Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia

 , ,
, ,  and
and
Abstract
1. Introduction
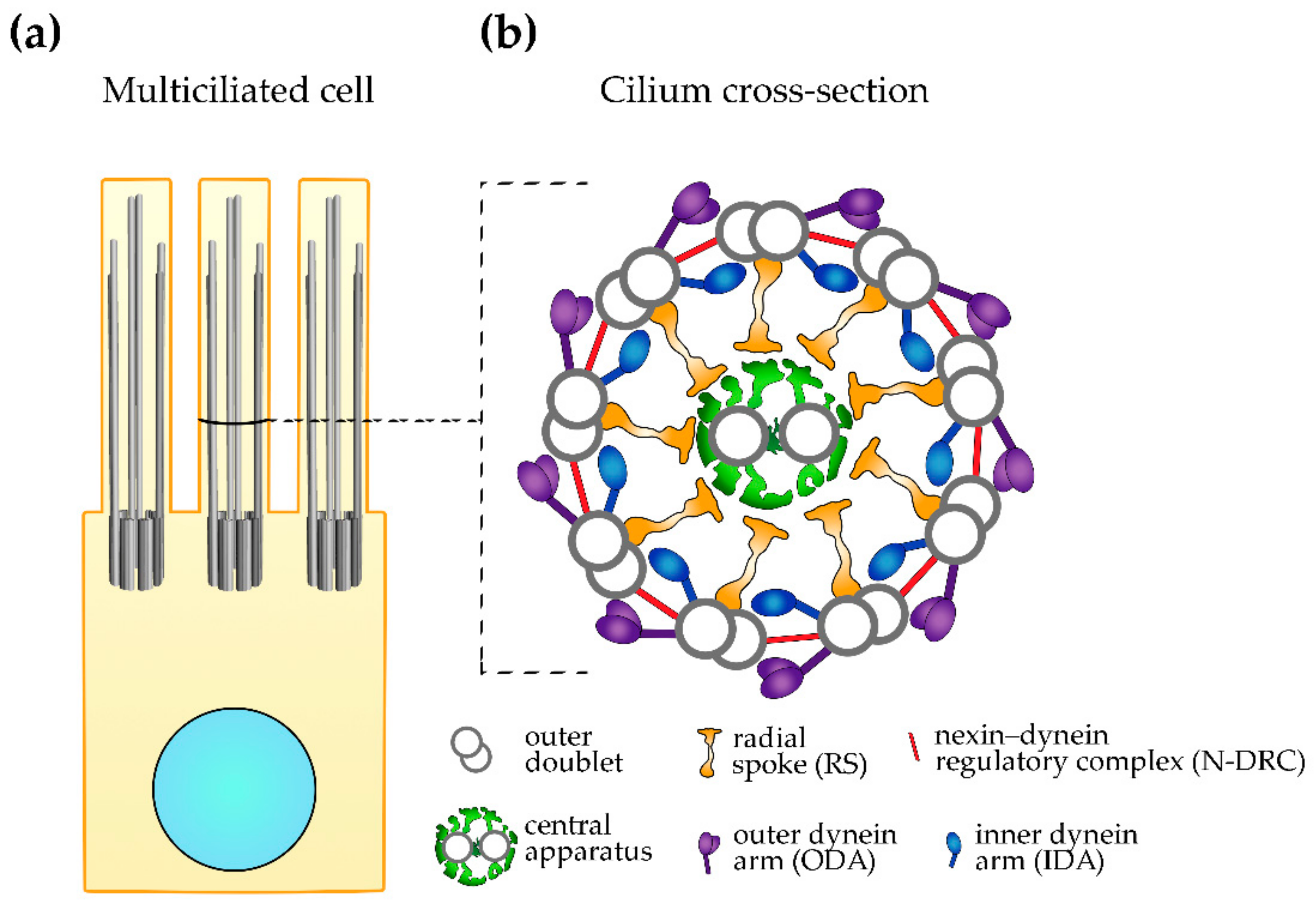
2. Ciliary Ultrastructure
3. Primary Ciliary Dyskinesia (PCD)
4. Advantages of the Model Organisms
4.1. Unicellular Models—the Power of Being Small
4.2. Vertebrate Models
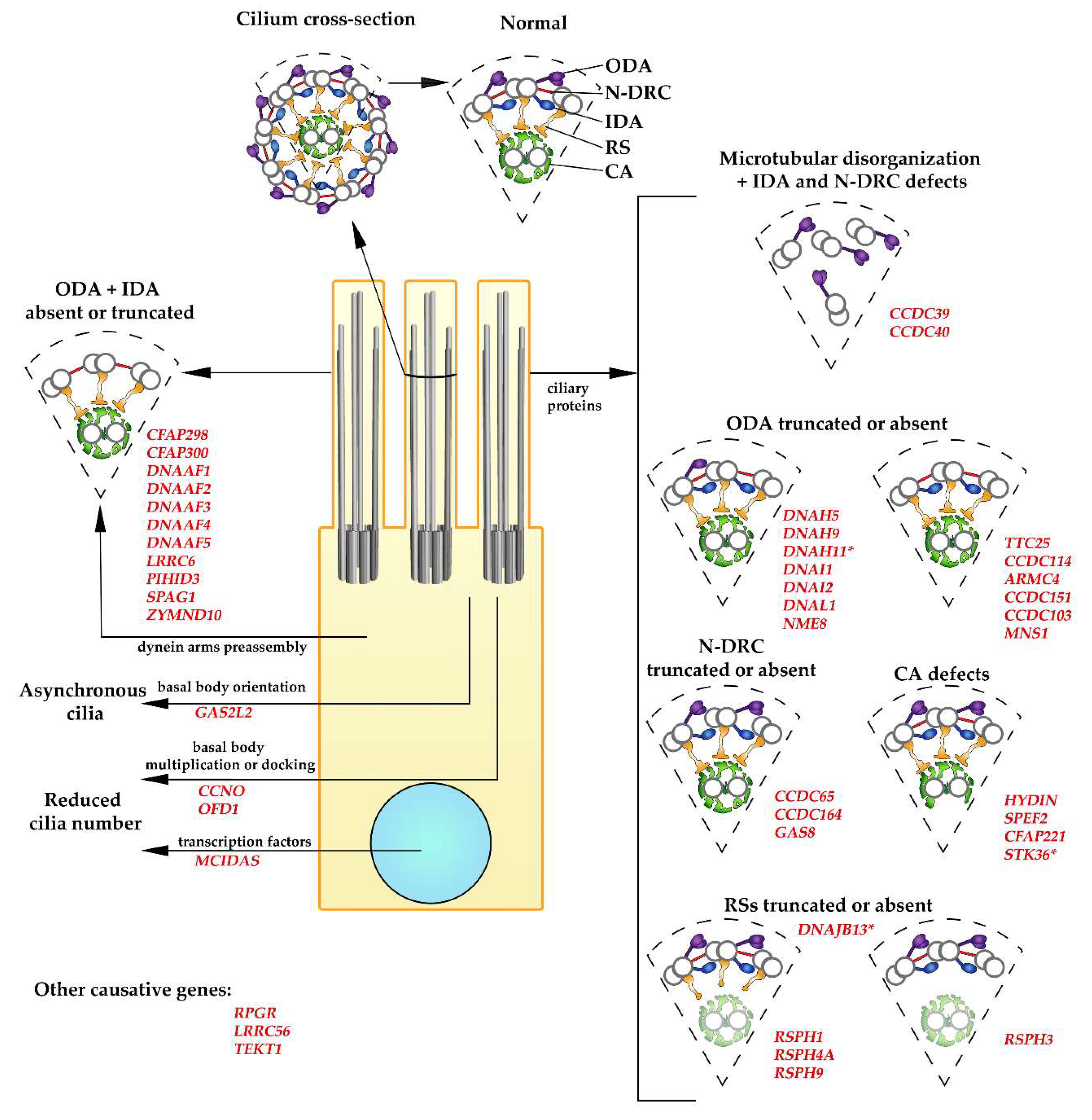
5. Causative Genes and Ultrastructural Changes
5.1. PCD Caused by a Reduced Number of Cilia
5.2. CCDC39 and CCDC40
5.3. Dynein Arms
5.3.1. Dynein Arm Subunits
5.3.2. Dynein Docking Proteins
5.3.3. Factors Involved in Dynein Arms Preassembly
5.4. Nexin–Dynein Regulatory Complex (N-DRC)
5.5. Radial Spokes
5.6. Central Apparatus
6. Other Proteins Causing PCD-Like Symptoms in Humans
7. Other Proteins Causing PCD in Model Organisms: Novel Candidate Genes in Humans?
8. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Mitchell, D.R. The evolution of eukaryotic cilia and flagella as motile and sensory organelles. Adv. Exp. Med. Biol. 2007, 607, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Falk, N.; Lösl, M.; Schröder, N.; Gießl, A. Specialized cilia in mammalian sensory systems. Cells 2015, 4, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Tasouri, E.; Tucker, K.L. Primary cilia and organogenesis: Is Hedgehog the only sculptor? Cell Tissue Res. 2011, 345, 21–40. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef]
- Christensen, S.T.; Pedersen, L.B.; Schneider, L.; Satir, P. Sensory cilia and integration of signal transduction in human health and disease. Traffic 2007, 8, 97–109. [Google Scholar] [CrossRef]
- Shah, A.S.; Ben-Shahar, Y.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile cilia of human airway epithelia are chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef]
- Jain, R.; Javidan-Nejad, C.; Alexander-Brett, J.; Horani, A.; Cabellon, M.C.; Walter, M.J.; Brody, S.L. Sensory functions of motile cilia and implication for bronchiectasis. Front Biosci. 2012, 4, 1088–1098. [Google Scholar]
- Noone, P.G.; Leigh, M.W.; Sannuti, A.; Minnix, S.L.; Carson, J.L.; Hazucha, M.; Zariwala, M.A.; Knowles, M.R. Primary ciliary dyskinesia: Diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004, 69, 459–467. [Google Scholar] [CrossRef]
- Shinohara, K.; Hamada, H. Cilia in left-right symmetry breaking. Cold Spring Harb. Perspect. Biol. 2017, 9, a028282. [Google Scholar] [CrossRef]
- Ishikawa, T. Axoneme structure from motile cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, a028076. [Google Scholar] [CrossRef]
- Loreng, T.D.; Smith, E.F. The central apparatus of cilia and eukaryotic flagella. Cold Spring Harb. Perspect. Biol. 2017, 9, a028118. [Google Scholar] [CrossRef] [PubMed]
- Carbajal-González, B.I.; Heuser, T.; Fu, X.; Lin, J.; Smith, B.W.; Mitchell, D.R.; Nicastro, D. Conserved structural motifs in the central pair complex of eukaryotic flagella. Cytoskeleton 2013, 70, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, D.; Schwartz, C.; Pierson, J.; Gaudette, R.; Porter, M.E.; McIntosh, J.R. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 2006, 313, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Osinka, A.; Poprzeczko, M.; Zielinska, M.M.; Fabczak, H.; Joachimiak, E.; Wloga, D. Ciliary proteins: Filling the gaps. recent advances in deciphering the protein composition of motile ciliary complexes. Cells 2019, 8, 730. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Song, K.; Yanagisawa, H.A.; Fox, L.; Yagi, T.; Wirschell, M.; Hirono, M.; Kamiya, R.; Nicastro, D.; Sale, W.S. The MIA complex is a conserved and novel dynein regulator essential for normal ciliary motility. J. Cell Biol. 2013, 201, 263–278. [Google Scholar] [CrossRef]
- Fu, G.; Wang, Q.; Phan, N.; Urbanska, P.; Joachimiak, E.; Lin, J.; Wloga, D.; Nicastro, D. The I1 dynein-associated tether and tether head complex is a conserved regulator of ciliary motility. Mol. Biol. Cell 2018, 29, 1048–1059. [Google Scholar] [CrossRef]
- Kubo, T.; Hou, Y.; Cochran, D.A.; Witman, G.B.; Oda, T. A microtubule-dynein tethering complex regulates the axonemal inner dynein f (I1). Mol. Biol. Cell 2018, 29, 1060–1074. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, Y.; Yang, P. Radial spokes-a snapshot of the motility regulation, assembly, and evolution of cilia and flagella. Cold Spring Harb. Perspect. Biol. 2017, 9, a028126. [Google Scholar] [CrossRef]
- Viswanadha, R.; Sale, W.S.; Porter, M.E. Ciliary motility: Regulation of axonemal dynein motors. Cold Spring Harb. Perspect. Biol. 2017, 9, a018325. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Agrin, N.; Leszyk, J.; Witman, G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005, 170, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Northey, J.G.; Garg, J.; Pearlman, R.E.; Siu, K.W. Robust method for proteome analysis by MS/MS using an entire translated genome: Demonstration on the ciliome of Tetrahymena thermophila. J. Proteome Res. 2005, 4, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, R.; Dawe, H.R.; Farr, H.; Griffiths, S.; Hart, S.R.; Portman, N.; Shaw, M.K.; Ginger, M.L.; Gaskell, S.J.; McKean, P.G.; et al. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 2006, 440, 224–227. [Google Scholar] [CrossRef]
- Leigh, M.W.; Horani, A.; Kinghorn, B.; O’Connor, M.G.; Zariwala, M.A.; Knowles, M.R. Primary Ciliary Dyskinesia (PCD): A genetic disorder of motile cilia. Transl. Sci. Rare Dis. 2019, 4, 51–75. [Google Scholar] [CrossRef]
- Leigh, M.W.; O’Callaghan, C.; Knowles, M.R. The challenges of diagnosing primary ciliary dyskinesia. Proc Am Thorac Soc. 2011, 8, 434–437. [Google Scholar] [CrossRef]
- Hayes, D., Jr.; Reynolds, S.D.; Tumin, D. Outcomes of lung transplantation for primary ciliary dyskinesia and Kartagener syndrome. J. Heart Lung Transplant. 2016, 35, 1377–1378. [Google Scholar] [CrossRef]
- Blyth, M.; Wellesley, D. Ectopic pregnancy in primary ciliary dyskinesia. J. Obstet. Gynaecol. 2008, 28, 358. [Google Scholar] [CrossRef]
- Vanaken, G.J.; Bassinet, L.; Boon, M.; Mani, R.; Honoré, I.; Papon, J.-F.; Cuppens, H.; Jaspers, M.; Lorent, N.; Coste, A.; et al. Infertility in an adult cohort with primary ciliary dyskinesia: Phenotype–gene association. Eur. Respir. J. 2017, 50, 1700314. [Google Scholar] [CrossRef]
- Goutaki, M.; Meier, A.B.; Halbeisen, F.S.; Lucas, J.S.; Dell, S.D.; Maurer, E.; Casaulta, C.; Jurca, M.; Spycher, B.D.; Kuehni, C.E. Clinical manifestations in primary ciliary dyskinesia: Systematic review and meta-analysis. Eur. Respir. J. 2016, 48, 1081–1095. [Google Scholar] [CrossRef]
- Lobo, J.; Zariwala, M.A.; Noone, P.G. Primary ciliary dyskinesia. Semin. Respir. Crit. Care Med. 2015, 36, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Davis, S.D.; Ferkol, T.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Wolf, W.; et al. Genetic disorders of mucociliary clearance consortium. laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest 2014, 146, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; le Borgne, P.; Louis, B.; Daudvohra, F.; Patel, M.P.; Thomas, L.; et al. Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.A.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef]
- Urbanska, P.; Joachimiak, E.; Bazan, R.; Fu, G.; Poprzeczko, M.; Fabczak, H.; Nicastro, D.; Wloga, D. Ciliary proteins Fap43 and Fap44 interact with each other and are essential for proper cilia and flagella beating. Cell. Mol. Life Sci. 2018, 75, 4479–4493. [Google Scholar] [CrossRef]
- Behan, L.; Dimitrov, B.D.; Kuehni, C.E.; Hogg, C.; Carroll, M.; Evans, H.J.; Goutaki, M.; Harris, A.; Packham, S.; Walker, W.T.; et al. PICADAR: A diagnostic predictive tool for primary ciliary dyskinesia. Eur. Respir. J. 2016, 47, 1103–1112. [Google Scholar] [CrossRef]
- Werner, C.; Onnebrink, J.G.; Omran, H. Diagnosis and management of primary ciliary dyskinesia. Cilia 2015, 4, 2. [Google Scholar] [CrossRef]
- Lucas, J.S.; Paff, T.; Goggin, P.; Haarman, E. Diagnostic Methods in Primary Ciliary Dyskinesia. Paediatr. Respir. Rev. 2016, 18, 8–17. [Google Scholar] [CrossRef]
- Vincensini, L.; Blisnick, T.; Bastin, P. 1001 model organisms to study cilia and flagella. Biol. Cell. 2011, 103, 109–130. [Google Scholar] [CrossRef]
- Brown, J.M.; Witman, G.B. Cilia and Diseases. Bioscience 2014, 64, 1126–1137. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, X.; Jia, S.; Yelick, P.C.; Zhao, C. Zebrafish as a Model for Human Ciliopathies. J. Genet. Genom. 2016, 43, 107–120. [Google Scholar] [CrossRef]
- Wingfield, J.L.; Lechtreck, K.F.; Lorentzen, E. Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery. Essays Biochem. 2018, 62, 753–763. [Google Scholar] [CrossRef]
- Blum, M.; Ott, T. Xenopus: An undervalued model organism to study and model human genetic disease. Cells Tissues Organs 2018, 205, 303–313. [Google Scholar] [CrossRef]
- Dave, D.; Wloga, D.; Gaertig, J. Manipulating ciliary protein-encoding genes in Tetrahymena thermophila. Methods Cell Biol. 2009, 93, 1–20. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Corpuz, E.O.; Hubenschmidt, M.J.; Townsend, C.R.; Asai, D.J.; Wilkes, D.E. Analysis of properties of cilia using Tetrahymena thermophila. Methods Mol. Biol. 2009, 586, 283–299. [Google Scholar] [CrossRef]
- Gaertig, J.; Wloga, D.; Vasudevan, K.K.; Guha, M.; Dentler, W. Discovery and functional evaluation of ciliary proteins in Tetrahymena thermophila. Methods Enzymol. 2013, 525, 265–284. [Google Scholar] [CrossRef]
- Lin, H.; Dutcher, S.K. Genetic and genomic approaches to identify genes involved in flagellar assembly in Chlamydomonas reinhardtii. Methods Cell Biol. 2015, 127, 349–386. [Google Scholar] [CrossRef]
- Santi-Rocca, J.; Chenouard, N.; Fort, C.; Lagache, T.; Olivo-Marin, J.C.; Bastin, P. Imaging intraflagellar transport in trypanosomes. Methods Cell Biol. 2015, 127, 487–508. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Kamiya, R. Axonemal motility in Chlamydomonas. Methods Cell Biol. 2015, 127, 387–402. [Google Scholar] [CrossRef]
- Nicastro, D. Cryo-electron microscope tomography to study axonemal organization. Methods Cell Biol. 2009, 91, 1–39. [Google Scholar] [CrossRef]
- Oda, T.; Kikkawa, M. Novel structural labeling method using cryo-electron tomography and biotin-streptavidin system. J. Struct. Biol. 2013, 183, 305–311. [Google Scholar] [CrossRef]
- Kramer-Zucker, A.G.; Olale, F.; Haycraft, C.J.; Yoder, B.K.; Schier, A.F.; Drummond, I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 2005, 132, 1907–1921. [Google Scholar] [CrossRef]
- Olstad, E.W.; Ringers, C.; Hansen, J.N.; Wens, A.; Brandt, C.; Wachten, D.; Yaksi, E.; Jurisch-Yaksi, N. Ciliary Beating Compartmentalizes Cerebrospinal Fluid Flow in the Brain and Regulates Ventricular Development. Curr. Biol. 2019, 29, 229–241.e6. [Google Scholar] [CrossRef]
- Choksi, S.P.; Babu, D.; Lau, D.; Yu, X.; Roy, S. Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development 2014, 141, 3410–3419. [Google Scholar] [CrossRef]
- Hartill, V.L.; van de Hoek, G.; Patel, M.P.; Little, R.; Watson, C.M.; Berry, I.R.; Shoemark, A.; Abdelmottaleb, D.; Parkes, E.; Bacchelli, C.; et al. DNAAF1 links heart laterality with the AAA+ ATPase RUVBL1 and ciliary intraflagellar transport. Hum. Mol. Genet. 2018, 27, 529–545. [Google Scholar] [CrossRef]
- Blum, M.; Beyer, T.; Weber, T.; Vick, P.; Andre, P.; Bitzer, E.; Schweickert, A. Xenopus, an ideal model system to study vertebrate left-right asymmetry. Dev. Dyn. 2009, 238, 1215–1225. [Google Scholar] [CrossRef]
- Billett, F.S.; Gould, R.P. Fine structural changes in the differentiating epidermis of Xenopus laevis embryos. J. Anat. 1971, 108, 465–480. [Google Scholar]
- Malicki, J.; Avanesov, A.; Li, J.; Yuan, S.; Sun, Z. Analysis of cilia structure and function in zebrafish. Methods Cell Biol. 2011, 101, 39–74. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Gonzales, A.P.; Joung, J.K.; Yeh, J.R. Targeted mutagenesis in zebrafish using crispr rna-guided nucleases. Methods Mol. Biol. 2015, 1311, 317–334. [Google Scholar] [CrossRef]
- Leventea, E.; Hazime, K.; Zhao, C.; Malicki, J. Analysis of cilia structure and function in zebrafish. Methods Cell Biol. 2016, 133, 179–227. [Google Scholar] [CrossRef] [PubMed]
- Albadri, S.; Del Bene, F.; Revenu, C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods 2017, 121–122, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Walentek, P.; Quigley, I.K. What we can learn from a tadpole about ciliopathies and airway diseases: Using systems biology in Xenopus to study cilia and mucociliary epithelia. Genesis 2017, 55. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.J.; Bian, W.P.; Liu, Y.; Huang, H.Y.; Yin, Q.; Yang, X.J.; Pei, D.S. CRISPR/Cas9-based genome engineering of zebrafish using a seamless integration strategy. FASEB J. 2018, 32, 5132–5142. [Google Scholar] [CrossRef]
- Naert, T.; Vleminckx, K. CRISPR/Cas9 disease models in zebrafish and Xenopus: The genetic renaissance of fish and frogs. Drug. Discov. Today Technol. 2018, 28, 41–52. [Google Scholar] [CrossRef]
- Norris, D.P.; Grimes, D.T. Mouse models of ciliopathies: The state of the art. Dis. Model Mech. 2012, 5, 299–312. [Google Scholar] [CrossRef]
- Boon, M.; Wallmeier, J.; Ma, L.; Loges, N.T.; Jaspers, M.; Olbrich, H.; Dougherty, G.W.; Raidt, J.; Werner, C.; Amirav, I.; et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Commun. 2014, 5, 4418. [Google Scholar] [CrossRef]
- Lu, H.; Anujan, P.; Zhou, F.; Zhang, Y.; Chong, Y.L.; Bingle, C.D.; Roy, S. Mcidas mutant mice reveal a two-step process for the specification and differentiation of multiciliated cells in mammals. Development 2019, 146, dev172643. [Google Scholar] [CrossRef]
- Wallmeier, J.; Al-Mutairi, D.A.; Chen, C.T.; Loges, N.T.; Pennekamp, P.; Menchen, T.; Ma, L.; Shamseldin, H.E.; Olbrich, H.; Dougherty, G.W.; et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 2014, 46, 646–651. [Google Scholar] [CrossRef]
- Amirav, I.; Wallmeier, J.; Loges, N.T.; Menchen, T.; Pennekamp, P.; Mussaffi, H.; Abitbul, R.; Avital, A.; Bentur, L.; Dougherty, G.W.; et al. Israeli PCD Consortium Investigators. Systematic analysis of Ccno variants in a defined population: Implications for clinical phenotype and differential diagnosis. Hum. Mutat. 2016, 37, 396–405. [Google Scholar] [CrossRef]
- Stubbs, J.L.; Vladar, E.K.; Axelrod, J.D.; Kintner, C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat. Cell Biol. 2012, 14, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.C.; Bera, A.N.; Menchen, T.; Kuales, G.; Thriene, K.; Lienkamp, S.S.; Dengjel, J.; Omran, H.; Frank, M.; Arnold, S.J. Cyclin O (Ccno) functions during deuterosome-mediated centriole amplification of multiciliated cells. EMBO J. 2015, 34, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Spassky, N.; Meunier, A. The development and functions of multiciliated epithelia. Nat. Rev. Mol. Cell Biol. 2017, 18, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A. Airway epithelial differentiation and mucociliary clearance. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. 3), S143–S148. [Google Scholar] [CrossRef]
- Arbi, M.; Pefani, D.E.; Taraviras, S.; Lygerou, Z. Controlling centriole numbers: Geminin family members as master regulators of centriole amplification and multiciliogenesis. Chromosoma 2018, 127, 151–174. [Google Scholar] [CrossRef]
- Shahid, U.; Singh, P. Emerging picture of deuterosome-dependent centriole amplification in mccs. Cells 2018, 7, 152. [Google Scholar] [CrossRef]
- Blanchon, S.; Legendre, M.; Copin, B.; Duquesnoy, P.; Montantin, G.; Kott, E.; Dastot, F.; Jeanson, L.; Cachanado, M.; Rousseau, A.; et al. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J. Med. Genet. 2012, 49, 410–416. [Google Scholar] [CrossRef]
- Merveille, A.C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef]
- Abdelhamed, Z.; Vuong, S.M.; Hill, L.; Shula, C.; Timms, A.; Beier, D.; Campbell, K.; Mangano, F.T.; Stottmann, R.W.; Goto, J. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 2018, 145, dev154500. [Google Scholar] [CrossRef]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Omran, H. Dynein dysfunction as a cause of primary ciliary dyskinesia and other ciliopathies. In Dyneins (Second Edition) Dynein Mechanics, Dysfunction, and Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 316–355. [Google Scholar] [CrossRef]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Omran, H.; Häffner, K.; Völkel, A.; Kuehr, J.; Ketelsen, U.P.; Ross, U.H.; Konietzko, N.; Wienker, T.; Brandis, M.; Hildebrandt, F. Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am. J. Respir. Cell Mol. Biol. 2000, 23, 696–702. [Google Scholar] [CrossRef]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef]
- Schwabe, G.C.; Hoffmann, K.; Loges, N.T.; Birker, D.; Rossier, C.; de Santi, M.M.; Olbrich, H.; Fliegauf, M.; Failly, M.; Liebers, U.; et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008, 29, 289–298. [Google Scholar] [CrossRef]
- Knowles, M.R.; Leigh, M.W.; Carson, J.L.; Davis, S.D.; Dell, S.D.; Ferkol, T.W.; Olivier, K.N.; Sagel, S.D.; Rosenfeld, M.; Burns, K.A.; et al. Genetic Disorders of Mucociliary Clearance Consortium. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 2012, 67, 433–441. [Google Scholar] [CrossRef]
- Raidt, J.; Wallmeier, J.; Hjeij, R.; Onnebrink, J.G.; Pennekamp, P.; Loges, N.T.; Olbrich, H.; Haffner, K.; Dougherty, G.W.; Omran, H.; et al. Ciliary beat pattern and frequency in genetic variants of primaryciliary dyskinesia. Eur. Respir. J. 2014, 44, 1579–1588. [Google Scholar] [CrossRef]
- Dougherty, G.W.; Loges, N.T.; Klinkenbusch, J.A.; Olbrich, H.; Pennekamp, P.; Menchen, T.; Raidt, J.; Wallmeier, J.; Werner, C.; Westermann, C.; et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell Mol. Biol. 2016, 55, 213–224. [Google Scholar] [CrossRef]
- Shoemark, A.; Burgoyne, T.; Kwan, R.; Dixon, M.; Patel, M.P.; Rogers, A.V.; Onoufriadis, A.; Scully, J.; Daudvohra, F.; Cullup, T.; et al. Primary ciliary dyskinesia with normal ultrastructure: Three-dimensional tomography detects absence of DNAH11. Eur. Respir. J. 2018, 51, 1701809. [Google Scholar] [CrossRef]
- Pennarun, G.; Escudier, E.; Chapelin, C.; Bridoux, A.M.; Cacheux, V.; Roger, G.; Clément, A.; Goossens, M.; Amselem, S.; Duriez, B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999, 65, 1508–1519. [Google Scholar] [CrossRef]
- Guichard, C.; Harricane, M.C.; Lafitte, J.J.; Godard, P.; Zaegel, M.; Tack, V.; Lalau, G.; Bouvagnet, P. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am. J. Hum. Genet. 2001, 68, 1030–1035. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Leigh, M.W.; Ceppa, F.; Kennedy, M.P.; Noone, P.G.; Carson, J.L.; Hazucha, M.J.; Lori, A.; Horvath, J.; Olbrich, H.; et al. Mutations of DNAI1 in primary ciliary dyskinesia: Evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med. 2006, 174, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Mazor, M.; Alkrinawi, S.; Chalifa-Caspi, V.; Manor, E.; Sheffield, V.C.; Aviram, M.; Parvari, R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011, 88, 599–607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duriez, B.; Duquesnoy, P.; Escudier, E.; Bridoux, A.M.; Escalier, D.; Rayet, I.; Marcos, E.; Vojtek, A.M.; Bercher, J.F.; Amselem, S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA 2007, 104, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Agrin, N.; Walker, B.L.; Witman, G.B. Identification of predicted human outer dynein arm genes: Candidates for primary ciliary dyskinesia genes. J. Med. Genet. 2006, 43, 62–73. [Google Scholar] [CrossRef]
- King, S.M. Axonemal Dynein Arms. Cold Spring Harb. Perspect. Biol. 2016, 8, a028100. [Google Scholar] [CrossRef]
- Brokaw, C.J.; Kamiya, R. Bending patterns of Chlamydomonas flagella: IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskeleton 1987, 8, 68–75. [Google Scholar] [CrossRef]
- Kamiya, R. Functional diversity of axonemal dyneins as studied in Chlamydomonas mutants. Int. Rev. Cytol. 2002, 219, 115–155. [Google Scholar] [CrossRef]
- King, S.M.; Kamiya, R. Axonemal dyneins: Assembly, structure, and force generation. In The Chlamydomonas Sourcebook: Cell Motility and Behavior, 2nd ed.; Witman, G.B., Ed.; Academic Press: San Diego, CA, USA, 2009; Volume 3, pp. 131–208. [Google Scholar]
- Ibañez-Tallon, I.; Gorokhova, S.; Heintz, N. Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet. 2002, 11, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, L.E.; Yin, W.; Rogers, T.D.; Busalacchi, K.B.; Chua, M.; O’Neal, W.K.; Grubb, B.R. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am. J. Respir. Cell. Mol. Biol. 2010, 43, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Adam, E.C.; Goggin, P.M.; Jackson, C.L.; Powles-Glover, N.; Patel, S.H.; Humphreys, J.; Fray, M.D.; Falconnet, E.; Blouin, J.L.; et al. Static respiratory cilia associated with mutations in Dnahc11/DNAH11: A mouse model of PCD. Hum. Mutat. 2012, 33, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Paff, T.; Antony, D.; Shoemark, A.; Micha, D.; Kuyt, B.; Schmidts, M.; Petridi, S.; Dankert-Roelse, J.E.; Haarman, E.G.; et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Leigh, M.W.; Ostrowski, L.E.; Huang, L.; Carson, J.L.; Hazucha, M.J.; Yin, W.; Berg, J.S.; Davis, S.D.; Dell, S.D.; et al. Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.H.; Singaraja, R.R. Loss-of-function mutations in CCDC114 cause primary ciliary dyskinesia. Clin. Genet. 2013, 83, 526–527. [Google Scholar] [CrossRef]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.; et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Shoemark, A.; Munye, M.M.; James, C.T.; Schmidts, M.; Patel, M.; Rosser, E.M.; Bacchelli, C.; Beales, P.L.; Scambler, P.J.; et al. Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 2014, 51, 61–67. [Google Scholar] [CrossRef]
- Wallmeier, J.; Shiratori, H.; Dougherty, G.W.; Edelbusch, C.; Hjeij, R.; Loges, N.T.; Menchen, T.; Olbrich, H.; Pennekamp, P.; Raidt, J.; et al. TTC25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am. J. Hum. Genet. 2016, 99, 460–469. [Google Scholar] [CrossRef]
- Jerber, J.; Baas, D.; Soulavie, F.; Chhin, B.; Cortier, E.; Vesque, C.; Thomas, J.; Durand, B. The coiled-coil domain containing protein CCDC151 is required for the function of IFT-dependent motile cilia in animals. Hum. Mol. Genet. 2014, 23, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cao, J.; Huang, S.; Feng, D.; Zhang, W.; Zhu, X.; Yan, X. Characterization of tetratricopeptide repeat-containing proteins critical for cilia formation and function. PLoS ONE 2015, 10, e0124378. [Google Scholar] [CrossRef] [PubMed]
- Koutoulis, A.; Pazour, G.J.; Wilkerson, C.G.; Inaba, K.; Sheng, H.; Takada, S.; Witman, G.B. The Chlamydomonas reinhardtii ODA3 gene encodes a protein of the outer dynein arm docking complex. J. Cell Biol. 1997, 137, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Wilkerson, C.G.; Wakabayashi, K.; Kamiya, R.; Witman, G.B. The outer dynein arm-docking complex: Composition and characterization of a subunit (oda1) necessary for outer arm assembly. Mol. Biol. Cell 2002, 13, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.M.; Inaba, K.; Pazour, G.J.; Takada, S.; Wakabayashi, K.; Wilkerson, C.G.; Kamiya, R.; Witman, G.B. DC3, the 21-kDa subunit of the outer dynein arm docking complex (ODA-DC), is a novel EF-hand protein important for assembly of both the outer arm and the ODA-DC. Mol. Biol. Cell 2003, 14, 3650–3663. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.M.; Yagi, T.; Kamiya, R.; Witman, G.B. DC3, the smallest subunit of the Chlamydomonas flagellar outer dynein arm-docking complex, is a redox-sensitive calcium-binding protein. J. Biol. Chem. 2003, 278, 42652–42659. [Google Scholar] [CrossRef]
- Dean, A.B.; Mitchell, D.R. Late steps in cytoplasmicmaturation of assembly-competent axonemal outer armdynein in Chlamydomonas require interaction of ODA5 and ODA10 in a complex. Mol. Biol. Cell 2015, 26, 3596–3605. [Google Scholar] [CrossRef]
- Hayes, J.M.; Kim, S.K.; Abitua, P.B.; Park, T.J.; Herrington, E.R.; Kitayama, A.; Grow, M.W.; Ueno, N.; Wallingford, J.B. Identification of novel ciliogenesis factors using a new in vivo model for mucociliary epithelial development. Dev. Biol. 2007, 312, 115–130. [Google Scholar] [CrossRef]
- Chung, M.I.; Peyrot, S.M.; LeBoeuf, S.; Park, T.J.; McGary, K.L.; Marcotte, E.M.; Wallingford, J.B. RFX2 is broadly required for ciliogenesis during vertebrate development. Dev. Biol. 2012, 363, 155–165. [Google Scholar] [CrossRef]
- Ta-Shma, A.; Hjeij, R.; Perles, Z.; Dougherty, G.W.; Abu Zahira, I.; Letteboer, S.J.F.; Antony, D.; Darwish, A.; Mans, D.A.; Spittler, S.; et al. Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet. 2018, 14, e1007602. [Google Scholar] [CrossRef]
- Leslie, J.S.; Rawlins, L.E.; Chioza, B.A.; Olubodun, O.R.; Salter, C.G.; Fasham, J.; Jones, H.F.; Cross, H.E.; Lam, S.; Harlalka, G.V.; et al. MNS1 variant associated with situs inversus and male infertility. Eur. J. Hum. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Panizzi, J.R.; Becker-Heck, A.; Castleman, V.H.; Al-Mutairi, D.A.; Liu, Y.; Loges, N.T.; Pathak, N.; Austin-Tse, C.; Sheridan, E.; Schmidts, M.; et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012, 44, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, A.; Moya, E.; Hirst, R.A.; Patel, M.P.; Robson, E.A.; Hayward, J.; Scully, J.; Fassad, M.R.; Lamb, W.; Schmidts, M.; et al. High prevalence of CCDC103 p.His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax 2018, 73, 157–166. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Patel-King, R.S. The oligomeric outer dynein arm assembly factor CCDC103 is tightly integrated within the ciliary axoneme and exhibits periodic binding to microtubules. J. Biol. Chem. 2015, 290, 7388–7401. [Google Scholar] [CrossRef] [PubMed]
- Duquesnoy, P.; Escudier, E.; Vincensini, L.; Freshour, J.; Bridoux, A.M.; Coste, A.; Deschildre, A.; de Blic, J.; Legendre, M.; Montantin, G.; et al. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009, 85, 890–896. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Häffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012, 44, 381–389, S1–S2. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, W.; Huang, J.; Wang, L.; Qian, L. Clinical and genetic analysis of patients with primary ciliary dyskinesia caused by novel DNAAF3 mutations. J. Hum. Genet. 2019, 64, 711–719. [Google Scholar] [CrossRef]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef]
- Casey, J.P.; McGettigan, P.A.; Healy, F.; Hogg, C.; Reynolds, A.; Kennedy, B.N.; Ennis, S.; Slattery, D.; Lynch, S.A. Unexpected genetic heterogeneity for primary ciliary dyskinesia in the Irish Traveller population. Eur. J. Hum. Genet. 2015, 23, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Moore, D.J.; Mali, G.; zur Lage, P.; Ait-Lounis, A.; Schmidts, M.; Shoemark, A.; Garcia Munoz, A.; Halachev, M.R.; Gautier, P.; et al. HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet. 2014, 10, e1004577. [Google Scholar] [CrossRef] [PubMed]
- Paff, T.; Loges, N.T.; Aprea, I.; Wu, K.; Bakey, Z.; Haarman, E.G.; Daniels, J.M.A.; Sistermans, E.A.; Bogunovic, N.; Dougherty, G.W.; et al. Mutations in PIH1D3 Cause X-Linked Primary Ciliary Dyskinesia with Outer and Inner Dynein Arm Defects. Am. J. Hum. Genet. 2017, 100, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Olcese, C.; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; Emes, R.D.; et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef]
- Fassad, M.R.; Shoemark, A.; le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 mutations disrupting the intraflagellar transport-dependent assembly of multiple axonemal dyneins cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef]
- Höben, I.M.; Hjeij, R.; Olbrich, H.; Dougherty, G.W.; Nöthe-Menchen, T.; Aprea, I.; Frank, D.; Pennekamp, P.; Dworniczak, B.; Wallmeier, J.; et al. Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. Am. J. Hum. Genet. 2018, 102, 973–984. [Google Scholar] [CrossRef]
- Zietkiewicz, E.; Bukowy-Bieryllo, Z.; Rabiasz, A.; Daca-Roszak, P.; Wojda, A.; Voelkel, K.; Rutkiewicz, E.; Pogorzelski, A.; Rasteiro, M.; Witt, M. cfap300: Mutations in slavic patients with primary ciliary dyskinesia and a role in ciliary dynein arms trafficking. Am. J. Respir. Cell Mol. Biol. 2019, 61, 440–449. [Google Scholar] [CrossRef]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.F.; Siffroi, J.P.; et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Oren, Y.S.; Kerem, B.; Amirav, I.; Cohen-Cymberknoh, M.; Dutcher, S.K.; Brody, S.L.; et al. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS ONE 2013, 8, e59436. [Google Scholar] [CrossRef]
- Liu, L.; Luo, H. Whole-exome sequencing identified a novel compound heterozygous mutation of lrrc6 in a chinese primary ciliary dyskinesia patient. Biomed. Res. Int. 2018, 1854269. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Loges, N.T.; Hurd, T.; Leigh, M.W.; Huang, L.; Wolf, W.E.; Carson, J.L.; Hazucha, M.J.; Yin, W.; et al. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am. J. Hum. Genet. 2013, 93, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Onoufriadis, A.; Shoemark, A.; Simpson, M.A.; zur Lage, P.I.; de Castro, S.C.; Bartoloni, L.; Gallone, G.; Petridi, S.; Woollard, W.J.; et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kurkowiak, M.; Ziętkiewicz, E.; Greber, A.; Voelkel, K.; Wojda, A.; Pogorzelski, A.; Witt, M. ZMYND10-Mutation analysis in Slavic patients with primary ciliary dyskinesia. PLoS ONE 2016, 11, e0148067. [Google Scholar] [CrossRef] [PubMed]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish ciliopathy screen plus human mutational analysis identifies c21orf59 and ccdc65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Takada, S.; Witman, G.B.; Kamiya, R. Transport and arrangement of the outer-dynein-arm docking complex in the flagella of Chlamydomonas mutants that lack outer dynein arms. Cell. Motil. Cytoskeleton 2001, 48, 277–286. [Google Scholar] [CrossRef]
- Mali, G.R.; Yeyati, P.L.; Mizuno, S.; Dodd, D.O.; Tennant, P.A.; Keighren, M.A.; Zur Lage, P.; Shoemark, A.; Garcia-Munoz, A.; Shimada, A.; et al. ZMYND10 functions in a chaperone relay during axonemal dynein assembly. eLife 2018, 7, e34389. [Google Scholar] [CrossRef]
- Liu, G.; Wang, L.; Pan, J. Chlamydomonas WDR92 in association with R2TP-like complex and multiple DNAAFs to regulate ciliary dynein preassembly. J. Mol. Cell Biol. 2018, 11, 770–780. [Google Scholar] [CrossRef]
- Warner, F.D. Ciliary inter-microtubule bridges. J. Cell Sci. 1976, 20, 101–114. [Google Scholar]
- Gibbons, I.R. Cilia and flagella of eukaryotes. J. Cell Biol. 1981, 91, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Wirschell, M.; Olbrich, H.; Werner, C.; Tritschler, D.; Bower, R.; Sale, W.S.; Loges, N.T.; Pennekamp, P.; Lindberg, S.; Stenram, U.; et al. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013, 45, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Hijikata, M.; Zariwala, M.A.; Nykamp, K.; Inaba, A.; Guo, T.C.; Yamada, H.; Truty, R.; Sasaki, Y.; Ohta, K.; et al. Recurring large deletion in DRC1 (CCDC164) identified as causing primary ciliary dyskinesia in two Asian patients. Mol. Genet. Genomic Med. 2019, 7, e838. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS ONE 2013, 26, e72299. [Google Scholar] [CrossRef]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-Function GAS8 Mutations Cause Primary Ciliary Dyskinesia and Disrupt the Nexin-Dynein Regulatory Complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef]
- Jeanson, L.; Thomas, L.; Copin, B.; Coste, A.; Sermet-Gaudelus, I.; Dastot-Le Moal, F.; Duquesnoy, P.; Montantin, G.; Collot, N.; Tissier, S.; et al. Mutations in Gas8, a gene encoding a nexin-dynein regulatory complex subunit, cause primary ciliary dyskinesia with axonemal disorganization. Hum. Mutat. 2016, 37, 776–785. [Google Scholar] [CrossRef]
- Lewis, W.R.; Malarkey, E.B.; Tritschler, D.; Bower, R.; Pasek, R.C.; Porath, J.D.; Birket, S.E.; Saunier, S.; Antignac, C.; Knowles, M.R.; et al. Mutation of growth arrest specific 8 reveals a role in motile cilia function and human disease. PLoS Genet. 2016, 12, e1006220. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Leigh, M.W. Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct. Pathol. 2017, 41, 373–385. [Google Scholar] [CrossRef]
- Carlen, B.; Lindberg, S.; Stenram, U. Absence of nexin links as a possible cause of primary ciliary dyskinesia. Ultrastruct. Pathol. 2003, 27, 123–126. [Google Scholar] [CrossRef]
- Cramnert, C.; Stenram, U. Number of nexin links detectable at standard electron microscopy of normal human nasal cilia and at nexin link deficiency. Ultrastruct. Pathol. 2014, 38, 377–381. [Google Scholar] [CrossRef]
- Heuser, T.; Raytchev, M.; Krell, J.; Porter, M.E.; Nicastro, D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 2009, 187, 921–933. [Google Scholar] [CrossRef]
- Lin, J.; Tritschler, D.; Song, K.; Barber, C.F.; Cobb, J.S.; Porter, M.E.; Nicastro, D. Building blocks of the nexin-dynein regulatory complex in Chlamydomonas flagella. J. Biol. Chem. 2011, 286, 29175–29191. [Google Scholar] [CrossRef]
- Bower, R.; Tritschler, D.; Vanderwaal, K.; Perrone, C.A.; Mueller, J.; Fox, L.; Sale, W.S.; Porter, M.E. The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol. Biol. Cell 2013, 24, 1134–1152. [Google Scholar] [CrossRef]
- Oda, T.; Yanagisawa, H.; Kikkawa, M. Detailed structural and biochemical characterization of the nexin-dynein regulatory complex. Mol. Biol. Cell 2015, 26, 294–304. [Google Scholar] [CrossRef]
- Song, K.; Awata, J.; Tritschler, D.; Bower, R.; Witman, G.B.; Porter, M.E.; Nicastro, D. In situ localization of N- and C-termini of subunits of the flagellar nexin-dynein regulatory complex (N-DRC) using SNAP-tag and cryo-electron tomography. J. Biol. Chem. 2015, 290, 5341–5353. [Google Scholar] [CrossRef]
- Bower, R.; Tritschler, D.; Mills, K.V.; Heuser, T.; Nicastro, D.; Porter, M.E. DRC2/CCDC65 is a central hub for assembly of the nexin-dynein regulatory complex and other regulators of ciliary and flagellar motility. Mol. Biol. Cell 2018, 29, 137–153. [Google Scholar] [CrossRef]
- Awata, J.; Song, K.; Lin, J.; King, S.M.; Sanderson, M.J.; Nicastro, D.; Witman, G.B. DRC3 connects the N-DRC to dynein g to regulate flagellar waveform. Mol. Biol. Cell 2015, 26, 2788–2800. [Google Scholar] [CrossRef]
- Ha, S.; Lindsay, A.M.; Timms, A.E.; Beier, D.R. Mutations in Dnaaf1 and Lrrc48 Cause Hydrocephalus, Laterality Defects, and Sinusitis in Mice. G3 2016, 6, 2479–2487. [Google Scholar] [CrossRef]
- Piperno, G.; Huang, B.; Ramanis, Z.; Luck, D.J. Radial spokes of Chlamydomonas flagella: Polypeptide composition and phosphorylation of stalk components. J. Cell Biol. 1981, 88, 73–79. [Google Scholar] [CrossRef]
- Diener, D.R.; Ang, L.H.; Rosenbaum, J.L. Assembly of flagellar radial spoke proteins in Chlamydomonas: Identification of the axoneme binding domain of radial spoke protein 3. J. Cell Biol. 1993, 123, 183–190. [Google Scholar] [CrossRef]
- Yang, P.; Diener, D.R.; Rosenbaum, J.L.; Sale, W.S. Localization of calmodulin and dynein light chain LC8 in flagellar radial spokes. J. Cell Biol. 2001, 153, 1315–1326. [Google Scholar] [CrossRef]
- Patel-King, R.S.; Gorbatyuk, O.; Takebe, S.; King, S.M. Flagellar radial spokes contain a Ca2+-stimulated nucleoside diphosphate kinase. Mol Biol Cell. 2004, 15, 3891–3902. [Google Scholar] [CrossRef]
- Yang, P.; Diener, D.R.; Yang, C.; Kohno, T.; Pazour, G.J.; Dienes, J.M.; Agrin, N.S.; King, S.M.; Sale, W.S.; Kamiya, R.; et al. Radial spoke proteins of Chlamydomonas flagella. J. Cell Sci. 2006, 119, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Wakabayashi, K.; Diener, D.R.; Rosenbaum, J.L.; Kamiya, R. Subunit interactions within the Chlamydomonas flagellar spokehead. Cytoskeleton 2011, 68, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Pigino, G.; Bui, K.H.; Maheshwari, A.; Lupetti, P.; Diener, D.; Ishikawa, T. Cryoelectron tomography of radial spokes in cilia and flagella. J. Cell Biol. 2011, 195, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Heuser, T.; Carbajal-Gonzalez, B.I.; Song, K.; Nicastro, D. The structural heterogeneity of radial spokes in cilia and flagella is conserved. Cytoskeleton 2012, 69, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Pigino, G.; Ishikawa, T. Axonemal radial spokes: 3D structure, function and assembly. Bioarchitecture 2012, 2, 50–58. [Google Scholar] [CrossRef]
- Padma, P.; Satouh, Y.; Wakabayashi, K.; Hozumi, A.; Ushimaru, Y.; Kamiya, R.; Inaba, K. Identification of a novel leucine-rich repeat protein as a component of flagellar radial spoke in the Ascidian Ciona intestinalis. Mol. Biol. Cell 2003, 14, 774–785. [Google Scholar] [CrossRef]
- Satouh, Y.; Padma, P.; Toda, T.; Satoh, N.; Ide, H.; Inaba, K. Molecular characterization of radial spoke subcomplex containing radial spoke protein 3 and heat shock protein 40 in sperm flagella of the ascidian Ciona intestinalis. Mol. Biol. Cell 2005, 16, 626–636. [Google Scholar] [CrossRef]
- Satouh, Y.; Inaba, K. Proteomic characterization of sperm radial spokes identifies a novel spoke protein with an ubiquitin domain. FEBS Lett. 2009, 583, 2201–2207. [Google Scholar] [CrossRef]
- Kott, E.; Legendre, M.; Copin, B.; Papon, J.F.; Dastot-Le Moal, F.; Montantin, G.; Duquesnoy, P.; Piterboth, W.; Amram, D.; Bassinet, L.; et al. Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am. J. Hum. Genet. 2013, 93, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Shoemark, A.; Schmidts, M.; Patel, M.; Jimenez, G.; Liu, H.; Thomas, B.; Dixon, M.; Hirst, R.A.; Rutman, A.; et al. Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum. Mol. Genet. 2014, 23, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Ziętkiewicz, E.; Bukowy-Bieryłło, Z.; Voelkel, K.; Klimek, B.; Dmeńska, H.; Pogorzelski, A.; Sulikowska-Rowińska, A.; Rutkiewicz, E.; Witt, M. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS ONE 2012, 7, e33667. [Google Scholar] [CrossRef]
- Daniels, M.L.; Leigh, M.W.; Davis, S.D.; Armstrong, M.C.; Carson, J.L.; Hazucha, M.; Dell, S.D.; Eriksson, M.; Collins, F.S.; Knowles, M.R.; et al. Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum. Mutat. 2013, 34, 1352–1356. [Google Scholar] [CrossRef]
- Alsaadi, M.M.; Gaunt, T.R.; Boustred, C.R.; Guthrie, P.A.; Liu, X.; Lenzi, L.; Rainbow, L.; Hall, N.; Alharbi, K.K.; Day, I.N. From a single whole exome read to notions of clinical screening: Primary ciliary dyskinesia and RSPH9 p.Lys268del in the Arabian Peninsula. Ann. Hum. Genet. 2012, 76, 211–220. [Google Scholar] [CrossRef]
- Frommer, A.; Hjeij, R.; Loges, N.T.; Edelbusch, C.; Jahnke, C.; Raidt, J.; Werner, C.; Wallmeier, J.; Große-Onnebrink, J.; Olbrich, H.; et al. Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 563–573. [Google Scholar] [CrossRef]
- Yiallouros, P.K.; Kouis, P.; Pirpa, P.; Michailidou, K.; Loizidou, M.A.; Potamiti, L.; Kalyva, M.; Koutras, G.; Kyriacou, K.; Hadjisavvas, A. Wide phenotypic variability in RSPH9-associated primary ciliary dyskinesia: Review of a case-series from Cyprus. J. Thorac. Dis. 2019, 11, 2067–2075. [Google Scholar] [CrossRef]
- Jeanson, L.; Copin, B.; Papon, J.F.; Dastot-Le Moal, F.; Duquesnoy, P.; Montantin, G.; Cadranel, J.; Corvol, H.; Coste, A.; Désir, J.; et al. RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes. Am. J. Hum. Genet. 2015, 97, 153–162. [Google Scholar] [CrossRef]
- El Khouri, E.; Thomas, L.; Jeanson, L.; Bequignon, E.; Vallette, B.; Duquesnoy, P.; Montantin, G.; Copin, B.; Dastot-Le Moal, F.; Blanchon, S.; et al. Mutations in dnajb13, encoding an hsp40 family member, cause primary ciliary dyskinesia and male infertility. Am. J. Hum. Genet. 2016, 99, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yin, W.; Smith, M.C.; Song, K.; Leigh, M.W.; Zariwala, M.A.; Knowles, M.R.; Ostrowski, L.E.; Nicastro, D. Cryo-electron tomography reveals ciliary defects underlying human RSPH1 primary ciliary dyskinesia. Nat. Commun. 2014, 5, 5727. [Google Scholar] [CrossRef] [PubMed]
- Witman, G.B.; Plummer, J.; Sander, G. Chlamydomonas flagellar mutants lacking radial spokes and central tubules. J. Cell Biol. 1978, 76, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Piperno, G.; Ramanis, Z.; Luck, D.J. Radial spokes of Chlamydomonas flagella: Genetic analysis of assembly and function. J. Cell Biol. 1981, 88, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Poghosyan, E.; Rezabkova, L.; Mehall, B.; Sakakibara, H.; Hirono, M.; Kamiya, R.; Ishikawa, T.; Yang, P. The roles of a flagellar HSP40 ensuring rhythmic beating. Mol. Biol. Cell 2019, 30, 228–241. [Google Scholar] [CrossRef]
- Yang, C.; Owen, H.A.; Yang, P. Dimeric heat shock protein 40 binds radial spokes for generating coupled power strokes and recovery strokes of 9 + 2 flagella. J. Cell Biol. 2008, 180, 403–415. [Google Scholar] [CrossRef]
- Olbrich, H.; Schmidts, M.; Werner, C.; Onoufriadis, A.; Loges, N.T.; Raidt, J.; Banki, N.F.; Shoemark, A.; Burgoyne, T.; Al Turki, S.; et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012, 91, 672–684. [Google Scholar] [CrossRef]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. spef2- and hydin-mutant cilia lack the central pair associated protein SPEF2 aiding PCD diagnostics. Am. J. Respir. Cell. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Shapiro, A.; Sears, P.R.; Charng, W.L.; Conrad, D.F.; Leigh, M.W.; Knowles, M.R.; Ostrowski, L.E.; Zariwala, M.A. Identification of genetic variants in CFAP221 as a cause of primary ciliary dyskinesia. J. Hum. Genet. 2019. [Google Scholar] [CrossRef]
- Davy, B.E.; Robinson, M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003, 12, 1163–1170. [Google Scholar] [CrossRef]
- Lechtreck, K.F.; Delmotte, P.; Robinson, M.L.; Sanderson, M.J.; Witman, G.B. Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 2008, 180, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Campagna, D.R.; Pinkus, J.L.; Mulhern, H.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Pinkus, G.S.; Fleming, M.D. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol. Cell Biol. 2008, 28, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Sironen, A.; Kotaja, N.; Mulhern, H.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Miiluniemi, M.; Fleming, M.D.; Lee, L. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol. Reprod. 2011, 85, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Lechtreck, K.F.; Witman, G.B. Chlamydomonas reinhardtii hydin is a central pair protein required for flagellar motility. J. Cell Biol. 2007, 176, 473–482. [Google Scholar] [CrossRef]
- Zhang, H.; Mitchell, D.R. Cpc1, a Chlamydomonas central pair protein with an adenylate kinase domain. J. Cell Sci. 2004, 117, 4179–4188. [Google Scholar] [CrossRef]
- DiPetrillo, C.G.; Smith, E.F. Pcdp1 is a central apparatus protein that binds Ca(2+)-calmodulin and regulates ciliary motility. J. Cell Biol. 2010, 189, 601–612. [Google Scholar] [CrossRef]
- Brown, J.M.; Dipetrillo, C.G.; Smith, E.F.; Witman, G.B. A FAP46 mutant provides new insights into the function and assembly of the C1d complex of the ciliary central apparatus. J. Cell Sci. 2012, 125, 3904–3913. [Google Scholar] [CrossRef]
- Smith, E.F.; Lefebvre, P.A. PF20 gene product contains WD repeats and localizes to the intermicrotubule bridges in Chlamydomonas flagella. Mol. Biol. Cell 1997, 8, 455–467. [Google Scholar] [CrossRef]
- Rupp, G.; O’Toole, E.; Porter, M.E. The Chlamydomonas PF6 locus encodes a large alanine/proline-rich polypeptide that is required for assembly of a central pair projection and regulates flagellar motility. Mol. Biol. Cell 2001, 12, 739–751. [Google Scholar] [CrossRef]
- Andjelkovic, M.; Minic, P.; Vreca, M.; Stojiljkovic, M.; Skakic, A.; Sovtic, A.; Rodic, M.; Skodric-Trifunovic, V.; Maric, N.; Visekruna, J.; et al. Genomic profiling supports the diagnosis of primary ciliary dyskinesia and reveals novel candidate genes and genetic variants. PLoS ONE 2018, 13, e0205422. [Google Scholar] [CrossRef]
- Teves, M.E.; Zhang, Z.; Costanzo, R.M.; Henderson, S.C.; Corwin, F.D.; Zweit, J.; Sundaresan, G.; Subler, M.; Salloum, F.N.; Rubin, B.K., 3rd; et al. Sperm-associated antigen-17 gene is essential for motile cilia function and neonatal survival. Am. J. Respir. Cell Mol. Biol. 2013, 48, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Edelbusch, C.; Cindrić, S.; Dougherty, G.W.; Loges, N.T.; Olbrich, H.; Rivlin, J.; Wallmeier, J.; Pennekamp, P.; Amirav, I.; Omran, H. Mutation of serine/threonine protein kinase 36 (STK36) causes primary ciliary dyskinesia with a central pair defect. Hum. Mutat. 2017, 38, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, K.; Yamazaki, Y.; Nishida, T.; Shinohara, K.; Ishikawa, H.; Hasegawa, T.; Okanoue, T.; Hamada, H.; Noda, T.; Tamura, A.; et al. Coordinated ciliary beating requires Odf2-mediated polarization of basal bodies via basal feet. Cell 2012, 148, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Nguyen, C.T.; Chen, M.H.; Yang, J.H.; Gacayan, R.; Huang, J.; Chen, J.N.; Chuang, P.T. Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 2009, 459, 98–102. [Google Scholar] [CrossRef]
- Nozawa, Y.I.; Yao, E.; Lin, C.; Yang, J.H.; Wilson, C.W.; Gacayan, R.; Chuang, P.T. Fused (Stk36) is a ciliary protein required for central pair assembly and motile cilia orientation in the mammalian oviduct. Dev. Dyn. 2013, 242, 1307–1319. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Yin, W.N.; Sears, P.R.; Werner, M.E.; Brotslaw, E.J.; Mitchell, B.J.; Jania, C.M.; Zeman, K.L.; Rogers, T.D.; Herring, L.E.; et al. Lack of GAS2L2 causes PCD by Impairing cilia orientation and mucociliary clearance. Am. J. Hum. Genet. 2019, 104, 229–245. [Google Scholar] [CrossRef]
- Bukowy-Bieryllo, Z.; Rabiasz, A.; Dabrowski, M.; Pogorzelski, A.; Wojda, A.; Dmenska, H.; Grzela, K.; Sroczynski, J.; Witt, M.; Zietkiewicz, E. Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms. J. Med. Genet. 2019, 56, 769–777. [Google Scholar] [CrossRef]
- Hannah, W.B.; DeBrosse, S.; Kinghorn, B.; Strausbaugh, S.; Aitken, M.L.; Rosenfeld, M.; Wolf, W.E.; Knowles, M.R.; Zariwala, M.A. The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol. Genet. Genomic. Med. 2019, 7, e911. [Google Scholar] [CrossRef]
- Bukowy-Bieryllo, Z.; Zietkiewicz, E.; Loges, N.T.; Wittmer, M.; Geremek, M.; Olbrich, H.; Fliegauf, M.; Voelkel, K.; Rutkiewicz, E.; Rutland, J.; et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 2013, 48, 352–363. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Fridman, G.; Cho, G.Y.; Buchovecky, C.; Tsang, S.H. Novel mutation in retinitis pigmentosa GTPase regulator gene causes primary ciliary dyskinesia and retinitis pigmentosa. Ophthalmic Surg. Lasers Imaging Retina 2018, 49, 548–552. [Google Scholar] [CrossRef]
- Ryan, R.; Failler, M.; Reilly, M.L.; Garfa-Traore, M.; Delous, M.; Filhol, E.; Reboul, T.; Bole-Feysot, C.; Nitschké, P.; Baudouin, V.; et al. Functional characterization of tektin-1 in motile cilia and evidence for TEKT1 as a new candidate gene for motile ciliopathies. Hum. Mol. Genet. 2018, 27, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, S.; Watson, C.M.; Kernohan, K.D.; Lemos, M.; Hutchinson, S.; Poulter, J.A.; Crinnion, L.A.; Berry, I.; Simmonds, J.; Vasudevan, P.; et al. Biallelic mutations in lrrc56, encoding a protein associated with intraflagellar transport, cause mucociliary clearance and laterality defects. Am. J. Hum. Genet. 2018, 103, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Stroud, M.J.; Nazgiewicz, A.; McKenzie, E.A.; Wang, Y.; Kammerer, R.A.; Ballestrem, C. GAS2-like proteins mediate communication between microtubules and actin through interactions with end-binding proteins. J. Cell Sci. 2014, 127, 2672–2682. [Google Scholar] [CrossRef]
- Desai, P.B.; Freshour, J.R.; Mitchell, D.R. Chlamydomonas axonemal dynein assembly locus ODA8 encodes a conserved flagellar protein needed for cytoplasmic maturation of outer dynein arm complexes. Cytoskeleton 2015, 72, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef] [PubMed]
- Toriello, H.V.; Franco, B.; Bruel, A.L.; Thauvin-Robinet, C. Oral-Facial-Digital Syndrome Type, I. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993–2019; 24 July 2002.
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019; 9 July 2003. [Google Scholar]
- Budny, B.; Chen, W.; Omran, H.; Fliegauf, M.; Tzschach, A.; Wisniewska, M.; Jensen, L.R.; Raynaud, M.; Shoichet, S.A.; Badura, M.; et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum. Genet. 2006, 120, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.A.; Prosser, S.L.; Romio, L.; Hirst, R.A.; O’Callaghan, C.; Woolf, A.S.; Fry, A.M. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 2011, 124, 600–612. [Google Scholar] [CrossRef]
- Bengueddach, H.; Lemullois, M.; Aubusson-Fleury, A.; Koll, F. Basal body positioning and anchoring in the multiciliated cell Paramecium tetraurelia: Roles of OFD1 and VFL3. Cilia 2017, 6, 6. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Romio, L.; Castro, S.; Collins, J.E.; Goulding, D.A.; Stemple, D.L.; Woolf, A.S.; Wilson, S.W. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 2009, 18, 289–303. [Google Scholar] [CrossRef]
- Anand, M.; Khanna, H. Ciliary transition zone (TZ) proteins RPGR and CEP290: Role in photoreceptor cilia and degenerative diseases. Expert. Opin. Ther. Targets 2012, 16, 541–551. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Remans, K.; Bürger, M.; Vetter, I.R.; Wittinghofer, A. C2 domains as protein-protein interaction modules in the ciliary transition zone. Cell Rep. 2014, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khanna, H. More Than Meets the Eye: Current Understanding of RPGR Function. Adv. Exp. Med. Biol. 2018, 1074, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Giacalone, J.C.; Searby, C.; Stone, E.M.; Tucker, B.A.; Sheffield, V.C. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc. Natl. Acad. Sci. USA 2019, 116, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Murga-Zamalloa, C.A.; Chan, L.; Hitchcock, P.F.; Swaroop, A.; Khanna, H. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum. Mol. Genet. 2010, 19, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Zeng, Z.; Gautier, P.; Lennon, A.; Gakovic, M.; Patton, E.E.; Wright, A.F. Zebrafish Rpgr is required for normal retinal development and plays a role in dynein-based retrograde transport processes. Hum. Mol. Genet. 2010, 19, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 165–174. [Google Scholar] [CrossRef]
- Fehrenbach, H.; Decker, C.; Eisenberger, T.; Frank, V.; Hampel, T.; Walden, U.; Amann, K.U.; Krüger-Stollfuß, I.; Bolz, H.J.; Häffner, K.; et al. Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr. Nephrol. 2014, 29, 1451–1456. [Google Scholar] [CrossRef]
- Li, Y.; Garrod, A.S.; Madan-Khetarpal, S.; Sreedher, G.; McGuire, M.; Yagi, H.; Klena, N.T.; Gabriel, G.C.; Khalifa, O.; Zahid, M.; et al. Respiratory motile cilia dysfunction in a patient with cranioectodermal dysplasia. Am. J. Med. Genet. A 2015, 167A, 2188–2196. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Kourembanas, S.; Wyatt, T.A.; Mitsialis, S.A. Mutation of murine adenylate kinase 7 underlies a primary ciliary dyskinesia phenotype. Am. J. Respir. Cell Mol. Biol. 2009, 40, 305–313. [Google Scholar] [CrossRef]
- McKenzie, C.W.; Craige, B.; Kroeger, T.V.; Finn, R.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Strittmatter, L.; Hendricks, G.M.; Witman, G.B.; et al. CFAP54 is required for proper ciliary motility and assembly of the central pair apparatus in mice. Mol. Biol. Cell 2015, 26, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Lefebvre, P.A. PF16 encodes a protein with armadillo repeats and localizes to a single microtubule of the central apparatus in Chlamydomonas flagella. J. Cell Biol. 1996, 132, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Teves, M.E.; Sears, P.R.; Li, W.; Zhang, Z.; Tang, W.; van Reesema, L.; Costanzo, R.M.; Davis, C.W.; Knowles, M.R.; Strauss, J.F., 3rd; et al. Sperm-associated antigen 6 (SPAG6) deficiency and defects in ciliogenesis and cilia function: Polarity, density, and beat. PLoS ONE 2014, 9, e107271. [Google Scholar] [CrossRef] [PubMed]
- Lee, L. Mechanisms of mammalian ciliary motility: Insights from primary ciliary dyskinesia genetics. Gene 2011, 473, 57–66. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mutated Gene | Model Organism | Localization in Cilia | Phenotype | Ref |
|---|---|---|---|---|
| AK7 | Mouse | n/d | Reduced ciliary beat frequency, significant number of cilia lacking CA (9 + 0), or with displaced peripheral doublet without CA (8 + 1) or with CA; hydrocephalus, mucus accumulation in the paranasal passages, exacerbated respiratory responses upon allergen challenge, male infertility, situs inversus not detected | [244] |
| CFAP54 | Mouse | C1d projection (based on studies in Chlamydomonas) [208,209] | Reduced ciliary beat frequency, lost C1d projection hydrocephalus, male infertility, and accumulation of mucus in the sinuses | [245] |
| SPAG6/PF16 | Mouse | Central apparatus (based on studies in Chlamydomonas) [246] | Reduced ciliary beat frequency, asynchronous beating, reduction in cilia density, normal axoneme structure but random orientation of CA hydrocephalus, male infertility random orientation of basal feet of the basal bodies | [247] |
| c15orf26/CFAP161 | Zebrafish | n/d | Missing outer dynein arms, pronephric cysts, axis curvature, laterality defects, hydrocephalus | [148] |
| LRRC48/FAP134/DRC3 | Mouse | N-DRC (based on studies in Chlamydomonas) [164] | Hydrocephalus, laterality defects, male infertility, accumulation of mucus in the sinuses | [170] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poprzeczko, M.; Bicka, M.; Farahat, H.; Bazan, R.; Osinka, A.; Fabczak, H.; Joachimiak, E.; Wloga, D. Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia. Cells 2019, 8, 1614. https://doi.org/10.3390/cells8121614
Poprzeczko M, Bicka M, Farahat H, Bazan R, Osinka A, Fabczak H, Joachimiak E, Wloga D. Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia. Cells. 2019; 8(12):1614. https://doi.org/10.3390/cells8121614
Chicago/Turabian StylePoprzeczko, Martyna, Marta Bicka, Hanan Farahat, Rafal Bazan, Anna Osinka, Hanna Fabczak, Ewa Joachimiak, and Dorota Wloga. 2019. "Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia" Cells 8, no. 12: 1614. https://doi.org/10.3390/cells8121614
APA StylePoprzeczko, M., Bicka, M., Farahat, H., Bazan, R., Osinka, A., Fabczak, H., Joachimiak, E., & Wloga, D. (2019). Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia. Cells, 8(12), 1614. https://doi.org/10.3390/cells8121614