Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fatty Acid Metabolism is Linked to Diseases
2.1. Fatty Acid Metabolic Pathways
2.2. Palmitic Acid-Regulated Innate Immune Responses
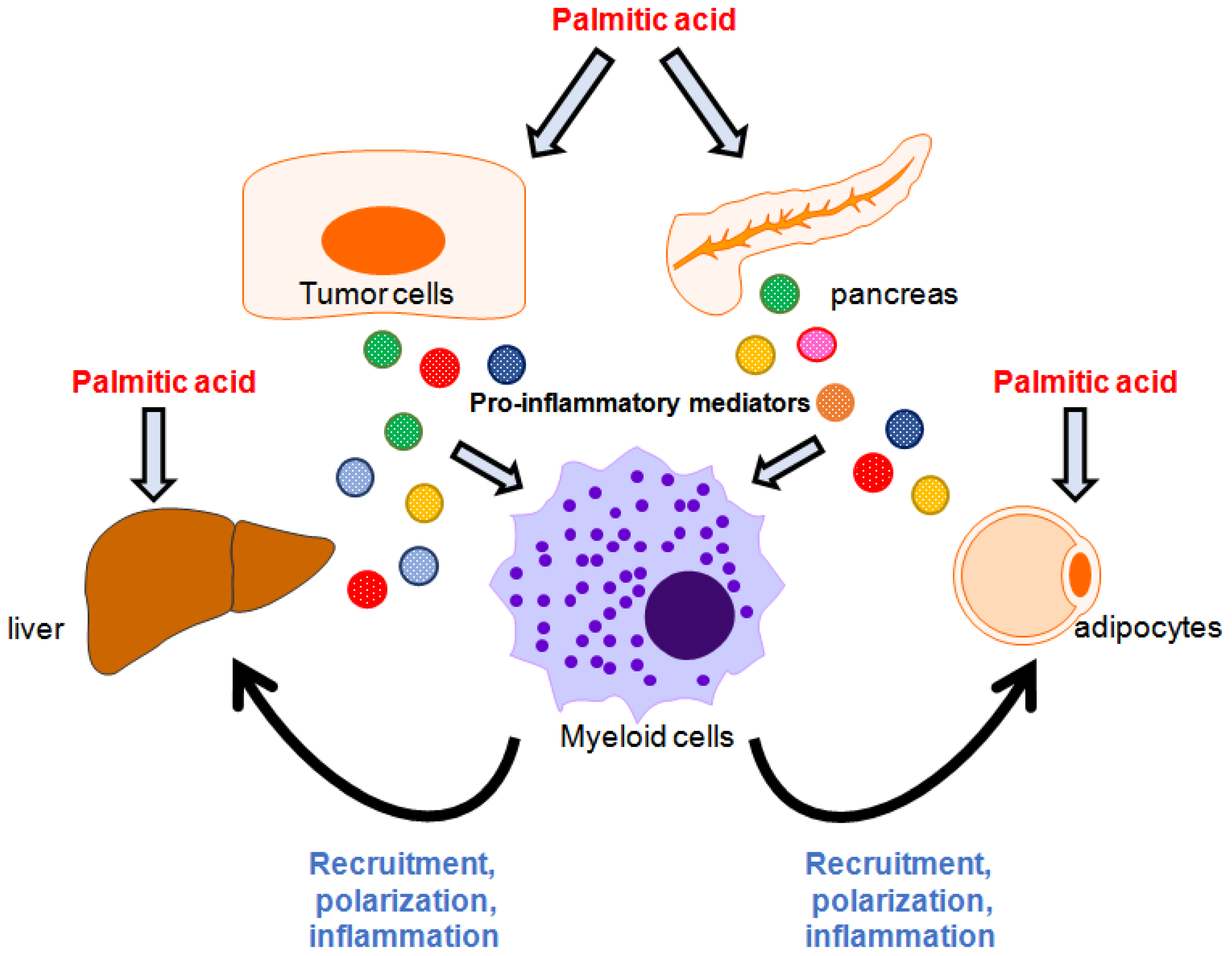
2.3. Palmitate Modulates Immune Responses Through Cooperation with Surrounding Cells
2.4. Palmitoylation
3. Palmitate in Metabolic Disorders
3.1. Palmitate in Diabetes
3.2. Non-Alcoholic Fatty Liver Disease (NAFLD)
4. Involvement of Palmitate in Immune-Related Cell Interactions during Cancer Development
5. Targeting of Palmitate-Induced Pathways as a Therapeutic Strategy
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Invest. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Price, N.L.; Suarez, Y.; Fernandez-Hernando, C. ANGPTL4 in Metabolic and Cardiovascular Disease. Trends Mol. Med. 2019, 25, 723–734. [Google Scholar] [CrossRef]
- Luci, C.; Vieira, E.; Perchet, T.; Gual, P.; Golub, R. Natural Killer Cells and Type 1 Innate Lymphoid Cells Are New Actors in Non-alcoholic Fatty Liver Disease. Front. Immunol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Skuratovskaia, D.; Vulf, M.; Komar, A.; Kirienkova, E.; Litvinova, L. Promising Directions in Atherosclerosis Treatment Based on Epigenetic Regulation Using MicroRNAs and Long Noncoding RNAs. Biomolecules 2019, 9, 226. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Exercise Metabolism: Fuels for the Fire. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef]
- McArthur, M.J.; Atshaves, B.P.; Frolov, A.; Foxworth, W.D.; Kier, A.B.; Schroeder, F. Cellular uptake and intracellular trafficking of long chain fatty acids. J. Lipid Res. 1999, 40, 1371–1383. [Google Scholar]
- Erridge, C.; Samani, N.J. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb. Vasc. Biol. 2009, 29, 1944–1949. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110 e1095. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Haas, J.T.; L’Homme, L.; Fleury, S.; Quemener, S.; Levavasseur, M.; Becquart, C.; Wartelle, J.; Bogomolova, A.; Pineau, L.; et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019, 177, 1201–1216.e19. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Kochumon, S.; Wilson, A.; Chandy, B.; Shenouda, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. Palmitate Activates CCL4 Expression in Human Monocytic Cells via TLR4/MyD88 Dependent Activation of NF-kappaB/MAPK/ PI3K Signaling Systems. Cell Physiol. Biochem. 2018, 46, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Akhter, N.; Madhoun, A.; Arefanian, H.; Wilson, A.; Kochumon, S.; Thomas, R.; Shenouda, S.; Al-Mulla, F.; Ahmad, R.; Sindhu, S. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol. Biochem. 2019, 53, 1–18. [Google Scholar] [CrossRef]
- Sindhu, S.; Akhter, N.; Kochumon, S.; Thomas, R.; Wilson, A.; Shenouda, S.; Tuomilehto, J.; Ahmad, R. Increased Expression of the Innate Immune Receptor TLR10 in Obesity and Type-2 Diabetes: Association with ROS-Mediated Oxidative Stress. Cell Physiol. Biochem. 2018, 45, 572–590. [Google Scholar] [CrossRef]
- Karasawa, T.; Kawashima, A.; Usui-Kawanishi, F.; Watanabe, S.; Kimura, H.; Kamata, R.; Shirasuna, K.; Koyama, Y.; Sato-Tomita, A.; Matsuzaka, T.; et al. Saturated Fatty Acids Undergo Intracellular Crystallization and Activate the NLRP3 Inflammasome in Macrophages. Arterioscler Thromb. Vasc. Biol. 2018, 38, 744–756. [Google Scholar] [CrossRef]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed]
- Kalugotla, G.; He, L.; Weber, K.J.; Daemen, S.; Reller, A.; Razani, B.; Schilling, J.D. Frontline Science: Acyl-CoA synthetase 1 exacerbates lipotoxic inflammasome activation in primary macrophages. J. Leukoc. Biol. 2019, 106, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Wang, Z.; Yang, Y.M.; Billet, S.; Tu, W.; Pimienta, M.; Cassel, S.L.; Pandol, S.J.; Lu, S.C.; Sutterwala, F.S.; et al. NOD-like receptor C4 Inflammasome Regulates the Growth of Colon Cancer Liver Metastasis in NAFLD. Hepatology 2019, 70, 1582–1599. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Schlutter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Cogolludo, A.; Villamor, E.; Perez-Vizcaino, F.; Moreno, L. Ceramide and Regulation of Vascular Tone. Int. J. Mol. Sci. 2019, 20, 411. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Soumura, M.; Kume, S.; Isshiki, K.; Takeda, N.; Araki, S.; Tanaka, Y.; Sugimoto, T.; Chin-Kanasaki, M.; Nishio, Y.; Haneda, M.; et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem. Biophys. Res. Commun. 2010, 402, 265–271. [Google Scholar] [CrossRef]
- Henique, C.; Mansouri, A.; Fumey, G.; Lenoir, V.; Girard, J.; Bouillaud, F.; Prip-Buus, C.; Cohen, I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem. 2010, 285, 36818–36827. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Koh, J.M.; So, J.S.; Jeon, Y.K.; Kim, H.Y.; Chung, D.H. Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1alpha-dependent decay. Sci. Rep. 2017, 7, 14940. [Google Scholar] [CrossRef] [PubMed]
- Torres-Hernandez, A.; Wang, W.; Nikiforov, Y.; Tejada, K.; Torres, L.; Kalabin, A.; Adam, S.; Wu, J.; Lu, L.; Chen, R.; et al. gammadelta T cells Promote Steatohepatitis by Orchestrating Innate and Adaptive Immune Programming. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Azizi, P.M.; Li, Y.E.; Liu, J.; Wang, C.; Chan, K.L.; Hopperton, K.E.; Bazinet, R.P.; Heit, B.; Bilan, P.J.; et al. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E35–E44. [Google Scholar] [CrossRef]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Chen, X.; Zhuo, S.; Zhu, T.; Yao, P.; Yang, M.; Mei, H.; Li, N.; Ma, F.; Wang, J.M.; Chen, S.; et al. Fpr2 Deficiency Alleviates Diet-Induced Insulin Resistance Through Reducing Body Weight Gain and Inhibiting Inflammation Mediated by Macrophage Chemotaxis and M1 Polarization. Diabetes 2019, 68, 1130–1142. [Google Scholar] [CrossRef]
- Dalmas, E.; Lehmann, F.M.; Dror, E.; Wueest, S.; Thienel, C.; Borsigova, M.; Stawiski, M.; Traunecker, E.; Lucchini, F.C.; Dapito, D.H.; et al. Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion through Myeloid Cell Retinoic Acid Production. Immunity 2017, 47, 928–942 e927. [Google Scholar] [CrossRef]
- Chenxu, G.; Minxuan, X.; Yuting, Q.; Tingting, G.; Jing, F.; Jinxiao, L.; Sujun, W.; Yongjie, M.; Deshuai, L.; Qiang, L.; et al. Loss of RIP3 initiates annihilation of high-fat diet initialized nonalcoholic hepatosteatosis: A mechanism involving Toll-like receptor 4 and oxidative stress. Free Radic. Biol. Med. 2019, 134, 23–41. [Google Scholar] [CrossRef]
- Shen, C.; Ma, W.; Ding, L.; Li, S.; Dou, X.; Song, Z. The TLR4-IRE1alpha pathway activation contributes to palmitate-elicited lipotoxicity in hepatocytes. J. Cell Mol. Med. 2018, 22, 3572–3581. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Mizukoshi, E.; Sunagozaka, H.; Arai, K.; Yamashita, T.; Takeshita, Y.; Misu, H.; Takamura, T.; Kitamura, S.; Zen, Y.; et al. Characteristics of hepatic fatty acid compositions in patients with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Song, M.J.; Gao, Y.; Mauer, A.S.; Revzin, A.; Malhi, H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front. Immunol. 2018, 9, 2980. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Ragan, M.A. Palmitoylation: A protein S-acylation with implications for breast cancer. NPJ Breast Cancer 2016, 2, 16028. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.E.; Yoshioka, C.; Mansoor, S.E. Full-Length P2X7 Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell 2019, 179, 659–670 e613. [Google Scholar] [CrossRef]
- Wiley, J.S.; Sluyter, R.; Gu, B.J.; Stokes, L.; Fuller, S.J. The human P2X7 receptor and its role in innate immunity. Tissue Antigens 2011, 78, 321–332. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Sarti, A.C.; Falzoni, S.; Di Virgilio, F. The P2X7 Receptor-Interleukin-1 Liaison. Front. Pharmacol. 2017, 8, 123. [Google Scholar] [CrossRef]
- Gentile, D.; Natale, M.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. The role of P2X7 receptors in tissue fibrosis: A brief review. Purinergic Signal. 2015, 11, 435–440. [Google Scholar] [CrossRef]
- Yang, S.; Harding, A.T.; Sweeney, C.; Miao, D.; Swan, G.; Zhou, C.; Jiang, Z.; Fitzgerald, K.A.; Hammer, G.; Bergo, M.O.; et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 2019, 5, eaav7999. [Google Scholar] [CrossRef]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERalpha36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef]
- Chaudhri, R.A.; Hadadi, A.; Lobachev, K.S.; Schwartz, Z.; Boyan, B.D. Estrogen receptor-alpha 36 mediates the anti-apoptotic effect of estradiol in triple negative breast cancer cells via a membrane-associated mechanism. Biochim. Biophys. Acta 2014, 1843, 2796–2806. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lv, D.; Li, H.; Shang, J.; Lu, J.; Zhou, L.; Bai, L.; Tang, H. Exosomal Interferon-Induced Transmembrane Protein 2 Transmitted to Dendritic Cells Inhibits Interferon Alpha Pathway Activation and Blocks Anti-Hepatitis B Virus Efficacy of Exogenous Interferon Alpha. Hepatology 2019, 69, 2396–2413. [Google Scholar] [CrossRef] [PubMed]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferre, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Chan, K.L.; Zhang, S.; Mejdani, M.; Jacobson, M.R.; Ducos, A.; Bilan, P.J.; Niu, W.; Klip, A. Saturated fatty acids activate caspase-4/5 in human monocytes, triggering IL-1beta and IL-18 release. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E825–E835. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Bandyopadhyay, G.K.; Luo, J.L.; Naugler, W.; Grivennikov, S.; Wynshaw-Boris, A.; Scadeng, M.; Olefsky, J.M.; et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef]
- Li, L.C.; Yang, J.L.; Lee, W.C.; Chen, J.B.; Lee, C.T.; Wang, P.W.; Vaghese, Z.; Chen, W.Y. Palmitate aggravates proteinuria-induced cell death and inflammation via CD36-inflammasome axis in the proximal tubular cells of obese mice. Am. J. Physiol. Renal Physiol. 2018, 315, F1720–F1731. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Sommerweiss, D.; Gorski, T.; Richter, S.; Garten, A.; Kiess, W. Oleate rescues INS-1E beta-cells from palmitate-induced apoptosis by preventing activation of the unfolded protein response. Biochem. Biophys. Res. Commun. 2013, 441, 770–776. [Google Scholar] [CrossRef] [PubMed]
- L’Homme, L.; Esser, N.; Riva, L.; Scheen, A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J. Lipid Res. 2013, 54, 2998–3008. [Google Scholar] [CrossRef]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovich, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Olson, L.K. Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic beta-cells by stearoyl-CoA desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E640–E649. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e4. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Cazanave, S.C.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Fingas, C.D.; Meng, X.W.; Finnberg, N.; El-Deiry, W.S.; Kaufmann, S.H.; Gores, G.J. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 2011, 286, 39336–39348. [Google Scholar] [CrossRef]
- Volkmann, X.; Fischer, U.; Bahr, M.J.; Ott, M.; Lehner, F.; Macfarlane, M.; Cohen, G.M.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 2007, 46, 1498–1508. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Bronk, S.F.; Yagita, H.; Gores, G.J. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e70599. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016, 63, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Schoenfield, L.; Plesec, T.; Saunthararajah, Y.; Tubbs, R.R.; Singh, A.D. Molecular profiling of primary uveal melanomas with tumor-infiltrating lymphocytes. Oncoimmunology 2019, 8, e947169. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Al-Roub, A.; Kochumon, S.; Akther, N.; Thomas, R.; Kumari, M.; Koshy, M.S.; Tiss, A.; Hannun, Y.A.; Tuomilehto, J.; et al. The Synergy between Palmitate and TNF-alpha for CCL2 Production Is Dependent on the TRIF/IRF3 Pathway: Implications for Metabolic Inflammation. J. Immunol. 2018, 200, 3599–3611. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.S.; Liu, R.; Wen, Y.F.; Liu, L.T.; Yuan, L.; Li, Y.X.; Li, Y.; Hao, W.W.; Peng, J.Y.; Chen, D.N.; et al. Endogenous production of C-C motif chemokine ligand 2 by nasopharyngeal carcinoma cells drives radioresistance-associated metastasis. Cancer Lett. 2020, 468, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Kim, T.H.; Yi, S.A.; Hwang, Y.C.; Hwang, W.S.; Choe, S.J.; Han, S.J.; Kim, H.J.; Kim, D.J.; Kang, Y.; et al. Capsaicin attenuates palmitate-induced expression of macrophage inflammatory protein 1 and interleukin 8 by increasing palmitate oxidation and reducing c-Jun activation in THP-1 (human acute monocytic leukemia cell) cells. Nutr. Res. 2011, 31, 468–478. [Google Scholar] [CrossRef]
- Schaller, T.H.; Batich, K.A.; Suryadevara, C.M.; Desai, R.; Sampson, J.H. Chemokines as adjuvants for immunotherapy: Implications for immune activation with CCL3. Expert Rev. Clin. Immunol. 2017, 13, 1049–1060. [Google Scholar] [CrossRef]
- Chan, K.L.; Pillon, N.J.; Sivaloganathan, D.M.; Costford, S.R.; Liu, Z.; Theret, M.; Chazaud, B.; Klip, A. Palmitoleate Reverses High Fat-induced Proinflammatory Macrophage Polarization via AMP-activated Protein Kinase (AMPK). J. Biol. Chem. 2015, 290, 16979–16988. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Iskandar, M.A.; Tuomilehto, J.; Al-Mulla, F.; Ahmad, R. TNF-alpha Induces a Pro-Inflammatory Phenotypic Shift in Monocytes through ACSL1: Relevance to Metabolic Inflammation. Cell Physiol. Biochem. 2019, 52, 397–407. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Rashed, F.; Akhter, N.; Al-Mulla, F.; Ahmad, R. ACSL1 Regulates TNFalpha-Induced GM-CSF Production by Breast Cancer MDA-MB-231 Cells. Biomolecules 2019, 9, 555. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.N.; Lin, J.J.; Chen, J.J.; Lee, P.H.; Yang, C.Y.; Kuo, M.L.; Chang, K.J.; Hsieh, F.J. Gene expression profile predicts patient survival of gastric cancer after surgical resection. J. Clin. Oncol. 2005, 23, 7286–7295. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef] [PubMed]
- Pararasa, C.; Bailey, C.; Griffiths, H. Macrophage polarisation by fatty acids is PPARgamma-dependent. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S31–S32. [Google Scholar] [CrossRef]
- Kitamura, T.; Doughty-Shenton, D.; Cassetta, L.; Fragkogianni, S.; Brownlie, D.; Kato, Y.; Carragher, N.; Pollard, J.W. Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front. Immunol. 2017, 8, 2004. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef]
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 1298–1307. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef]
- Kanter, J.E.; Kramer, F.; Barnhart, S.; Averill, M.M.; Vivekanandan-Giri, A.; Vickery, T.; Li, L.O.; Becker, L.; Yuan, W.; Chait, A.; et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. USA 2012, 109, E715–E724. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E. Growing evidence for a role for acyl-CoA synthetase 1 in immunometabolism. J. Leukoc. Biol. 2019, 106, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Rossi Sebastiano, M.; Konstantinidou, G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 3624. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Kumar, A.; Singh, S.M. Myelopoietic efficacy of orlistat in murine hosts bearing T cell lymphoma: Implication in macrophage differentiation and activation. PLoS ONE 2013, 8, e82396. [Google Scholar] [CrossRef] [PubMed][Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzeng, H.-T.; Chyuan, I.-T.; Chen, W.-Y. Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate. Cells 2019, 8, 1633. https://doi.org/10.3390/cells8121633
Tzeng H-T, Chyuan I-T, Chen W-Y. Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate. Cells. 2019; 8(12):1633. https://doi.org/10.3390/cells8121633
Chicago/Turabian StyleTzeng, Hong-Tai, I-Tsu Chyuan, and Wei-Yu Chen. 2019. "Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate" Cells 8, no. 12: 1633. https://doi.org/10.3390/cells8121633
APA StyleTzeng, H.-T., Chyuan, I.-T., & Chen, W.-Y. (2019). Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate. Cells, 8(12), 1633. https://doi.org/10.3390/cells8121633