Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Detection of Cell Proliferation
2.4. Apoptosis Assays
2.5. Immunofluorescence Assays
2.6. Endothelial Cell Isolation
2.7. RT-PCR and Quantitative RT-PCR
2.8. mRNA Sequencing
2.9. Bioinformatics
2.10. Tissue Samples and Immunohistology
2.11. Cloning and Transient Transfection Experiments
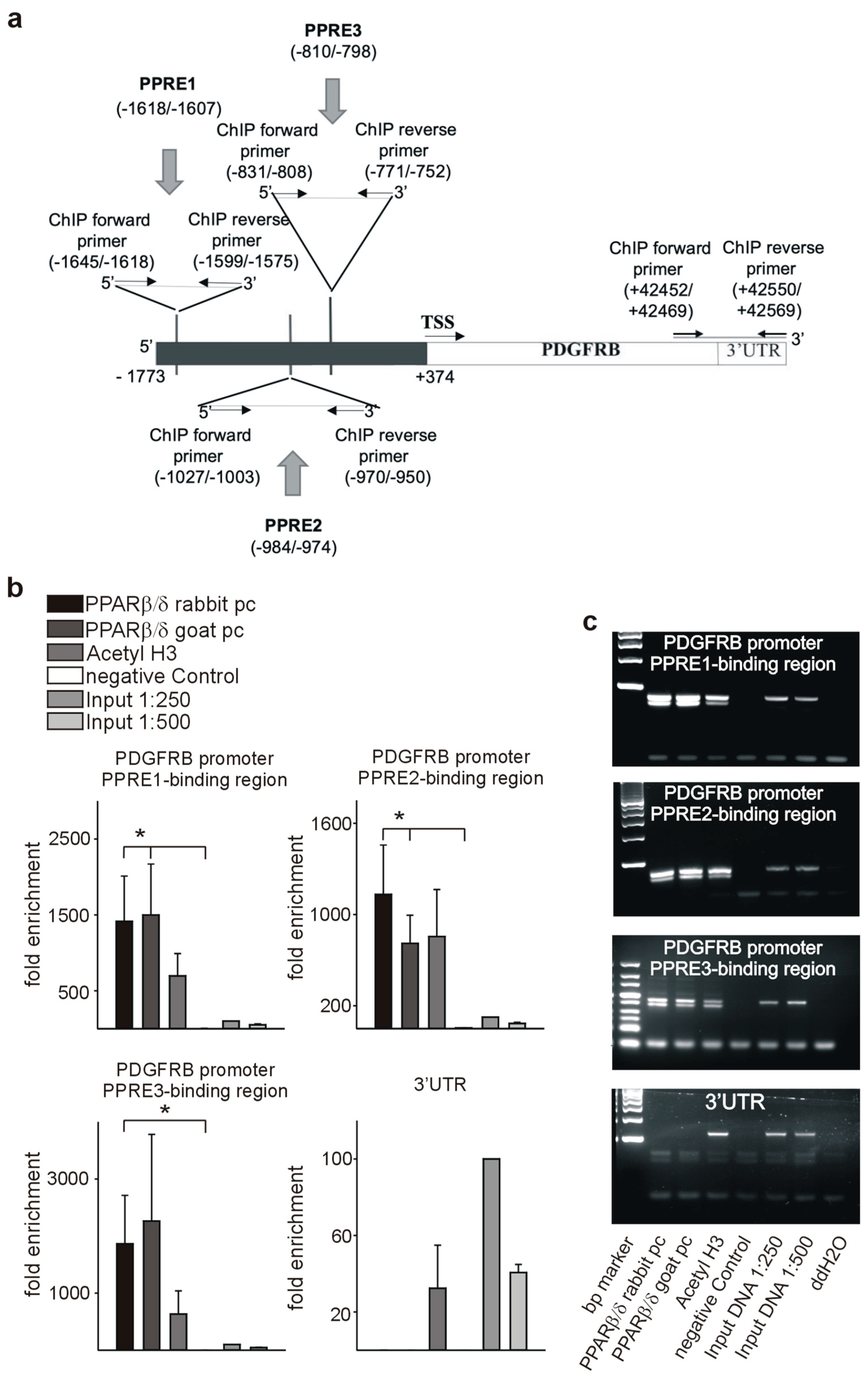
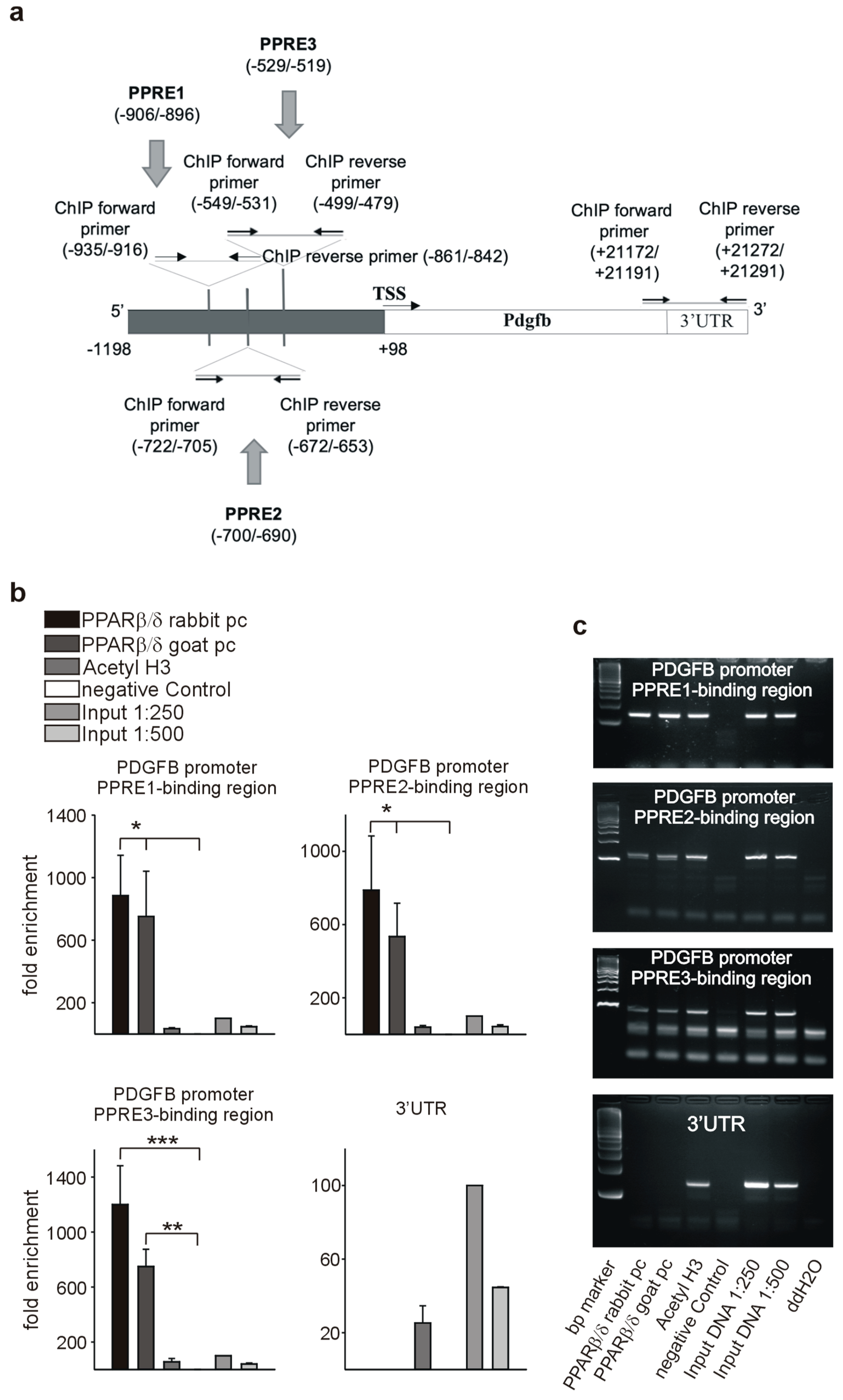
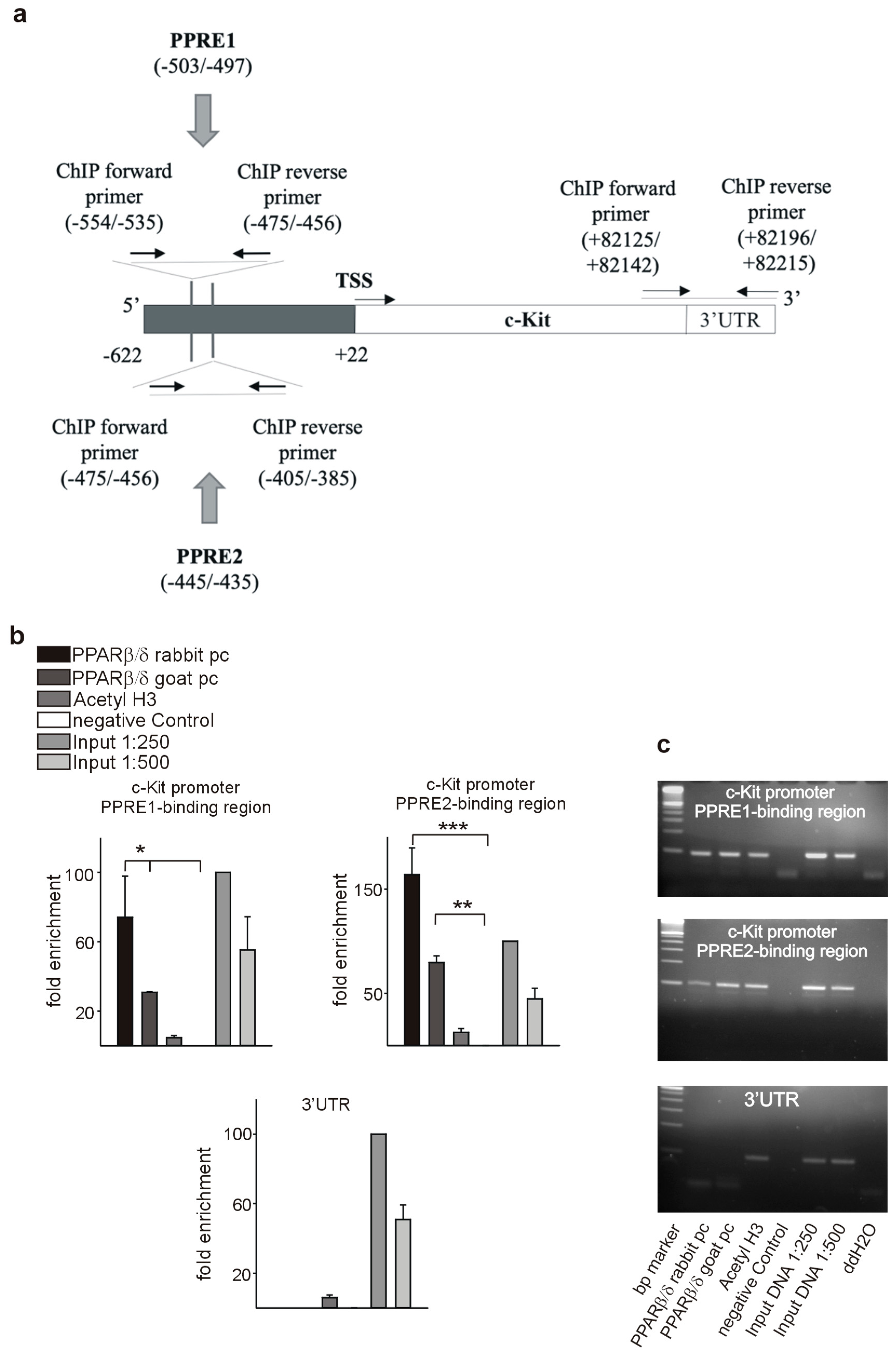
2.12. Chromatin Immunoprecipitation Assay
2.13. Statistical Analysis
3. Results and Discussion
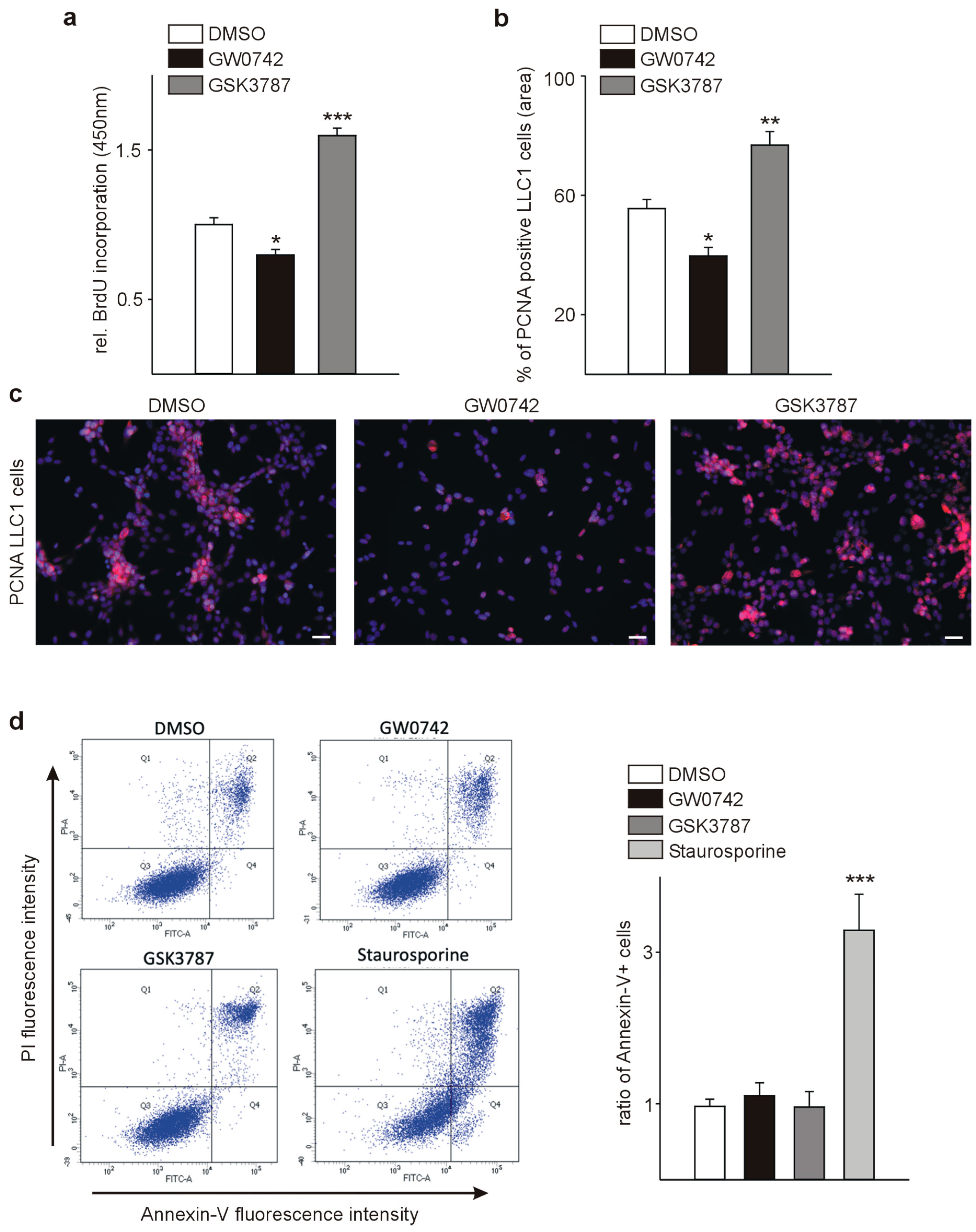
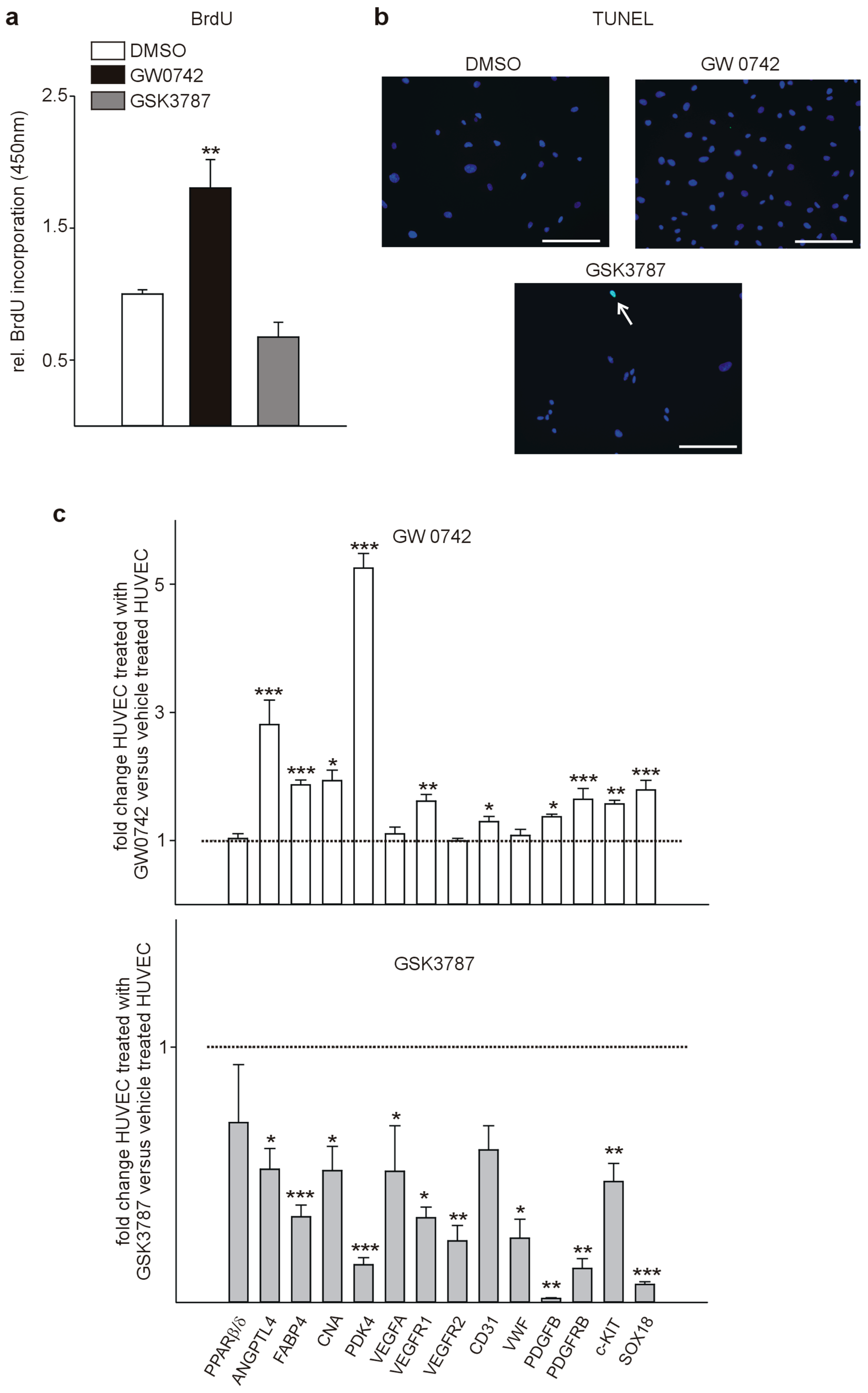
3.1. The PPARβ/δ Agonist GW0742 Decreases LLC1 Lewis Lung Cancer Cell Proliferation In Vitro
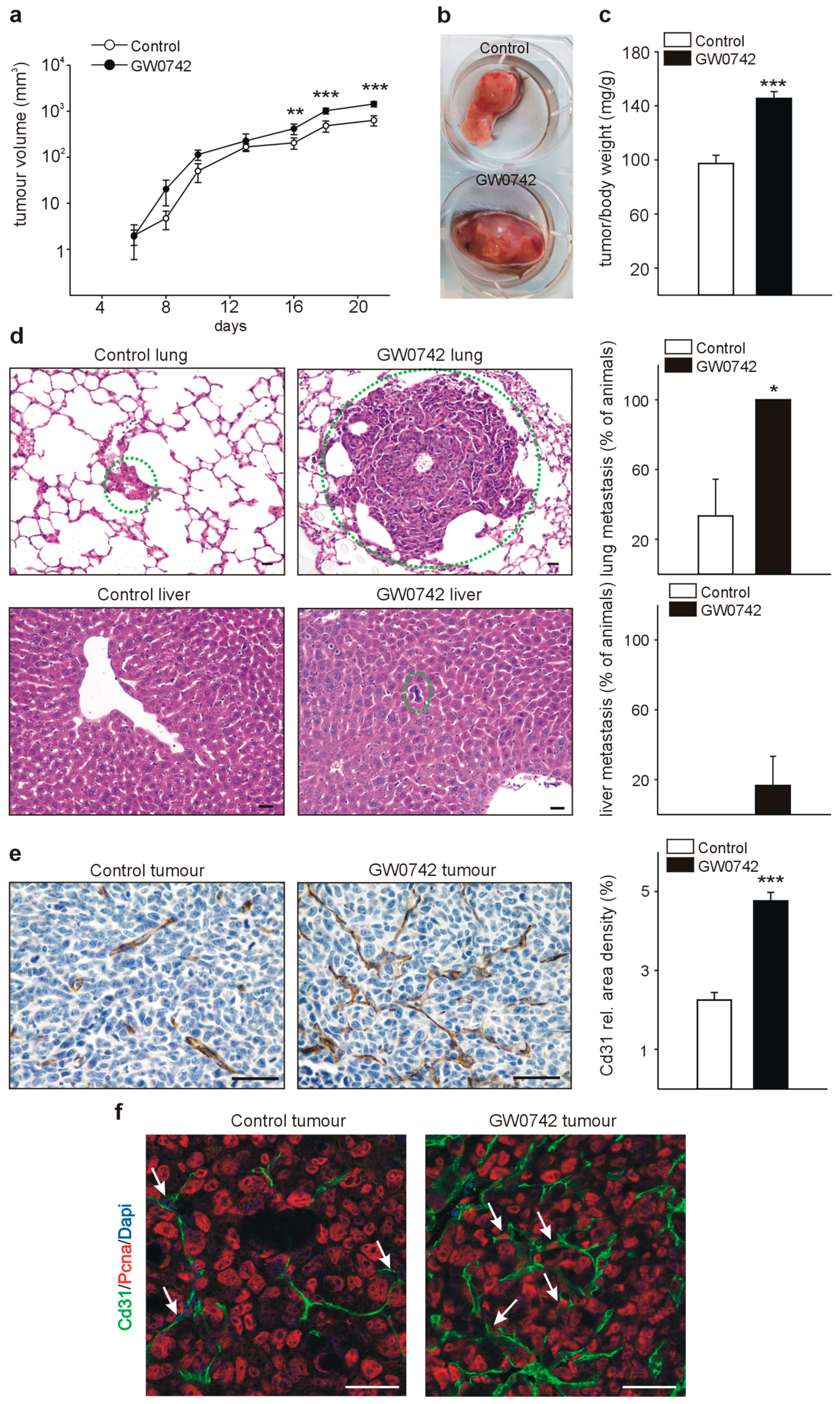
3.2. The PPARβ/δ Agonist GW0742 Increases LLC1 Lewis Lung Cancer Progression In Vivo
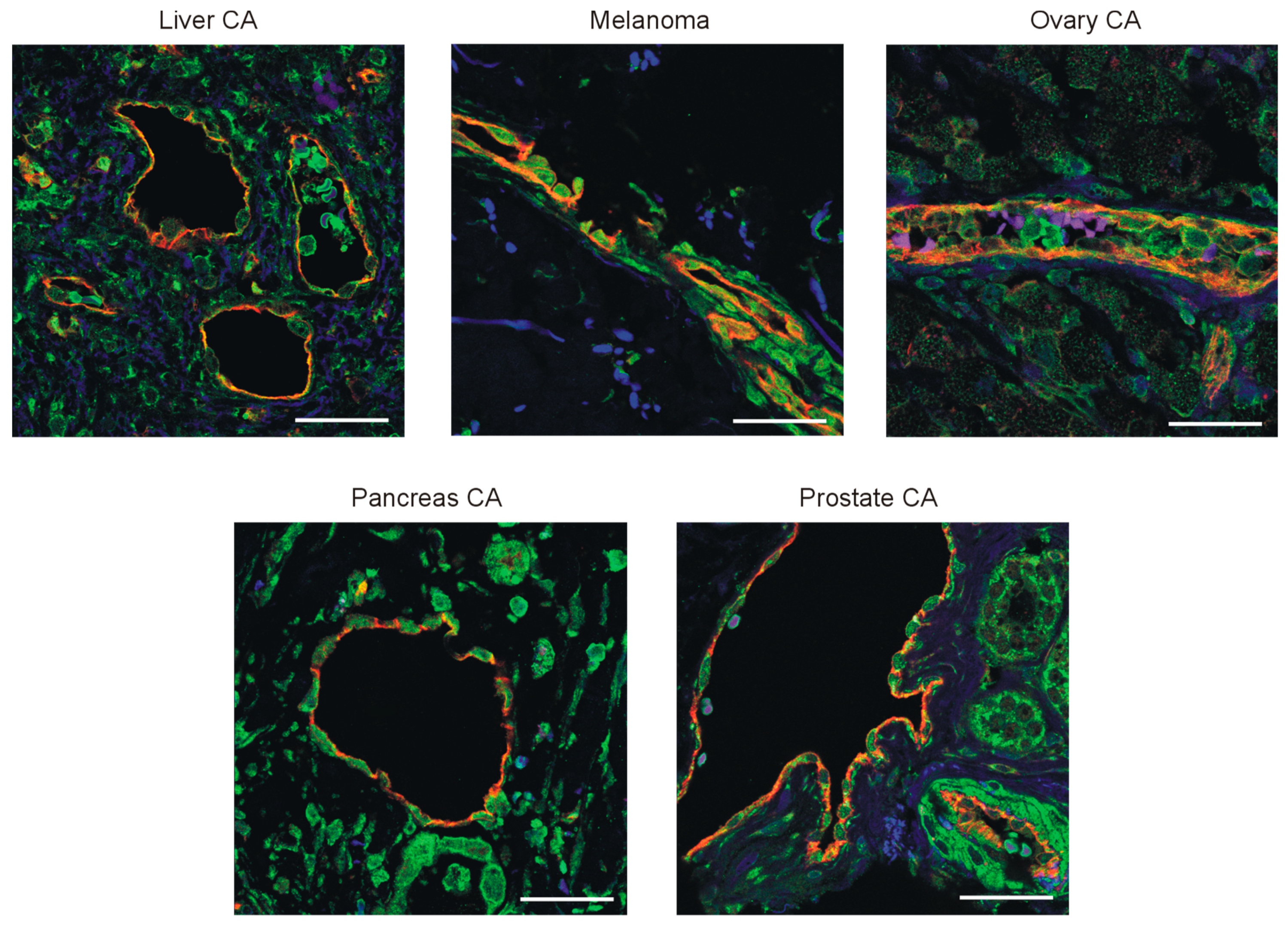
3.3. PPARβ/δ Is Expressed in the Vasculature of Human Tumors and PPARβ/δ Modulation Impacts Proliferation and Expression of Pro-Angiogenic Factors in Human Endothelial Cells
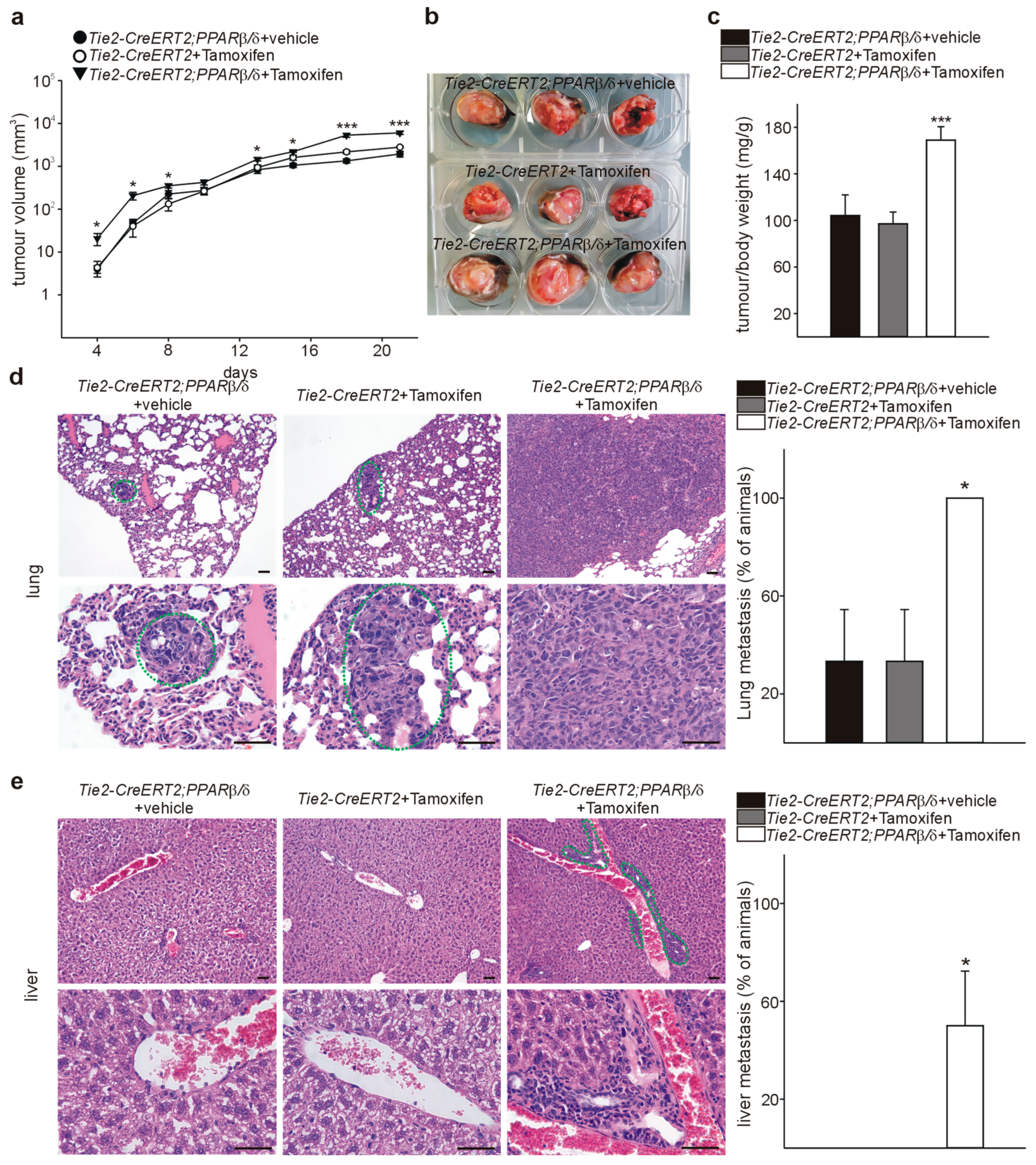
3.4. Inducible Vascular-Specific Overexpression of PPARβ/δ Promotes Tumor Angiogenesis, Growth, and Spontaneous Metastases Formation In Vivo
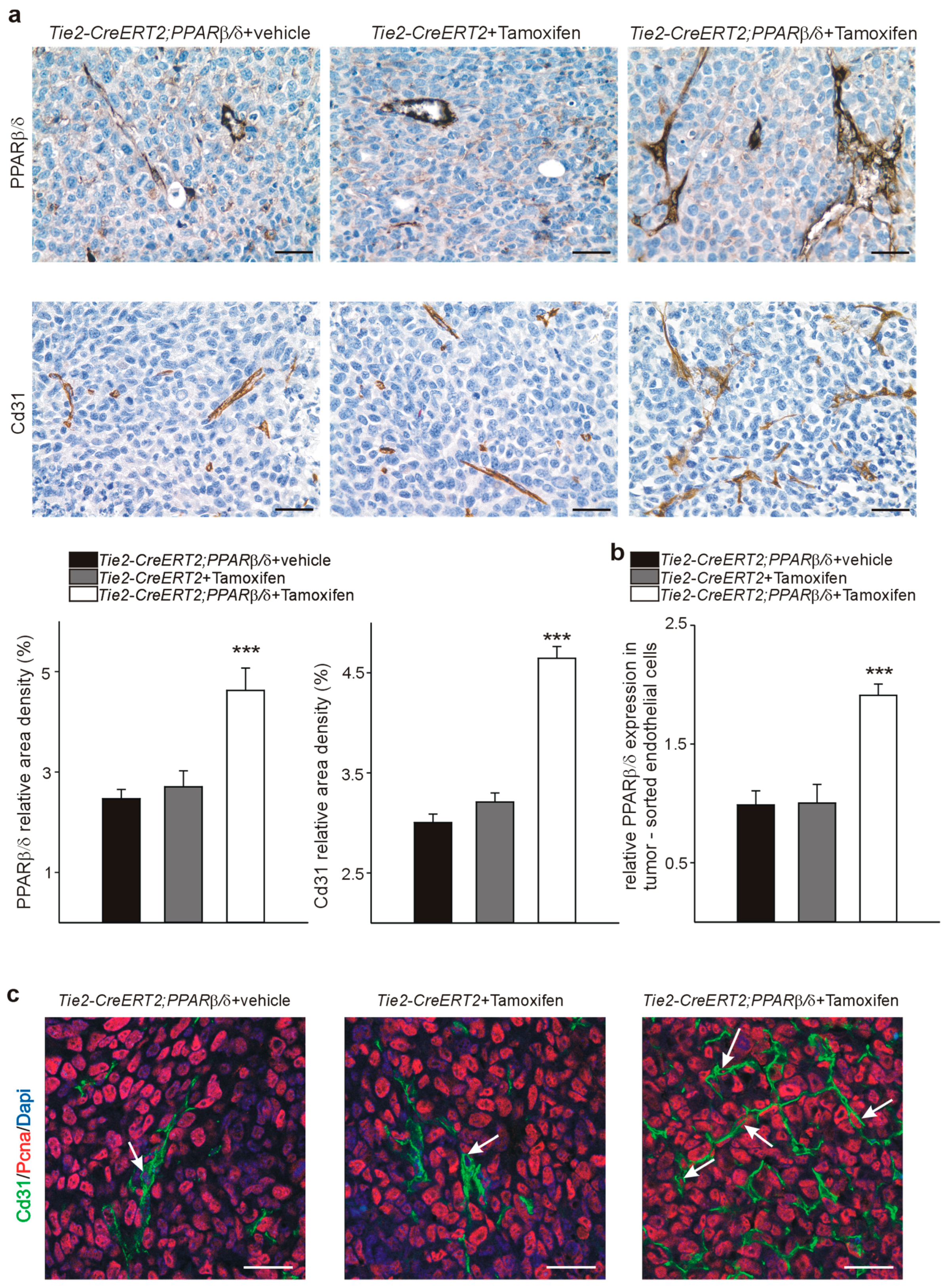
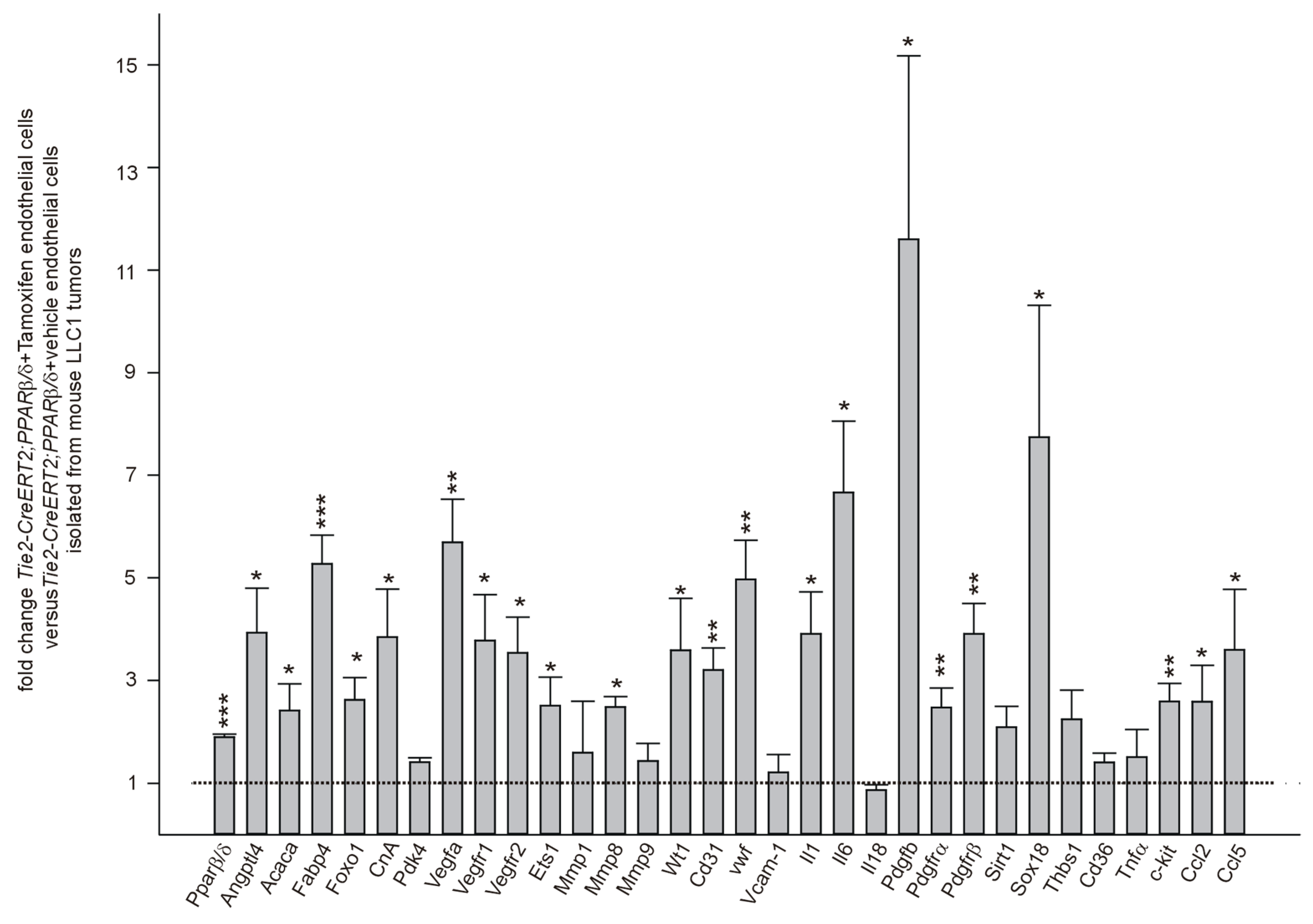
3.5. PPARβ/δ Overexpression Upregulates Angiogenic and Metastases Promoting Molecules in Endothelial Cells In Vivo
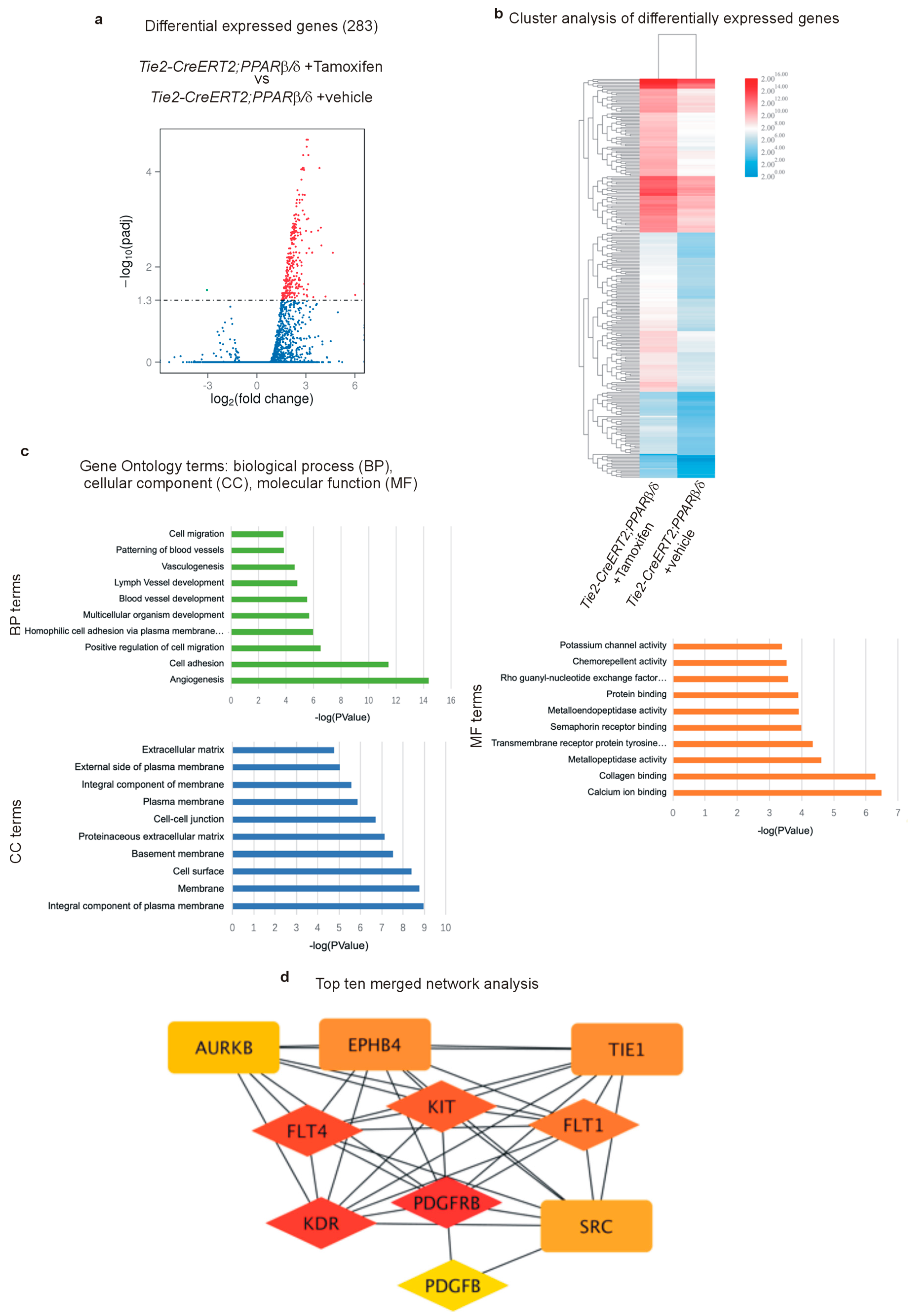
3.6. RNA Sequencing Further Certifies the Acquisition of a Highly angiogenic Endothelial Phenotype Upon PPARβ/δ Overexpression and Identifies Pro-Tumor-Angiogenic Signaling Networks
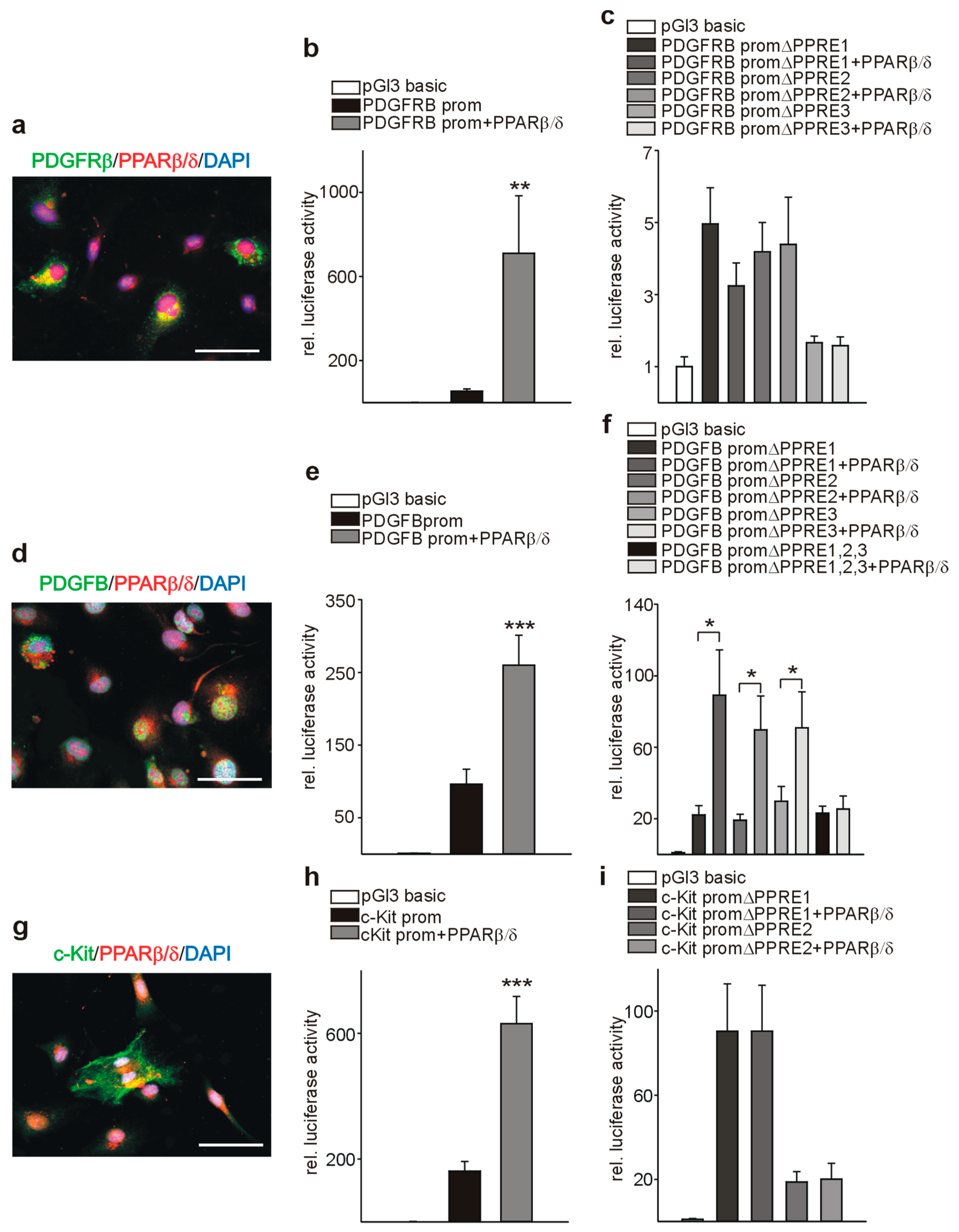
3.7. PPARβ/δ Directly Activates PDGFRβ, PDGFB, and c-Kit
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, P.A. Regulatory functions of PPARbeta in metabolism: Implications for the treatment of metabolic syndrome. Biochim. Biophys. Acta 2007, 1771, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D.; Swales, K.E. The Role of PPARs in the Endothelium: Implications for Cancer Therapy. PPAR Res. 2008, 2008, 904251. [Google Scholar] [CrossRef]
- Michiels, J.F.; Perrin, C.; Leccia, N.; Massi, D.; Grimaldi, P.; Wagner, N. PPARbeta activation inhibits melanoma cell proliferation involving repression of the Wilms’ tumour suppressor WT1. Pflügers Arch. 2010, 459, 689–703. [Google Scholar] [CrossRef][Green Version]
- Wagner, K.D.; Benchetrit, M.; Bianchini, L.; Michiels, J.F.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) is highly expressed in liposarcoma and promotes migration and proliferation. J. Pathol. 2011, 224, 575–588. [Google Scholar] [CrossRef]
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arter. Thromb. Vasc. Biol. 2007, 27, 63–69. [Google Scholar] [CrossRef]
- Wagner, N.; Jehl-Piétri, C.; Lopez, P.; Murdaca, J.; Giordano, C.; Schwartz, C.; Gounon, P.; Hatem, S.N.; Grimaldi, P.; Wagner, K.D. Peroxisome proliferator-activated receptor beta stimulation induces rapid cardiac growth and angiogenesis via direct activation of calcineurin. Cardiovasc. Res. 2009, 83, 61–71. [Google Scholar] [CrossRef]
- Gaudel, C.; Schwartz, C.; Giordano, C.; Abumrad, N.A.; Grimaldi, P.A. Pharmacological activation of PPARbeta promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E297–E304. [Google Scholar] [CrossRef]
- Wagner, K.D.; Vukolic, A.; Baudouy, D.; Michiels, J.F.; Wagner, N. Inducible Conditional Vascular-Specific Overexpression of Peroxisome Proliferator-Activated Receptor Beta/Delta Leads to Rapid Cardiac Hypertrophy. PPAR Res. 2016, 2016, 7631085. [Google Scholar] [CrossRef]
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Forde, A.; Constien, R.; Gröne, H.J.; Hämmerling, G.; Arnold, B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis 2002, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N.; Guo, J.K.; Elger, M.; Dallman, M.J.; Bugeon, L.; Schedl, A. An inducible mouse model for PAX2-dependent glomerular disease: Insights into a complex pathogenesis. Curr. Biol. 2006, 16, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Panelos, J.; Massi, D.; Wagner, K.D. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflügers Arch. 2008, 455, 839–847. [Google Scholar] [CrossRef] [PubMed]
- El Maï, M.; Wagner, K.D.; Michiels, J.F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef]
- Wagner, K.D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Beisser, D.; Klau, G.W.; Dandekar, T.; Müller, T.; Dittrich, M.T. BioNet: An R-Package for the functional analysis of biological networks. Bioinformatics 2010, 26, 1129–1130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Carlin, D.E.; Yu, M.K.; Zhang, W.; Kreisberg, J.F.; Tamayo, P.; Ideker, T. Systematic Evaluation of Molecular Networks for Discovery of Disease Genes. Cell Syst. 2018, 6, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Pillich, R.T.; Chen, J.; Rynkov, V.; Welker, D.; Pratt, D. NDEx: A Community Resource for Sharing and Publishing of Biological Networks. Methods Mol. Biol. 2017, 1558, 271–301. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.T.; Grimes, M.; Kutlu, B.; Bot, J.J.; Galas, D.J. RCytoscape: Tools for exploratory network analysis. BMC Bioinform. 2013, 14, 217. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. Genes Genomes Genet. 2012, 2, 987–1002. [Google Scholar] [CrossRef]
- Maeda, K.; Nishiyama, C.; Ogawa, H.; Okumura, K. GATA2 and Sp1 positively regulate the c-kit promoter in mast cells. J. Immunol. 2010, 185, 4252–4260. [Google Scholar] [CrossRef]
- Lim, J.C.W.; Kwan, Y.P.; Tan, M.S.; Teo, M.H.Y.; Chiba, S.; Wahli, W.; Wang, X. The Role of PPARβ/δ in Melanoma Metastasis. Int. J. Mol. Sci. 2018, 19, 2860. [Google Scholar] [CrossRef]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am. J. Respir. Cell Mol. Biol. 2008, 39, 689–696. [Google Scholar] [CrossRef]
- Genini, D.; Garcia-Escudero, R.; Carbone, G.M.; Catapano, C.V. Transcriptional and Non-Transcriptional Functions of PPARβ/δ in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e46009. [Google Scholar] [CrossRef]
- Fukumoto, K.; Yano, Y.; Virgona, N.; Hagiwara, H.; Sato, H.; Senba, H.; Suzuki, K.; Asano, R.; Yamada, K.; Yano, T. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005, 579, 3829–3836. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Borland, M.G.; Zhu, B.; Sharma, A.K.; Amin, S.; El-Bayoumy, K.; Gonzalez, F.J.; Peters, J.M. Effect of ligand activation of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in human lung cancer cell lines. Toxicology 2008, 254, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Müller-Brüsselbach, S.; Kömhoff, M.; Rieck, M.; Meissner, W.; Kaddatz, K.; Adamkiewicz, J.; Keil, B.; Klose, K.J.; Moll, R.; Burdick, A.D.; et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. EMBO J. 2007, 26, 3686–3698. [Google Scholar] [CrossRef] [PubMed]
- Stephen, R.L.; Gustafsson, M.C.; Jarvis, M.; Tatoud, R.; Marshall, B.R.; Knight, D.; Ehrenborg, E.; Harris, A.L.; Wolf, C.R.; Palmer, C.N. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004, 64, 3162–3170. [Google Scholar] [CrossRef]
- Hatae, T.; Wada, M.; Yokoyama, C.; Shimonishi, M.; Tanabe, T. Prostacyclin-dependent apoptosis mediated by PPAR delta. J. Biol. Chem. 2001, 276, 46260–46267. [Google Scholar] [CrossRef]
- Staiger, H.; Haas, C.; Machann, J.; Werner, R.; Weisser, M.; Schick, F.; Machicao, F.; Stefan, N.; Fritsche, A.; Häring, H.U. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes 2009, 58, 579–589. [Google Scholar] [CrossRef]
- Shin, J.; Li, B.; Davis, M.E.; Suh, Y.; Lee, K. Comparative analysis of fatty acid-binding protein 4 promoters: Conservation of peroxisome proliferator-activated receptor binding sites. J. Anim. Sci. 2009, 87, 3923–3934. [Google Scholar] [CrossRef]
- Degenhardt, T.; Saramäki, A.; Malinen, M.; Rieck, M.; Väisänen, S.; Huotari, A.; Herzig, K.H.; Müller, R.; Carlberg, C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 2007, 372, 341–355. [Google Scholar] [CrossRef]
- Zhang, Z.; Neiva, K.G.; Lingen, M.W.; Ellis, L.M.; Nör, J.E. VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell Death Differ. 2010, 17, 499–512. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C.; Kovach, N.L.; Bennett, L.G.; Lin, N.; Jacobsen, F.W.; Kidd, P.G. Human umbilical vein endothelial cells display high-affinity c-kit receptors and produce a soluble form of the c-kit receptor. Blood 1994, 83, 2145–2152. [Google Scholar] [CrossRef]
- Sihto, H.; Tynninen, O.; Bützow, R.; Saarialho-Kere, U.; Joensuu, H. Endothelial cell KIT expression in human tumours. J. Pathol. 2007, 211, 481–488. [Google Scholar] [CrossRef]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef]
- Young, N.; Hahn, C.N.; Poh, A.; Dong, C.; Wilhelm, D.; Olsson, J.; Muscat, G.E.; Parsons, P.; Gamble, J.R.; Koopman, P. Effect of disrupted SOX18 transcription factor function on tumor growth, vascularization, and endothelial development. J. Natl. Cancer Inst. 2006, 98, 1060–1067. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Guo, Y.; Ning, W.; Katkuri, S.; Wahli, W.; Desvergne, B.; Dey, S.K.; DuBois, R.N. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA 2006, 103, 19069–19074. [Google Scholar] [CrossRef]
- Randi, A.M.; Laffan, M.A. Von Willebrand factor and angiogenesis: Basic and applied issues. J. Thromb. Haemost. 2017, 15, 13–20. [Google Scholar] [CrossRef]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef]
- Oettgen, P. The role of ets factors in tumor angiogenesis. J. Oncol. 2010, 2010, 767384. [Google Scholar] [CrossRef][Green Version]
- Fang, C.; Wen, G.; Zhang, L.; Lin, L.; Moore, A.; Wu, S.; Ye, S.; Xiao, Q. An important role of matrix metalloproteinase-8 in angiogenesis in vitro and in vivo. Cardiovasc. Res. 2013, 99, 146–155. [Google Scholar] [CrossRef]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol. Cell Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [PubMed]
- Roblek, M.; Protsyuk, D.; Becker, P.F.; Stefanescu, C.; Gorzelanny, C.; Glaus Garzon, J.F.; Knopfova, L.; Heikenwalder, M.; Luckow, B.; Schneider, S.W.; et al. CCL2 Is a Vascular Permeability Factor Inducing CCR2-Dependent Endothelial Retraction during Lung Metastasis. Mol. Cancer Res. 2019, 17, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Spanaus, K.S.; Borsig, L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood 2009, 114, 4583–4591. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Bishop-Bailey, D. PPARβ/δ a potential target in pulmonary hypertension blighted by cancer risk. Pulm. Circ. 2019, 9. [Google Scholar] [CrossRef]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef]
- Sadremomtaz, A.; Mansouri, K.; Alemzadeh, G.; Safa, M.; Rastaghi, A.E.; Asghari, S.M. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF-driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Biochim. Biophys. Acta 2018, 1862, 2688–2700. [Google Scholar] [CrossRef]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Accession Number | Primer Sequences | Amplicon Size (bp) 1 |
|---|---|---|---|
| PPARβ/δ | NM_011145.3 | F: ATGGGGGACCAGAACACAC R: GGAGGAATTCTGGGAGAGGT | 62 |
| Angptl4 | NM_020581.2 | F: CACCCACTTACACAGGCCG R: GAAGTCCACAGAGCCGTTCA | 178 |
| Acaca | NM_133360.2 | F: GCCTTTCACATGAGATCCAGC R: CTGCAATACCATTGTTGGCGA | 175 |
| Fabp4 | NM_024406.3 | F: TGAAATCACCGCAGACGACA R: ACACATTCCACCACCAGCTT | 141 |
| Foxo1 | NM_019739.3 | F: CAAGGCCATCGAGAGCTCAG R: AATTGAATTCTTCCAGCCCGCC | 130 |
| Cna | NM_008913.5 | F: AAAGCGCTACTGTTGAGGCT R: ATTCGGTCTAAGCCCTTGGC | 103 |
| Pdk4 | NM_013743.2 | F: TTCCAGGCCAACCAATCCAC R: TGGCCCTCATGGCATTCTTG | 87 |
| Vegf | NM_001025250.3 | F: CTCACCAAAGCCAGCACATA R: AATGCTTTCTCCGCTCTGAA | 198 |
| Vegfr1 | NM_010228.4 | F: TACCTCACCGTGCAAGGAAC R: AAGGAGCCAAAAGAGGGTCG | 93 |
| Vegfr2 | NM_010612.3 | F: AGTGGTACTGGCAGCTAGAAG R: ACAAGCATACGGGCTTGTTT | 65 |
| Ets1 | NM_011808.3 | F: CTGACCTCAACAAGGACAAGC R: AGAAACTGCCACAGCTGGAT | 88 |
| Mmp1 | NM_008607.2 | F: GGCCAGAACTTCCCAACCAT R: AGCCCAGAATTTTCTCCCTCT | 89 |
| Mmp8 | NM_008611.4 | F: CCTGCAGGACTCCTTCTTCCT R: CCTCATAGGGTGCGTGCAA | 156 |
| Mmp9 | NM_013599.4 | F: CCATGCACTGGGCTTAGATCA R: GGCCTTGGGTCAGGCTTAGA | 147 |
| Wt1 | NM_144783.2 | F: CCAGCTCAGTGAAATGGACA R: CTGTACTGGGCACCACAGAG | 97 |
| Cd31 | NM_008816.3 | F: CGGTGTTCAGCGAGATCC R: CGACAGGATGGAAATCACAA | 71 |
| Vwf | NM_011708.4 | F: TGTGACACATGTGAGGAGCC R: CTTTGCTGGCACACTTTCCC | 127 |
| Vcam1 | NM_011693.3 | F: TATGTCAACGTTGCCCCCAA R: CAGGACTGCCCTCCTCTAGT | 73 |
| Il-1 | NM_008361.4 | F: GCCACCTTTTGACAGTGATGAG R: AGCTTCTCCACAGCCACAAT | 186 |
| Il-6 | NM_031168.2 | F: CACTTCACAAGTCGGAGGCT R: TGCCATTGCACAACTCTTTTCT | 86 |
| Il-18 | NM_008360.2 | F: CAAAGTGCCAGTGAACCCCA R: TTCACAGAGAGGGTCACAGC | 89 |
| Pdgfb | NM_011057.4 | F: GGAGTCGGCATGAATCGCT R: GCCCCATCTTCATCTACGGA | 182 |
| Pdgfra | NM_001083316.2 | F: ATGAGAGTGAGATCGAAGGCA R: CGGCAAGGTATGATGGCAGAG | 130 |
| Pdgfrb | NM_001146268.1 | F: CCAGCACCTTTGTTCTGACCT R: TGCCGTCCTGATTCATGGC | 99 |
| Sirt1 | NM_019812.3 | F: GCCGCGGATAGGTCCATA R: AACAATCTGCCACAGCGTCA | 136 |
| Sox18 | NM_009236.2 | F: ACTGGCGCAACAAAATCC R: CTTCTCCGCCGTGTTCAG | 88 |
| Thbs1 | NM_011580.4 | F: CCTGCCAGGGAAGCAACAA R: ACAGTCTATGTAGAGTTGAGCCC | 115 |
| Cd36 | NM_001159555.1 | F: GTGTGGAGCAACTGGTGGAT R: ACGTGGCCCGGTTCTAATTC | 147 |
| Tnfα | NM_013693.3 | F: GTAGCCCACGTCGTAGCAAA R: ACAAGGTACAACCCATCGGC | 137 |
| c-kit | NM_001122733.1 | F: GCCTGACGTGCATTGATCC R: AGTGGCCTCGGCTTTTTCC | 110 |
| Ccl2 | NM_011333.3 | F: AGCTGTAGTTTTTGTCACCAAGC R: GTGCTGAAGACCTTAGGGCA | 155 |
| Ccl5 | NM_013653.3 | F: TGCAGTCGTGTTTGTCACTC R: AGAGCAAGCAATGACAGGGA | 152 |
| Actnb | NM_007393.5 | F: CTTCCTCCCTGGAGAAGAGC R: ATGCCACAGGATTCCATACC | 124 |
| Gapdh | NM_001289726.1 | F: AGGTCGGTGTGAACGGATTTG R: TGTAGACCATGTAGTTGAGGTCA | 123 |
| Rplp0 | NM_007475.5 | F: CACTGGTCTAGGACCCGAGAAG R: GGTGCCTCTGGAGATTTTCG | 73 |
| Name | Accession Number | Primer Sequences | Amplicon Size (bp) 1 |
|---|---|---|---|
| PPARβ/δ | NM_006238.5 | F: TCAGAAGAAGAACCGCAACAAGTG R: CCTGCCACCAGCTTCCTCTT | 126 |
| ANGPTL4 | NM_001039667.3 | F: ATCCAGCAACTCTTCCACAAGGT R: TTGAAGTCCACTGAGCCATCGT | 254 |
| FABP4 | NM_001442.3 | F: AAGTCAAGAGCACCATAACCTTAGATG R: TGACGCATTCCACCACCAGTT | 120 |
| CNA | NM_000944.5 | F: AAAGCGCTACTGTTGAGGCT R: ATTCGGTCTAAGCCCTTGGC | 103 |
| PDK4 | NM_002612.4 | F: CCACATTGGAAGCATTGATCCTAACT R: TCACAGAGCATCCTTGAACACTCA | 81 |
| VEGFA | NM_001025366.3 | F: GAGGAGTCCAACATCACCATGC R: CTTGCAACGCGAGTCTGTGTT | 351 |
| VEGFR1 | NM_002019.4 | F: GCCCGGGATATTTATAAGAAC R: CCATCCATTTTAGGGGAAGTC | 70 |
| VEGFR2 | NM_002253.3 | F: CAGAGTGAGGAAGGAGGACGAAGG R: GATGATGACAAGAAGTAGCCAGAAGAACA | 181 |
| CD31 | NM_000442.5 | F: GCCCGAAGGCAGAACTAAC R: AACAGAGCAGAAGGGTCAG | 111 |
| VWF | NM_000552.4 | F: TGTGACACATGTGAGGAGCC R: CTTTGCTGGCACACTTTCCC | 127 |
| PDGFB | NM_002608.4 | F: TCCGCTCCTTTGATGATCTCCAA R: GGTCATGTTCAGGTCCAACTCG | 83 |
| c-KIT | NM_000222.2 | F: GCTCTGCTTCTGTACTGCC R: TAGGCAGAAGTCTTGCCCAC | 160 |
| SOX18 | NM_018419.3 | F: ATGGTGTGGGCAAAGGAC R: GCGTTCAGCTCCTTCCAC | 107 |
| ACTNB | NM_001101.5 | F: CTCCTTAATGTCACGCACGAT R: CATGTACGTTGCTATCCAGGC | 250 |
| GAPDH | NM_002046.7 | F: AGCCACATCGCTCAGACAC R: GCCCAATACGACCAAATCC | 66 |
| RPLP0 | NM_001002.4 | F: CAGATTGGCTACCCAACTGTT R: GGCCAGGACTCGTTTGTACC | 69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, K.-D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.-F.; Wagner, N. Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623. https://doi.org/10.3390/cells8121623
Wagner K-D, Du S, Martin L, Leccia N, Michiels J-F, Wagner N. Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells. 2019; 8(12):1623. https://doi.org/10.3390/cells8121623
Chicago/Turabian StyleWagner, Kay-Dietrich, Siyue Du, Luc Martin, Nathalie Leccia, Jean-François Michiels, and Nicole Wagner. 2019. "Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression" Cells 8, no. 12: 1623. https://doi.org/10.3390/cells8121623
APA StyleWagner, K.-D., Du, S., Martin, L., Leccia, N., Michiels, J.-F., & Wagner, N. (2019). Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells, 8(12), 1623. https://doi.org/10.3390/cells8121623