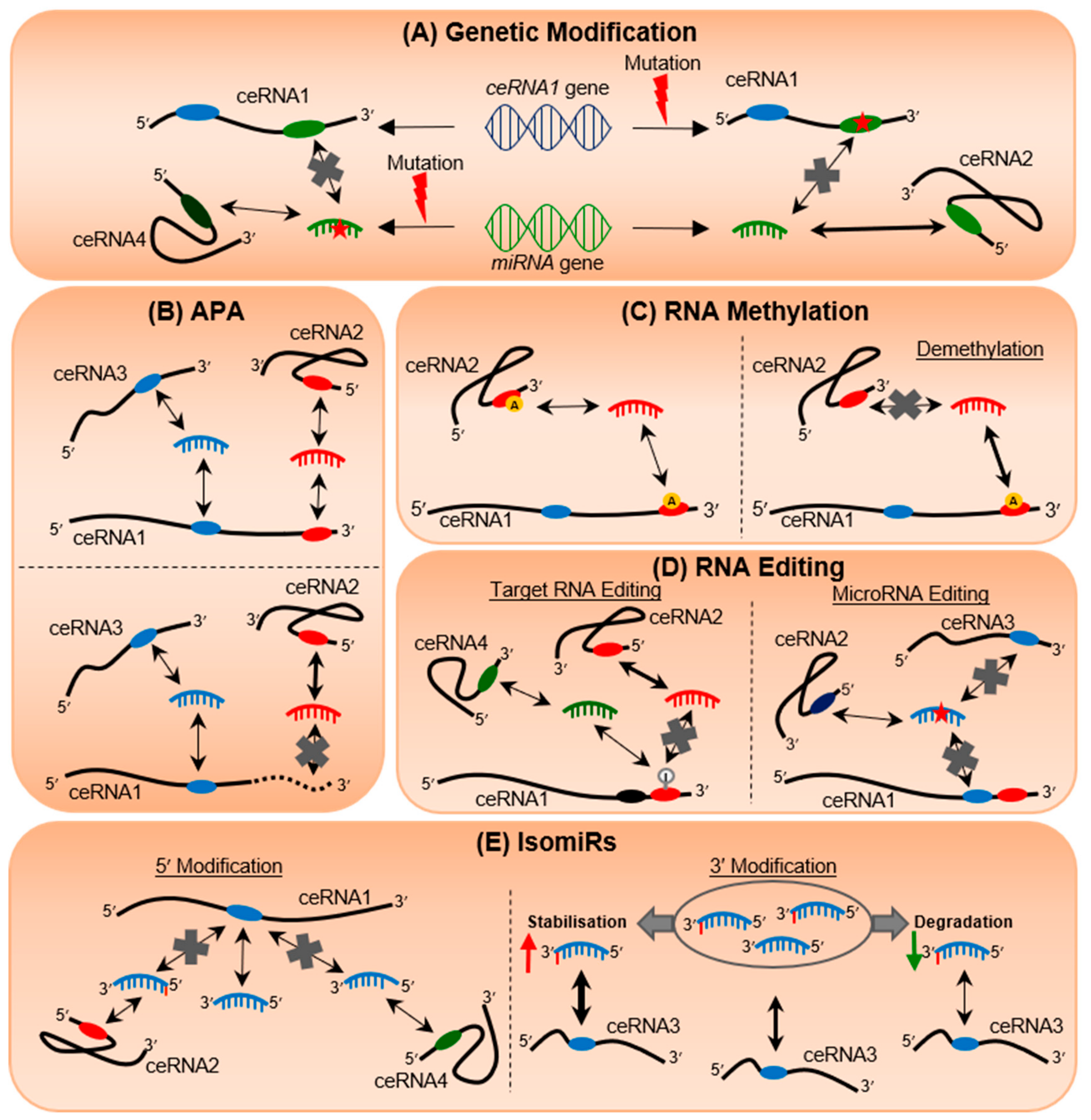
4. Genetic Mutations Can Alter Post-Transcriptional Regulation
5. 3′UTR Shortening Transforms the miRNA Regulatory Landscape
6. RNA Methylation Regulates Epitranscriptomic Plasticity
7. RNA Editing Rewires RNA Communication Networks
8. IsomiRs Add Tremendous Diversity to miRNA Species and Targetome Regulation
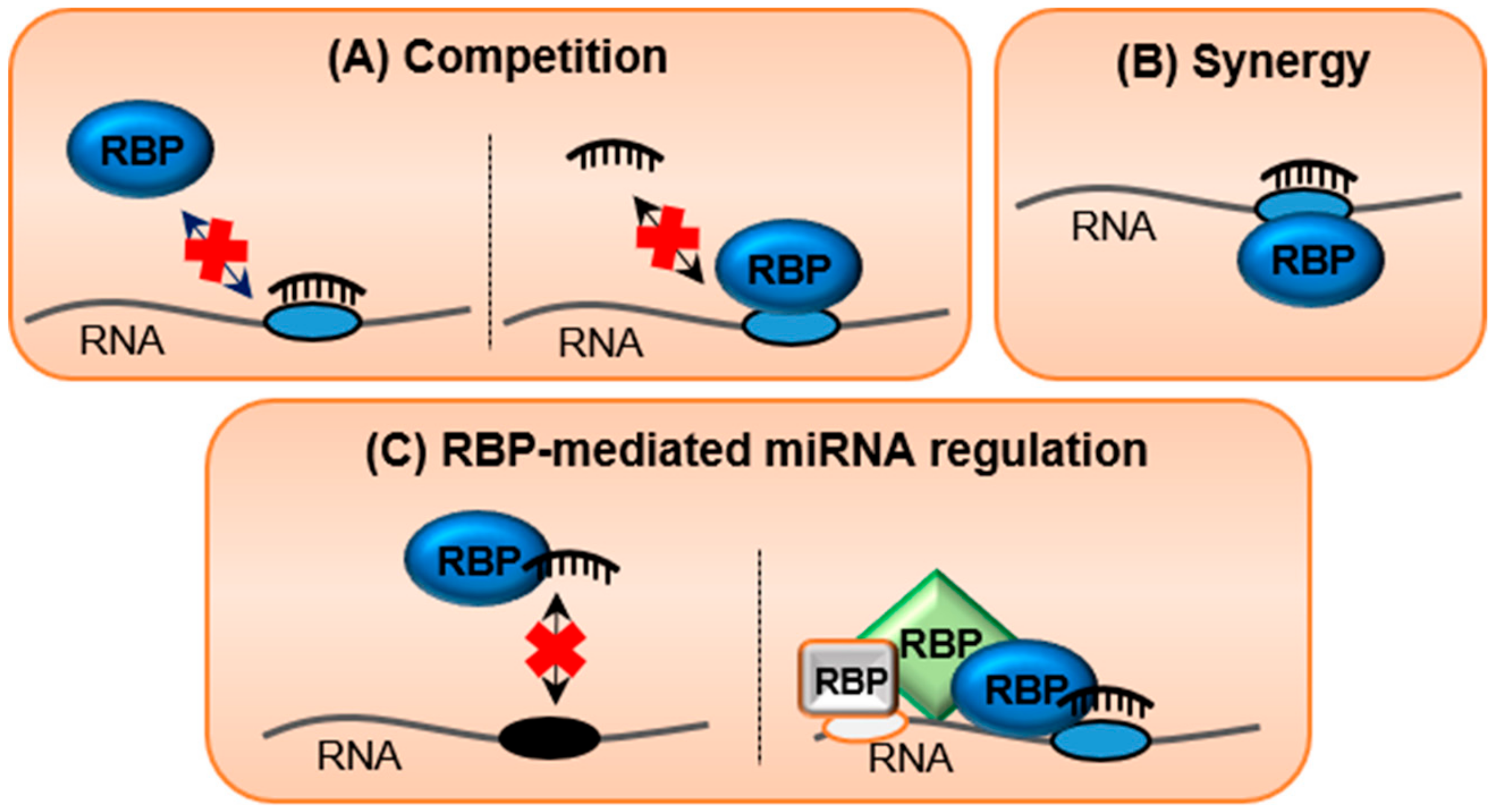
9. RNA-Binding Proteins (RBPs), the Silent Guardians of the Transcriptome
10. Crosstalk between RNA/RNA and RNA/Protein Interactions in Regulating Transcriptomic Equilibrium
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar]
- Smith, D.R.; Myint, T.; Goh, H.S. Over-expression of the c-myc proto-oncogene in colorectal carcinoma. Br. J. Cancer 1993, 68, 407–413. [Google Scholar] [CrossRef][Green Version]
- Rochlitz, C.F.; Herrmann, R.; de Kant, E. Overexpression and amplification of c-myc during progression of human colorectal cancer. Oncology 1996, 53, 448–454. [Google Scholar] [CrossRef]
- Wurth, L. Versatility of rna-binding proteins in cancer. Comp. Funct. Genom. 2012, 2012, 178525. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. Micrornas in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The c. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in c. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- rna genes mir15 and mir16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Su, J.L.; Chen, P.S.; Johansson, G.; Kuo, M.L. Function and regulation of let-7 family micrornas. Microrna 2012, 1, 34–39. [Google Scholar] [CrossRef]
- Hermeking, H. The mir-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef]
- Sha, H.H.; Wang, D.D.; Chen, D.; Liu, S.W.; Wang, Z.; Yan, D.L.; Dong, S.C.; Feng, J.F. Mir-138: A promising therapeutic target for cancer. Tumour. Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.H.; Tsao, C.J. Emerging role of microrna-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The mir-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Mehlich, D.; Garbicz, F.; Wlodarski, P.K. The emerging roles of the polycistronic mir-106b approximately 25 cluster in cancer—A comprehensive review. Biomed. Pharmacother. 2018, 107, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Song, M.L.; Min, H.; Hwang, I.; Baek, S.K.; Kwon, T.K.; Park, J.W. Mirna biogenesis-associated rnase iii nucleases drosha and dicer are upregulated in colorectal adenocarcinoma. Oncol. Lett. 2017, 14, 4379–4383. [Google Scholar] [CrossRef]
- Lai, H.H.; Li, J.N.; Wang, M.Y.; Huang, H.Y.; Croce, C.M.; Sun, H.L.; Lyu, Y.J.; Kang, J.W.; Chiu, C.F.; Hung, M.C.; et al. Hif-1alpha promotes autophagic proteolysis of dicer and enhances tumor metastasis. J. Clin. Investig. 2018, 128, 625–643. [Google Scholar] [CrossRef]
- Jansson, M.D.; Lund, A.H. Microrna and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary micrornas by the drosha-dgcr8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef]
- Zeng, Y.; Cullen, B.R. Structural requirements for pre-microrna binding and nuclear export by exportin 5. Nucleic Acids Res. 2004, 32, 4776–4785. [Google Scholar] [CrossRef]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for dicer-independent, ago2-mediated microrna biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. Trna-derived microrna modulates proliferation and the DNA damage response and is down-regulated in b cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microrna target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Composition of seed sequence is a major determinant of microrna targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef]
- Yekta, S.; Shih, I.H.; Bartel, D.P. Microrna-directed cleavage of hoxb8 mrna. Science 2004, 304, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian rnai. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of sirnas for mediating efficient rnai in drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human mirna interactome by clash reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. Mir-24 inhibits cell proliferation by targeting e2f2, myc, and other cell-cycle genes via binding to “seedless” 3’utr microrna recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef]
- Loeb, G.B.; Khan, A.A.; Canner, D.; Hiatt, J.B.; Shendure, J.; Darnell, R.B.; Leslie, C.S.; Rudensky, A.Y. Transcriptome-wide mir-155 binding map reveals widespread noncanonical microrna targeting. Mol. Cell 2012, 48, 760–770. [Google Scholar] [CrossRef]
- Park, J.H.; Shin, S.Y.; Shin, C. Non-canonical targets destabilize micrornas in human argonautes. Nucleic Acids Res. 2017, 45, 1569–1583. [Google Scholar] [CrossRef]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of cds-located mirna target sites suggests that they can effectively inhibit translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. Microrna-10a binds the 5’utr of ribosomal protein mrnas and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: Micrornas can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. Micrornas to nanog, oct4 and sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mrnas regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular rnas are a large class of animal rnas with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microrna activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microrna by a herpesvirus saimiri noncoding rna. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor pten by competing endogenous mrnas. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microrna-mediated network of rna-rna interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding rna controls muscle differentiation by functioning as a competing endogenous rna. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H. Redefining microrna targets. Curr. Biol. 2009, 19, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A cerna hypothesis: The rosetta stone of a hidden rna language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ala, U.; Karreth, F.A.; Bosia, C.; Pagnani, A.; Taulli, R.; Leopold, V.; Tay, Y.; Provero, P.; Zecchina, R.; Pandolfi, P.P. Integrated transcriptional and competitive endogenous rna networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. USA 2013, 110, 7154–7159. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. Emerging roles for natural microrna sponges. Curr. Biol. 2010, 20, R858–R861. [Google Scholar] [CrossRef]
- Bosson, A.D.; Zamudio, J.R.; Sharp, P.A. Endogenous mirna and target concentrations determine susceptibility to potential cerna competition. Mol. Cell 2014, 56, 347–359. [Google Scholar] [CrossRef]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute divides its rna guide into domains with distinct functions and rna-binding properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of microrna levels, target-site complementarity, and cooperativity on competing endogenous rna-regulated gene expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef]
- Sanchez-Mejias, A.; Tay, Y. Competing endogenous rna networks: Tying the essential knots for cancer biology and therapeutics. J. Hematol. Oncol. 2015, 8, 30. [Google Scholar] [CrossRef]
- Shuwen, H.; Qing, Z.; Yan, Z.; Xi, Y. Competitive endogenous rna in colorectal cancer: A systematic review. Gene 2018, 645, 157–162. [Google Scholar] [CrossRef]
- Abdollahzadeh, R.; Daraei, A.; Mansoori, Y.; Sepahvand, M.; Amoli, M.M.; Tavakkoly-Bazzaz, J. Competing endogenous rna (cerna) cross talk and language in cerna regulatory networks: A new look at hallmarks of breast cancer. J. Cell Physiol. 2019, 234, 10080–10100. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lin, Y.; Chen, J.; Shen, B. Decoding competing endogenous rna networks for cancer biomarker discovery. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kotani, A.; Ha, D.; Schotte, D.; den Boer, M.L.; Armstrong, S.A.; Lodish, H.F. A novel mutation in the mir-128b gene reduces mirna processing and leads to glucocorticoid resistance of mll-af4 acute lymphocytic leukemia cells. Cell Cycle 2010, 9, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.A.; Liang, H.; Li, W.H. Human polymorphism at micrornas and microrna target sites. Proc. Natl. Acad. Sci. USA 2007, 104, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of snps in microrna genes and the snp effects on microrna target binding and biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Gu, H.; Zeng, Y.; Xia, Y.; Wang, Y.; Jing, Y.; Yang, L.; Wang, B. Hsa-mir-27a genetic variant contributes to gastric cancer susceptibility through affecting mir-27a and target gene expression. Cancer Sci. 2010, 101, 2241–2247. [Google Scholar] [CrossRef]
- Saetrom, P.; Biesinger, J.; Li, S.M.; Smith, D.; Thomas, L.F.; Majzoub, K.; Rivas, G.E.; Alluin, J.; Rossi, J.J.; Krontiris, T.G.; et al. A risk variant in an mir-125b binding site in bmpr1b is associated with breast cancer pathogenesis. Cancer Res. 2009, 69, 7459–7465. [Google Scholar] [CrossRef]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.U.; Straka, E.; Su, L.; Burki, E.A.; et al. A snp in a let-7 microrna complementary site in the kras 3’ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008, 68, 8535–8540. [Google Scholar] [CrossRef]
- Acunzo, M.; Romano, G.; Nigita, G.; Veneziano, D.; Fattore, L.; Lagana, A.; Zanesi, N.; Fadda, P.; Fassan, M.; Rizzotto, L.; et al. Selective targeting of point-mutated kras through artificial micrornas. Proc. Natl. Acad. Sci. USA 2017, 114, E4203–E4212. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mrna polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3’utrs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.R.; Bos, A.; Diosdado, B.; Rooijers, K.; Elkon, R.; Bolijn, A.S.; Carvalho, B.; Meijer, G.A.; Agami, R. Alternative cleavage and polyadenylation during colorectal cancer development. Clin. Cancer Res. 2012, 18, 5256–5266. [Google Scholar] [CrossRef]
- Andres, S.F.; Williams, K.N.; Plesset, J.B.; Headd, J.J.; Mizuno, R.; Chatterji, P.; Lento, A.A.; Klein-Szanto, A.J.; Mick, R.; Hamilton, K.E.; et al. Imp1 3’ utr shortening enhances metastatic burden in colorectal cancer. Carcinogenesis 2019, 40, 569–579. [Google Scholar] [CrossRef]
- Sun, M.; Ding, J.; Li, D.; Yang, G.; Cheng, Z.; Zhu, Q. Nudt21 regulates 3’-utr length and microrna-mediated gene silencing in hepatocellular carcinoma. Cancer Lett. 2017, 410, 158–168. [Google Scholar] [CrossRef]
- Tan, S.; Li, H.; Zhang, W.; Shao, Y.; Liu, Y.; Guan, H.; Wu, J.; Kang, Y.; Zhao, J.; Yu, Q.; et al. Nudt21 negatively regulates psmb2 and cxxc5 by alternative polyadenylation and contributes to hepatocellular carcinoma suppression. Oncogene 2018, 37, 4887–4900. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Yan, W.; Han, P.; Liu, J.; Gong, J.; Li, D.; Ding, X.; Wang, H.; Lin, Z.; et al. Cfim25 inhibits hepatocellular carcinoma metastasis by suppressing the p38 and jnk/c-jun signaling pathways. Oncotarget 2018, 9, 11783–11793. [Google Scholar] [CrossRef]
- Masamha, C.P.; Xia, Z.; Yang, J.; Albrecht, T.R.; Li, M.; Shyu, A.B.; Li, W.; Wagner, E.J. Cfim25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014, 510, 412–416. [Google Scholar] [CrossRef]
- Han, T.; Kim, J.K. Driving glioblastoma growth by alternative polyadenylation. Cell Res. 2014, 24, 1023–1024. [Google Scholar] [CrossRef][Green Version]
- Akman, H.B.; Oyken, M.; Tuncer, T.; Can, T.; Erson-Bensan, A.E. 3’utr shortening and egf signaling: Implications for breast cancer. Hum. Mol. Genet. 2015, 24, 6910–6920. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Weng, T.; Ko, J.; Chen, N.Y.; Xiang, Y.; Volcik, K.; Han, L.; Blackburn, M.R.; Lu, X. Suppression of cleavage factor im 25 promotes the proliferation of lung cancer cells through alternative polyadenylation. Biochem. Biophys. Res. Commun. 2018, 503, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Alley, T.L.; Wright, S.M.; Kamdar, S.; Schott, W.; Wilpan, R.Y.; Mills, K.D.; Graber, J.H. Global changes in processing of mrna 3’ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009, 69, 9422–9430. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Li, Y.Q.; Liu, N.; He, Q.M.; Tang, X.R.; Wen, X.; Yang, X.J.; Sun, Y.; Ma, J.; Tang, L.L. Differential genome-wide profiling of alternative polyadenylation sites in nasopharyngeal carcinoma by high-throughput sequencing. J. Biomed. Sci. 2018, 25, 74. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, A.; Plass, M.; Dossing, K.; Knigge, U.; Kjaer, A.; Krogh, A.; Friis-Hansen, L. Alternative polyadenylation of tumor suppressor genes in small intestinal neuroendocrine tumors. Front. Endocrinol. (Lausanne) 2014, 5, 46. [Google Scholar] [CrossRef][Green Version]
- Zhu, Z.J.; Huang, P.; Chong, Y.X.; Kang, L.X.; Huang, X.; Zhu, Z.X.; Nie, L. Microrna-181a promotes proliferation and inhibits apoptosis by suppressing cfim25 in osteosarcoma. Mol. Med. Rep. 2016, 14, 4271–4278. [Google Scholar] [CrossRef]
- Misiewicz-Krzeminska, I.; Sarasquete, M.E.; Vicente-Duenas, C.; Krzeminski, P.; Wiktorska, K.; Corchete, L.A.; Quwaider, D.; Rojas, E.A.; Corral, R.; Martin, A.A.; et al. Post-transcriptional modifications contribute to the upregulation of cyclin d2 in multiple myeloma. Clin. Cancer Res. 2016, 22, 207–217. [Google Scholar] [CrossRef]
- Akman, B.H.; Can, T.; Erson-Bensan, A.E. Estrogen-induced upregulation and 3’-utr shortening of cdc6. Nucleic Acids Res. 2012, 40, 10679–10688. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mrnas with shortened 3’ untranslated regions and fewer microrna target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Masamha, C.P.; Wagner, E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 2018, 39, 2–10. [Google Scholar] [CrossRef]
- Chen, R.W.; Bemis, L.T.; Amato, C.M.; Myint, H.; Tran, H.; Birks, D.K.; Eckhardt, S.G.; Robinson, W.A. Truncation in ccnd1 mrna alters mir-16-1 regulation in mantle cell lymphoma. Blood 2008, 112, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mrna stability factor hur inhibits microrna-16 targeting of cox-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhu, X.; Wang, H.; Jin, X. A dynamic interplay between alternative polyadenylation and microrna regulation: Implications for cancer (review). Int. J. Oncol. 2013, 43, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microrna let-7 represses the hmga2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef]
- Park, H.J.; Ji, P.; Kim, S.; Xia, Z.; Rodriguez, B.; Li, L.; Su, J.; Chen, K.; Masamha, C.P.; Baillat, D.; et al. 3’ utr shortening represses tumor-suppressor genes in trans by disrupting cerna crosstalk. Nat. Genet. 2018, 50, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3’ utrs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef]
- Li, L.; Wang, D.; Xue, M.; Mi, X.; Liang, Y.; Wang, P. 3’utr shortening identifies high-risk cancers with targeted dysregulation of the cerna network. Sci. Rep. 2014, 4, 5406. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger rna from novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Modified nucleosides and bizarre 5’-termini in mouse myeloma mrna. Nature 1975, 255, 28–33. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E.; Friderici, K.; Rottman, F. The methylated constituents of l cell messenger rna: Evidence for an unusual cluster at the 5’ terminus. Cell 1975, 4, 387–394. [Google Scholar] [CrossRef]
- Dubin, D.T.; Taylor, R.H. The methylation state of poly a-containing messenger rna from cultured hamster cells. Nucleic Acids Res. 1975, 2, 1653–1668. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5’ terminus of hela cell messenger rna. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Lewis, C.J.; Pan, T.; Kalsotra, A. Rna modifications and structures cooperate to guide rna-protein interactions. Nat. Rev. Mol. Cell Biol. 2017, 18, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Camper, S.A.; Albers, R.J.; Coward, J.K.; Rottman, F.M. Effect of undermethylation on mrna cytoplasmic appearance and half-life. Mol. Cell Biol. 1984, 4, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. Rna n6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through ythdf2-dependent posttranscriptional silencing of socs2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The n(6)-methyladenosine (m(6)a)-forming enzyme mettl3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, X.; Cao, C.; Gao, Y.; Zhang, S.; Yang, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Ye, L. Hbxip-elevated methyltransferase mettl3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018, 415, 11–19. [Google Scholar] [CrossRef]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. Mrna circularization by mettl3-eif3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef]
- Pinello, N.; Sun, S.; Wong, J.J. Aberrant expression of enzymes regulating m(6)a mrna methylation: Implication in cancer. Cancer Biol. Med. 2018, 15, 323–334. [Google Scholar]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear rna is a major substrate of the obesity-associated fto. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. Rna n6-methyladenosine demethylase fto promotes breast tumor progression through inhibiting bnip3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. Fto plays an oncogenic role in acute myeloid leukemia as a n(6)-methyladenosine rna demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, W.; Jiang, Y.; Wang, X.; Liu, Y.; Liu, X. Fto expression is associated with the occurrence of gastric cancer and prognosis. Oncol. Rep. 2017, 38, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the rna m(6)a reader ythdf2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell 2019, 25, 137–148.e136. [Google Scholar] [CrossRef] [PubMed]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the epitranscriptome: Deciphering the function of rna modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mrna methylation reveals enrichment in 3’ utrs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6a residues are in the last exons, allowing the potential for 3’ utr regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef]
- Chen, T.; Hao, Y.J.; Zhang, Y.; Li, M.M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. M(6)a rna methylation is regulated by micrornas and promotes reprogramming to pluripotency. Cell Stem Cell 2015, 16, 289–301. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m(6)am in the 5’ cap controls mrna stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. Hnrnpa2b1 is a mediator of m(6)a-dependent nuclear rna processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Berulava, T.; Rahmann, S.; Rademacher, K.; Klein-Hitpass, L.; Horsthemke, B. N6-adenosine methylation in mirnas. PLoS ONE 2015, 10, e0118438. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct methylation levels of mature micrornas in gastrointestinal cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef] [PubMed]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Hobartner, C.; Sloan, K.E.; Bohnsack, M.T. Human mettl16 is a n(6)-methyladenosine (m(6)a) methyltransferase that targets pre-mrnas and various non-coding rnas. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef]
- Huang, L.; Ashraf, S.; Wang, J.; Lilley, D.M. Control of box c/d snornp assembly by n(6)-methylation of adenine. EMBO Rep. 2017, 18, 1631–1645. [Google Scholar] [CrossRef]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N(6)-methyladenosine modification in a long noncoding rna hairpin predisposes its conformation to protein binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef]
- He, Y.; Hu, H.; Wang, Y.; Yuan, H.; Lu, Z.; Wu, P.; Liu, D.; Tian, L.; Yin, J.; Jiang, K.; et al. Alkbh5 inhibits pancreatic cancer motility by decreasing long non-coding rna kcnk15-as1 methylation. Cell Physiol. Biochem. 2018, 48, 838–846. [Google Scholar] [CrossRef]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by rna editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Maas, S.; Rich, A. Changing genetic information through rna editing. Bioessays 2000, 22, 790–802. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Lin, C.H.; Chan, T.H.; Chow, R.K.; Song, Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding rna editing of azin1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, H. When micrornas meet rna editing in cancer: A nucleotide change can make a difference. Bioessays 2018, 40. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-i rna editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human cancer tissues exhibit reduced a-to-i editing of mirnas coupled with elevated editing of their targets. Nucleic Acids Res. 2018, 46, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated rna editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-i rna editing contributes to proteomic diversity in cancer. Cancer Cell 2018, 33, 817–828.e817. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The genomic landscape and clinical relevance of a-to-i rna editing in human cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of rna-seq data reveals extensive rna editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef]
- Bass, B.L.; Nishikura, K.; Keller, W.; Seeburg, P.H.; Emeson, R.B.; O’Connell, M.A.; Samuel, C.E.; Herbert, A. A standardized nomenclature for adenosine deaminases that act on rna. RNA 1997, 3, 947–949. [Google Scholar]
- Liu, X.; Fu, Y.; Huang, J.; Wu, M.; Zhang, Z.; Xu, R.; Zhang, P.; Zhao, S.; Liu, L.; Jiang, H. Adar1 promotes the epithelial-to-mesenchymal transition and stem-like cell phenotype of oral cancer by facilitating oncogenic microrna maturation. J. Exp. Clin. Cancer Res. 2019, 38, 315. [Google Scholar] [CrossRef]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific rna processing produces apolipoprotein-b48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Shigeyasu, K.; Okugawa, Y.; Toden, S.; Miyoshi, J.; Toiyama, Y.; Nagasaka, T.; Takahashi, N.; Kusunoki, M.; Takayama, T.; Yamada, Y.; et al. Azin1 rna editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight 2018, 3, 99976. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Shigeyasu, K.; Okugawa, Y.; Yoshida, K.; Mori, Y.; Yano, S.; Noma, K.; Umeda, Y.; Kondo, Y.; Kishimoto, H.; et al. Activation of azin1 rna editing is a novel mechanism that promotes invasive potential of cancer-associated fibroblasts in colorectal cancer. Cancer Lett. 2019, 444, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, Y.; Toiyama, Y.; Shigeyasu, K.; Yamamoto, A.; Shigemori, T.; Yin, C.; Ichikawa, T.; Yasuda, H.; Fujikawa, H.; Yoshiyama, S.; et al. Enhanced azin1 rna editing and overexpression of its regulatory enzyme adar1 are important prognostic biomarkers in gastric cancer. J. Transl. Med. 2018, 16, 366. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, J.; Shi, X.; Feng, F.; Lau, K.W.; Chen, Y.; Chen, Y.; Jiang, L.; Cui, F.; Zhang, Y.; et al. Rna editing of azin1 induces the malignant progression of non-small-cell lung cancers. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef]
- Qin, Y.R.; Qiao, J.J.; Chan, T.H.; Zhu, Y.H.; Li, F.F.; Liu, H.; Fei, J.; Li, Y.; Guan, X.Y.; Chen, L. Adenosine-to-inosine rna editing mediated by adars in esophageal squamous cell carcinoma. Cancer Res. 2014, 74, 840–851. [Google Scholar] [CrossRef]
- Borchert, G.M.; Gilmore, B.L.; Spengler, R.M.; Xing, Y.; Lanier, W.; Bhattacharya, D.; Davidson, B.L. Adenosine deamination in human transcripts generates novel microrna binding sites. Hum. Mol. Genet. 2009, 18, 4801–4807. [Google Scholar] [CrossRef]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant a-to-i editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef]
- Wang, Q.; Hui, H.; Guo, Z.; Zhang, W.; Hu, Y.; He, T.; Tai, Y.; Peng, P.; Wang, L. Adar1 regulates arhgap26 gene expression through rna editing by disrupting mir-30b-3p and mir-573 binding. RNA 2013, 19, 1525–1536. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, C.S.; Varelas, X.; Monti, S. Altered rna editing in 3’ utr perturbs microrna-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 23226. [Google Scholar] [CrossRef]
- Gong, J.; Wu, Y.; Zhang, X.; Liao, Y.; Sibanda, V.L.; Liu, W.; Guo, A.Y. Comprehensive analysis of human small rna sequencing data provides insights into expression profiles and mirna editing. RNA Biol. 2014, 11, 1375–1385. [Google Scholar] [CrossRef]
- Gallego, A.; Hartasanchez, D.A.; Braso-Vives, M.; Garcia-Ramallo, E.; Lopez-Valenzuela, M.; Baena, N.; Guitart, M.; Fernandez-Bellon, H.; Kondova, I.; Bontrop, R.; et al. Rna editing independently occurs at three mir-376a-1 sites and may compromise the stability of the microrna hairpin. Gene 2017, 628, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microrna editing, expression and processing by adar2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of mirnas. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic characterization of a-to-i rna editing hotspots in micrornas across human cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Liu, C.; Liu, W.; Xiang, Y.; Diao, L.; Guo, A.Y.; Han, L. Lncediting: A database for functional effects of rna editing in lncrnas. Nucleic Acids Res. 2017, 45, D79–D84. [Google Scholar] [CrossRef]
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microrna profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. Micrornas and their isomirs function cooperatively to target common biological pathways. Genome Biol. 2011, 12, R126. [Google Scholar] [CrossRef]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. Isomirs--the overlooked repertoire in the dynamic micrornaome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Telonis, A.G.; Loher, P.; Jing, Y.; Londin, E.; Rigoutsos, I. Beyond the one-locus-one-mirna paradigm: Microrna isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res. 2015, 43, 9158–9175. [Google Scholar] [CrossRef]
- Telonis, A.G.; Magee, R.; Loher, P.; Chervoneva, I.; Londin, E.; Rigoutsos, I. Knowledge about the presence or absence of mirna isoforms (isomirs) can successfully discriminate amongst 32 tcga cancer types. Nucleic Acids Res. 2017, 45, 2973–2985. [Google Scholar] [CrossRef]
- Li, S.C.; Liao, Y.L.; Ho, M.R.; Tsai, K.W.; Lai, C.H.; Lin, W.C. Mirna arm selection and isomir distribution in gastric cancer. BMC Genom. 2012, 13 (Suppl. 1), S13. [Google Scholar] [CrossRef]
- Boele, J.; Persson, H.; Shin, J.W.; Ishizu, Y.; Newie, I.S.; Sokilde, R.; Hawkins, S.M.; Coarfa, C.; Ikeda, K.; Takayama, K.; et al. Papd5-mediated 3’ adenylation and subsequent degradation of mir-21 is disrupted in proliferative disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11467–11472. [Google Scholar] [CrossRef] [PubMed]
- Loher, P.; Londin, E.R.; Rigoutsos, I. Isomir expression profiles in human lymphoblastoid cell lines exhibit population and gender dependencies. Oncotarget 2014, 5, 8790–8802. [Google Scholar] [CrossRef] [PubMed]
- Magee, R.G.; Telonis, A.G.; Loher, P.; Londin, E.; Rigoutsos, I. Profiles of mirna isoforms and trna fragments in prostate cancer. Sci. Rep. 2018, 8, 5314. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Z.; Chen, P.; Wu, M. Tumor classification and biomarker discovery based on the 5’isomir expression level. BMC Cancer 2019, 19, 127. [Google Scholar] [CrossRef]
- Lan, C.; Peng, H.; McGowan, E.M.; Hutvagner, G.; Li, J. An isomir expression panel based novel breast cancer classification approach using improved mutual information. BMC Med. Genom. 2018, 11, 118. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Hackenberg, M.; de Menezes, R.; Misovic, B.; Wachalska, M.; Geldof, A.; Zini, N.; de Reijke, T.; Wurdinger, T.; Vis, A.; et al. Noninvasive prostate cancer detection by measuring mirna variants (isomirs) in urine extracellular vesicles. Oncotarget 2016, 7, 22566–22578. [Google Scholar] [CrossRef]
- Tan, G.C.; Chan, E.; Molnar, A.; Sarkar, R.; Alexieva, D.; Isa, I.M.; Robinson, S.; Zhang, S.; Ellis, P.; Langford, C.F.; et al. 5′ isomir variation is of functional and evolutionary importance. Nucleic Acids Res. 2014, 42, 9424–9435. [Google Scholar] [CrossRef]
- Salem, O.; Erdem, N.; Jung, J.; Munstermann, E.; Worner, A.; Wilhelm, H.; Wiemann, S.; Korner, C. The highly expressed 5’isomir of hsa-mir-140-3p contributes to the tumor-suppressive effects of mir-140 by reducing breast cancer proliferation and migration. BMC Genom. 2016, 17, 566. [Google Scholar] [CrossRef]
- Katoh, T.; Sakaguchi, Y.; Miyauchi, K.; Suzuki, T.; Kashiwabara, S.; Baba, T.; Suzuki, T. Selective stabilization of mammalian micrornas by 3’ adenylation mediated by the cytoplasmic poly(a) polymerase gld-2. Genes Dev. 2009, 23, 433–438. [Google Scholar] [CrossRef]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.; Tomaru, Y.; Nishibu, T.; Ukekawa, R.; Funakoshi, T.; Kurokawa, T.; Suzuki, H.; Hayashizaki, Y.; et al. A comprehensive survey of 3’ animal mirna modification events and a possible role for 3’ adenylation in modulating mirna targeting effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Bofill-De Ros, X.; Shao, T.J.; Jiang, M.; Li, K.; Villanueva, P.; Dai, L.; Gu, S. 3’ uridylation confers mirnas with non-canonical target repertoires. Mol. Cell 2019, 75, 511–522.e514. [Google Scholar] [CrossRef] [PubMed]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. Rna-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.N.; Sonenberg, N. Mammalian poly(a)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Thakor, N.; Smith, M.D.; Roberts, L.; Faye, M.D.; Patel, H.; Wieden, H.J.; Cate, J.H.D.; Holcik, M. Cellular mrna recruits the ribosome via eif3-pabp bridge to initiate internal translation. RNA Biol. 2017, 14, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ouyang, M.; Rao, J.N.; Zou, T.; Xiao, L.; Chung, H.K.; Wu, J.; Donahue, J.M.; Gorospe, M.; Wang, J.Y. Competition between rna-binding proteins celf1 and hur modulates myc translation and intestinal epithelium renewal. Mol. Biol. Cell 2015, 26, 1797–1810. [Google Scholar] [CrossRef]
- Hong, S. Rna binding protein as an emerging therapeutic target for cancer prevention and treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef]
- Bousquet-Antonelli, C.; Deragon, J.M. A comprehensive analysis of the la-motif protein superfamily. RNA 2009, 15, 750–764. [Google Scholar] [CrossRef]
- Hopkins, T.G.; Mura, M.; Al-Ashtal, H.A.; Lahr, R.M.; Abd-Latip, N.; Sweeney, K.; Lu, H.; Weir, J.; El-Bahrawy, M.; Steel, J.H.; et al. The rna-binding protein larp1 is a post-transcriptional regulator of survival and tumorigenesis in ovarian cancer. Nucleic Acids Res. 2016, 44, 1227–1246. [Google Scholar] [CrossRef]
- Mura, M.; Hopkins, T.G.; Michael, T.; Abd-Latip, N.; Weir, J.; Aboagye, E.; Mauri, F.; Jameson, C.; Sturge, J.; Gabra, H.; et al. Larp1 post-transcriptionally regulates mtor and contributes to cancer progression. Oncogene 2015, 34, 5025–5036. [Google Scholar] [CrossRef]
- Xie, C.; Huang, L.; Xie, S.; Xie, D.; Zhang, G.; Wang, P.; Peng, L.; Gao, Z. Larp1 predict the prognosis for early-stage and afp-normal hepatocellular carcinoma. J. Transl. Med. 2013, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lin, S.T.; Mi, Y.S.; Liu, Y.; Ma, Y.; Sun, H.M.; Peng, Z.H.; Fan, J.W. Overexpression of larp1 predicts poor prognosis of colorectal cancer and is expected to be a potential therapeutic target. Tumour Biol. 2016, 37, 14585–14594. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Scully, S.J., Jr.; Yan, W.; Bentley, B.; Mueller, J.; Brown, C.; Bigelow, C.; Schwartz, L.M. The novel lupus antigen related protein acheron enhances the development of human breast cancer. Int. J. Cancer 2012, 130, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, S.; Flemyng, E.; Shen, J.; Conte, M.R.; Ridley, A.J. The rna-binding protein larp4 regulates cancer cell migration and invasion. Cytoskeleton (Hoboken) 2016, 73, 680–690. [Google Scholar] [CrossRef]
- Koso, H.; Yi, H.; Sheridan, P.; Miyano, S.; Ino, Y.; Todo, T.; Watanabe, S. Identification of rna-binding protein larp4b as a tumor suppressor in glioma. Cancer Res. 2016, 76, 2254–2264. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute hits-clip decodes microrna-mrna interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of rna-binding protein binding sites with enhanced clip (eclip). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef]
- Spitzer, J.; Hafner, M.; Landthaler, M.; Ascano, M.; Farazi, T.; Wardle, G.; Nusbaum, J.; Khorshid, M.; Burger, L.; Zavolan, M.; et al. Par-clip (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation): A step-by-step protocol to the transcriptome-wide identification of binding sites of rna-binding proteins. Methods Enzymol. 2014, 539, 113–161. [Google Scholar]
- Dang, H.; Takai, A.; Forgues, M.; Pomyen, Y.; Mou, H.; Xue, W.; Ray, D.; Ha, K.C.H.; Morris, Q.D.; Hughes, T.R.; et al. Oncogenic activation of the rna binding protein nelfe and myc signaling in hepatocellular carcinoma. Cancer Cell 2017, 32, 101–114.e108. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into rna biology from an atlas of mammalian mrna-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human rna-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of rna-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Ciafre, S.A.; Galardi, S. Micrornas and rna-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013, 10, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Connick, M.C.; Vanderhoof, J.; Ishak, M.A.; Hartley, R.S. Microrna-16 modulates hur regulation of cyclin e1 in breast cancer cells. Int. J. Mol. Sci. 2015, 16, 7112–7132. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microrna-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef]
- Tominaga, K.; Srikantan, S.; Lee, E.K.; Subaran, S.S.; Martindale, J.L.; Abdelmohsen, K.; Gorospe, M. Competitive regulation of nucleolin expression by hur and mir-494. Mol. Cell Biol. 2011, 31, 4219–4231. [Google Scholar] [CrossRef]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. Crd-bp protects the coding region of betatrcp1 mrna from mir-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef]
- Kedde, M.; Strasser, M.J.; Boldajipour, B.; Oude Vrielink, J.A.; Slanchev, K.; le Sage, C.; Nagel, R.; Voorhoeve, P.M.; van Duijse, J.; Orom, U.A.; et al. Rna-binding protein dnd1 inhibits microrna access to target mrna. Cell 2007, 131, 1273–1286. [Google Scholar] [CrossRef]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. Hur recruits let-7/risc to repress c-myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef]
- Leibovich, L.; Mandel-Gutfreund, Y.; Yakhini, Z. A structural-based statistical approach suggests a cooperative activity of pum1 and mir-410 in human 3’-untranslated regions. Silence 2010, 1, 17. [Google Scholar] [CrossRef][Green Version]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Oude Vrielink, J.A.; Elkon, R.; Agami, R. A pumilio-induced rna structure switch in p27-3’ utr controls mir-221 and mir-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Frohn, A.; Eberl, H.C.; Stohr, J.; Glasmacher, E.; Rudel, S.; Heissmeyer, V.; Mann, M.; Meister, G. Dicer-dependent and -independent argonaute2 protein interaction networks in mammalian cells. Mol. Cell Proteom. 2012, 11, 1442–1456. [Google Scholar] [CrossRef] [PubMed]
- Poria, D.K.; Guha, A.; Nandi, I.; Ray, P.S. Rna-binding protein hur sequesters microrna-21 to prevent translation repression of proinflammatory tumor suppressor gene programmed cell death 4. Oncogene 2016, 35, 1703–1715. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Spengler, R.M.; Grigelioniene, G.; Kobayashi, T.; Sharp, P.A. Deconvolution of seed and rna-binding protein crosstalk in rnai-based functional genomics. Nat. Genet. 2018, 50, 657–661. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microrna delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic delivery of a mir34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microrna mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desi, N.; Tay, Y. The Butterfly Effect of RNA Alterations on Transcriptomic Equilibrium. Cells 2019, 8, 1634. https://doi.org/10.3390/cells8121634
Desi N, Tay Y. The Butterfly Effect of RNA Alterations on Transcriptomic Equilibrium. Cells. 2019; 8(12):1634. https://doi.org/10.3390/cells8121634
Chicago/Turabian StyleDesi, Ng, and Yvonne Tay. 2019. "The Butterfly Effect of RNA Alterations on Transcriptomic Equilibrium" Cells 8, no. 12: 1634. https://doi.org/10.3390/cells8121634
APA StyleDesi, N., & Tay, Y. (2019). The Butterfly Effect of RNA Alterations on Transcriptomic Equilibrium. Cells, 8(12), 1634. https://doi.org/10.3390/cells8121634