Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis

 , ,
, ,  and
and 
Abstract
1. Chronic Liver Disease (CLD)
1.1. Etiology and Pathophysiology of Chronic Liver Disease (CLD)
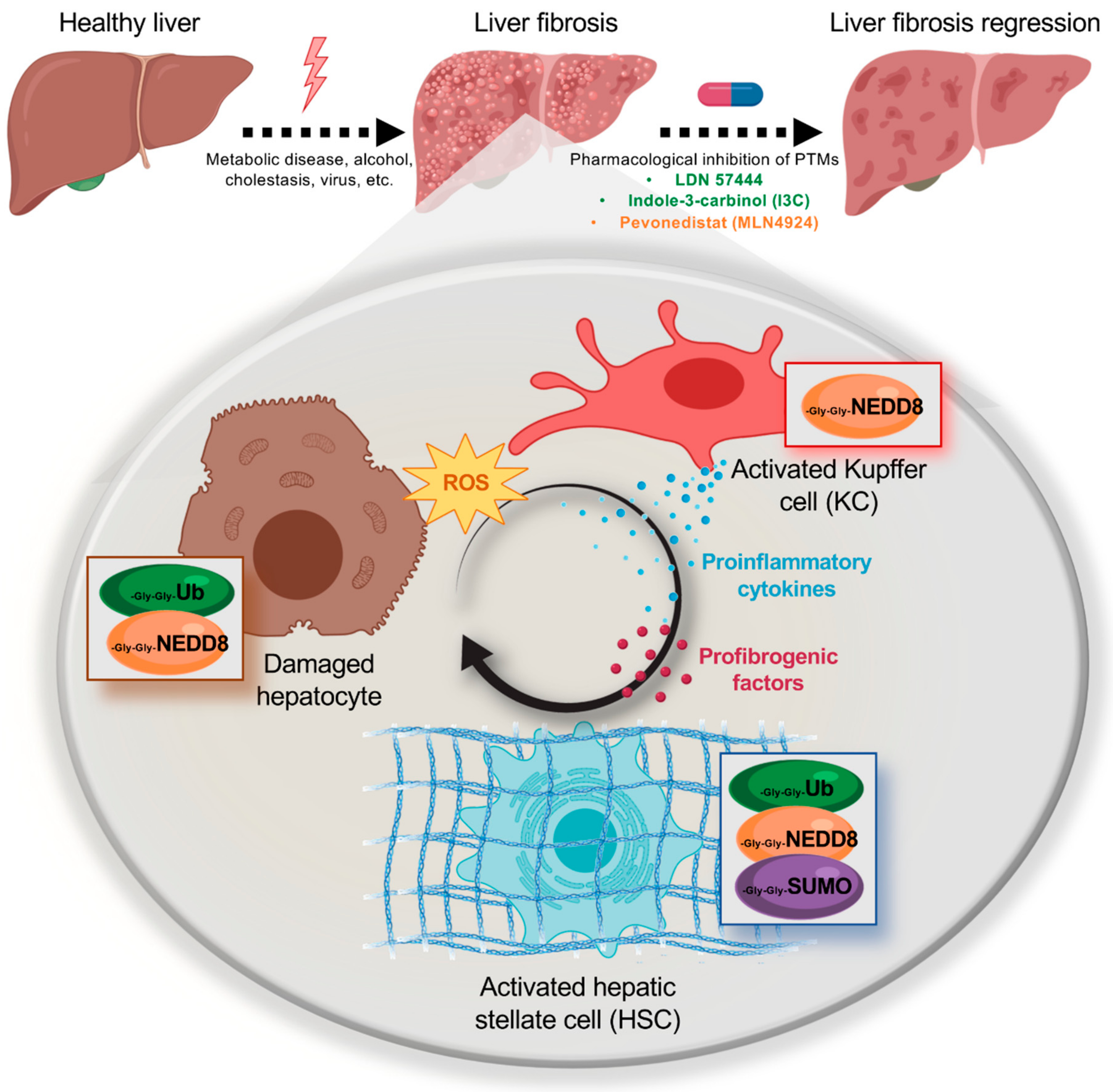
1.2. Liver Fibrosis and Cell Types
1.3. Overview of the Current Treatment Options for Chronic Liver Disease and Liver Fibrosis
2. Post-Translational Modifications (PTMs) by Ubiquitin-Like (Ubl) Proteins
2.1. NEDDylation in Liver Fibrosis
2.2. SUMOylation in Liver Fibrosis
2.3. Therapeutic Strategies Targeting Ubls Modifications in Liver Fibrosis
3. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [PubMed]
- Yoon, Y.J.; Friedman, S.L.; Lee, Y.A. Antifibrotic Therapies: Where Are We Now? Semin. Liver Dis. 2016, 36, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Byass, P. The global burden of liver disease: A challenge for methods and for public health. BMC Med. 2014, 12, 159. [Google Scholar] [CrossRef]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38 (Suppl. 1), 2–6. [Google Scholar] [CrossRef]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549–6570. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Rombouts, K. Liver fibrosis: From the bench to clinical targets. Dig. Liver Dis. 2004, 36, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Arndtz, K.; Hirschfield, G.M. The Pathogenesis of Autoimmune Liver Disease. Dig. Dis. 2016, 34, 327–333. [Google Scholar] [CrossRef]
- Iwaisako, K.; Haimerl, M.; Paik, Y.H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor delta agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef]
- Wells, R.G.; Schwabe, R.F. Origin and function of myofibroblasts in the liver. Semin. Liver Dis. 2015, 35, e1. [Google Scholar] [CrossRef]
- Karin, D.; Koyama, Y.; Brenner, D.; Kisseleva, T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation 2016, 92, 84–92. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Perez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Chen, C.R.; Massague, J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 2003, 11, 915–926. [Google Scholar] [CrossRef]
- Okada, M.; Enomoto, M.; Kawada, N.; Nguyen, M.H. Effects of antiviral therapy in patients with chronic hepatitis B and cirrhosis. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 1095–1104. [Google Scholar] [CrossRef]
- Navasa, M.; Forns, X. Antiviral therapy in HCV decompensated cirrhosis: To treat or not to treat? J. Hepatol. 2007, 46, 185–188. [Google Scholar] [CrossRef]
- Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M.; Adams, D.; Alpini, G.; Banales, J.M.; Beuers, U.; Bjornsson, E.; Bowlus, C.; et al. The challenges of primary biliary cholangitis: What is new and what needs to be done. J. Autoimmun. 2019, 102328. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Trauner, M.; Nevens, F.; Shiffman, M.L.; Drenth, J.P.H.; Bowlus, C.L.; Vargas, V.; Andreone, P.; Hirschfield, G.M.; Pencek, R.; Malecha, E.S.; et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol. Hepatol. 2019, 4, 445–453. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- Banner, B.F.; Savas, L.; Zivny, J.; Tortorelli, K.; Bonkovsky, H.L. Ubiquitin as a marker of cell injury in nonalcoholic steatohepatitis. Am. J. Clin. Pathol. 2000, 114, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Burchette, J.L.; Brunt, E.M.; Abdelmalek, M.F.; Cardona, D.; McCall, S.J.; Unalp, A.; Belt, P.; Ferrell, L.D.; et al. Costaining for keratins 8/18 plus ubiquitin improves detection of hepatocyte injury in nonalcoholic fatty liver disease. Hum. Pathol. 2012, 43, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.Z.; Zhou, C.H.; Wang, J.Y. Abnormal expression of Smurf2 during the process of rat liver fibrosis. Chin. J. Dig. Dis. 2006, 7, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kho, D.H.; Wang, Y.; Harazono, Y.; Nakajima, K.; Xie, Y.; Raz, A. Gp78, an E3 ubiquitin ligase acts as a gatekeeper suppressing nonalcoholic steatohepatitis (NASH) and liver cancer. PLoS ONE 2015, 10, e0118448. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Murphy, L.B.; Leslie, J.; Kendrick, S.; French, J.; Fox, C.R.; Sheerin, N.S.; Fisher, A.; Robinson, J.H.; Tiniakos, D.G.; et al. Ubiquitin C-terminal hydrolase 1: A novel functional marker for liver myofibroblasts and a therapeutic target in chronic liver disease. J. Hepatol. 2015, 63, 1421–1428. [Google Scholar] [CrossRef]
- Hasegawa, D.; Fujii, R.; Yagishita, N.; Matsumoto, N.; Aratani, S.; Izumi, T.; Azakami, K.; Nakazawa, M.; Fujita, H.; Sato, T.; et al. E3 ubiquitin ligase synoviolin is involved in liver fibrogenesis. PLoS ONE 2010, 5, e13590. [Google Scholar] [CrossRef]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Kim, K.M.; Han, C.Y.; Kim, J.Y.; Cho, S.S.; Kim, Y.S.; Koo, J.H.; Lee, J.M.; Lim, S.C.; Kang, K.W.; Kim, J.S.; et al. Galpha12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 2018, 68, 493–504. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Tillman, B.; French, B.A.; French, S.W. Ufmylation and FATylation pathways are downregulated in human alcoholic and nonalcoholic steatohepatitis, and mice fed DDC, where Mallory-Denk bodies (MDBs) form. Exp. Mol. Pathol. 2014, 97, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lebosse, F.; Testoni, B.; Fresquet, J.; Facchetti, F.; Galmozzi, E.; Fournier, M.; Hervieu, V.; Berthillon, P.; Berby, F.; Bordes, I.; et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2017, 66, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tomooka, Y.; Noda, M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem. Biophys. Res. Commun. 1992, 185, 1155–1161. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef]
- Abidi, N.; Xirodimas, D.P. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr. Relat. Cancer 2015, 22, T55–T70. [Google Scholar] [CrossRef]
- Mendoza, H.M.; Shen, L.N.; Botting, C.; Lewis, A.; Chen, J.; Ink, B.; Hay, R.T. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J. Biol. Chem. 2003, 278, 25637–25643. [Google Scholar] [CrossRef]
- Sundqvist, A.; Liu, G.; Mirsaliotis, A.; Xirodimas, D.P. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Rep. 2009, 10, 1132–1139. [Google Scholar] [CrossRef]
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.T.; Schulman, B.A. Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu Rev. Biophys. Biomol. Struct. 2007, 36, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Leidecker, O.; Matic, I.; Mahata, B.; Pion, E.; Xirodimas, D.P. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle 2012, 11, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Neve, R.L.; Liu, H. Neddylation dysfunction in Alzheimer’s disease. J. Cell Mol. Med. 2012, 16, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Barbier-Torres, L.; Delgado, T.C.; Garcia-Rodriguez, J.L.; Zubiete-Franco, I.; Fernandez-Ramos, D.; Buque, X.; Cano, A.; Gutierrez-de Juan, V.; Fernandez-Dominguez, I.; Lopitz-Otsoa, F.; et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget 2015, 6, 2509–2523. [Google Scholar] [CrossRef]
- Yu, J.; Huang, W.L.; Xu, Q.G.; Zhang, L.; Sun, S.H.; Zhou, W.P.; Yang, F. Overactivated neddylation pathway in human hepatocellular carcinoma. Cancer Med. 2018, 7, 3363–3372. [Google Scholar] [CrossRef]
- Delgado, T.C.; Barbier-Torres, L.; Zubiete-Franco, I.; Lopitz-Otsoa, F.; Varela-Rey, M.; Fernandez-Ramos, D.; Martinez-Chantar, M.L. Neddylation, a novel paradigm in liver cancer. Transl. Gastroenterol. Hepatol. 2018, 3, 37. [Google Scholar] [CrossRef]
- Gao, Q.; Yu, G.Y.; Shi, J.Y.; Li, L.H.; Zhang, W.J.; Wang, Z.C.; Yang, L.X.; Duan, M.; Zhao, H.; Wang, X.Y.; et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget 2014, 5, 7820–7832. [Google Scholar] [CrossRef]
- Embade, N.; Fernandez-Ramos, D.; Varela-Rey, M.; Beraza, N.; Sini, M.; Gutierrez de Juan, V.; Woodhoo, A.; Martinez-Lopez, N.; Rodriguez-Iruretagoyena, B.; Bustamante, F.J.; et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 2012, 55, 1237–1248. [Google Scholar] [CrossRef]
- Bailly, A.P.; Perrin, A.; Serrano-Macia, M.; Maghames, C.; Leidecker, O.; Trauchessec, H.; Martinez-Chantar, M.L.; Gartner, A.; Xirodimas, D.P. The Balance between Mono- and NEDD8-Chains Controlled by NEDP1 upon DNA Damage Is a Regulatory Module of the HSP70 ATPase Activity. Cell Rep. 2019, 29, 212–224. [Google Scholar] [CrossRef]
- Zubiete-Franco, I.; Fernandez-Tussy, P.; Barbier-Torres, L.; Simon, J.; Fernandez-Ramos, D.; Lopitz-Otsoa, F.; Gutierrez-de Juan, V.; de Davalillo, S.L.; Duce, A.M.; Iruzubieta, P.; et al. Deregulated neddylation in liver fibrosis. Hepatology 2017, 65, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Das, M.; Sauceda, C.; Ellies, L.G.; Kuo, K.; Parwal, P.; Kaur, M.; Jih, L.; Bandyopadhyay, G.K.; Burton, D.; et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Investig. 2019, 130, 4477–4491. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418. [Google Scholar] [CrossRef]
- Eisenhardt, N.; Chaugule, V.K.; Koidl, S.; Droescher, M.; Dogan, E.; Rettich, J.; Sutinen, P.; Imanishi, S.Y.; Hofmann, K.; Palvimo, J.J.; et al. A new vertebrate SUMO enzyme family reveals insights into SUMO-chain assembly. Nat. Struct. Mol. Biol. 2015, 22, 959–967. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Muller, S.; Ledl, A.; Schmidt, D. SUMO: A regulator of gene expression and genome integrity. Oncogene 2004, 23, 1998–2008. [Google Scholar] [CrossRef]
- Jackson, S.P.; Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef]
- Yang, W.; Thompson, J.W.; Wang, Z.; Wang, L.; Sheng, H.; Foster, M.W.; Moseley, M.A.; Paschen, W. Analysis of oxygen/glucose-deprivation-induced changes in SUMO3 conjugation using SILAC-based quantitative proteomics. J. Proteome Res. 2012, 11, 1108–1117. [Google Scholar] [CrossRef]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Melchior, F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 2006, 21, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Saunier, E.F.; Akhurst, R.J.; Derynck, R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 2008, 10, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, D.; Qiu, F.; Zhang, L.L.; Liu, S.K.; Li, Y.Y.; Liu, M.T.; Wu, D.; Wang, J.X.; Ding, X.Q.; et al. Manipulating PML SUMOylation via Silencing UBC9 and RNF4 Regulates Cardiac Fibrosis. Mol. Ther. 2017, 25, 666–678. [Google Scholar] [CrossRef]
- Ahner, A.; Gong, X.; Frizzell, R.A. Divergent signaling via SUMO modification: Potential for CFTR modulation. Am. J. Physiol. Cell Physiol. 2016, 310, C175–C180. [Google Scholar] [CrossRef][Green Version]
- Arvaniti, E.; Vakrakou, A.; Kaltezioti, V.; Stergiopoulos, A.; Prakoura, N.; Politis, P.K.; Charonis, A. Nuclear receptor NR5A2 is involved in the calreticulin gene regulation during renal fibrosis. Biochim. Biophys. Acta 2016, 1862, 1774–1785. [Google Scholar] [CrossRef]
- Fang, S.; Yuan, J.; Shi, Q.; Xu, T.; Fu, Y.; Wu, Z.; Guo, W. Downregulation of UBC9 promotes apoptosis of activated human LX-2 hepatic stellate cells by suppressing the canonical NF-kappaB signaling pathway. PLoS ONE 2017, 12, e0174374. [Google Scholar]
- Bu, F.T.; Chen, Y.; Yu, H.X.; Chen, X.; Yang, Y.; Pan, X.Y.; Wang, Q.; Wu, Y.T.; Huang, C.; Meng, X.M.; et al. SENP2 alleviates CCl4-induced liver fibrosis by promoting activated hepatic stellate cell apoptosis and reversion. Toxicol. Lett. 2018, 289, 86–98. [Google Scholar] [CrossRef]
- Ramani, K.; Tomasi, M.L.; Yang, H.; Ko, K.; Lu, S.C. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J. Biol. Chem. 2012, 287, 36341–36355. [Google Scholar] [CrossRef]
- Lin, X.; Liang, M.; Liang, Y.Y.; Brunicardi, F.C.; Feng, X.H. SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J. Biol. Chem. 2003, 278, 31043–31048. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Long, J.; Yuan, B.; Zheng, M.; Xiao, M.; Xu, J.; Lin, X.; Feng, X.H. SUMO Modification Reverses Inhibitory Effects of Smad Nuclear Interacting Protein-1 in TGF-beta Responses. J. Biol. Chem. 2016, 291, 24418–24430. [Google Scholar] [CrossRef] [PubMed]
- Janka, C.; Selmi, C.; Gershwin, M.E.; Will, H.; Sternsdorf, T. Small ubiquitin-related modifiers: A novel and independent class of autoantigens in primary biliary cirrhosis. Hepatology 2005, 41, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Gerpe, M.C.R.; Sidhu, S.S.; Zhang, W. Emerging drug development technologies targeting ubiquitination for cancer therapeutics. Pharmacol. Ther. 2019, 199, 139–154. [Google Scholar] [CrossRef]
- Wertz, I.E.; Murray, J.M. Structurally-defined deubiquitinase inhibitors provide opportunities to investigate disease mechanisms. Drug Discov. Today Technol. 2019, 31, 109–123. [Google Scholar] [CrossRef]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef]
- Zhou, Y.; Ji, C.; Cao, M.; Guo, M.; Huang, W.; Ni, W.; Meng, L.; Yang, H.; Wei, J.F. Inhibitors targeting the SUMOylation pathway: A patent review 20122015 (Review). Int. J. Mol. Med. 2018, 41, 3–12. [Google Scholar] [CrossRef]
- Li, B.; Cong, M.; Zhu, Y.; Xiong, Y.; Jin, W.; Wan, Y.; Zhou, Y.; Ao, Y.; Wang, H. Indole-3-Carbinol Induces Apoptosis of Hepatic Stellate Cells through K63 De-Ubiquitination of RIP1 in Rats. Cell Physiol. Biochem. 2017, 41, 1481–1490. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef]
- Da Silva-Ferrada, E.; Xolalpa, W.; Lang, V.; Aillet, F.; Martin-Ruiz, I.; de la Cruz-Herrera, C.F.; Lopitz-Otsoa, F.; Carracedo, A.; Goldenberg, S.J.; Rivas, C.; et al. Analysis of SUMOylated proteins using SUMO-traps. Sci. Rep. 2013, 3, 1690. [Google Scholar] [CrossRef] [PubMed]
- Zubiete-Franco, I.; Garcia-Rodriguez, J.L.; Lopitz-Otsoa, F.; Serrano-Macia, M.; Simon, J.; Fernandez-Tussy, P.; Barbier-Torres, L.; Fernandez-Ramos, D.; Gutierrez-de-Juan, V.; Lopez de Davalillo, S.; et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine 2019, 40, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; Menendez-Benito, V.; Masucci, M.G.; Dantuma, N.P. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 2003, 21, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef]
- Pirone, L.; Xolalpa, W.; Mayor, U.; Barrio, R.; Sutherland, J.D. Analysis of SUMOylated Proteins in Cells and In Vivo Using the bioSUMO Strategy. Methods Mol. Biol. 2016, 1475, 161–169. [Google Scholar] [CrossRef]


{kind=link}
| Ubls | Structure | Identity with Ubiquitin | Size (kDa) | Amino Acid | Highly Conserved between Species |
|---|---|---|---|---|---|
| Ubiquitin |  | 100 | 8.6 | 76 |  |
| NEDD8 |  | 59 | 8 | 81 |  |
| SUMO |  SUMO 1  SUMO 2 | 18 | ≈12 | ≈100 |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachiondo-Ortega, S.; Mercado-Gómez, M.; Serrano-Maciá, M.; Lopitz-Otsoa, F.; Salas-Villalobos, T.B.; Varela-Rey, M.; Delgado, T.C.; Martínez-Chantar, M.L. Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis. Cells 2019, 8, 1575. https://doi.org/10.3390/cells8121575
Lachiondo-Ortega S, Mercado-Gómez M, Serrano-Maciá M, Lopitz-Otsoa F, Salas-Villalobos TB, Varela-Rey M, Delgado TC, Martínez-Chantar ML. Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis. Cells. 2019; 8(12):1575. https://doi.org/10.3390/cells8121575
Chicago/Turabian StyleLachiondo-Ortega, Sofia, Maria Mercado-Gómez, Marina Serrano-Maciá, Fernando Lopitz-Otsoa, Tanya B Salas-Villalobos, Marta Varela-Rey, Teresa C. Delgado, and María Luz Martínez-Chantar. 2019. "Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis" Cells 8, no. 12: 1575. https://doi.org/10.3390/cells8121575
APA StyleLachiondo-Ortega, S., Mercado-Gómez, M., Serrano-Maciá, M., Lopitz-Otsoa, F., Salas-Villalobos, T. B., Varela-Rey, M., Delgado, T. C., & Martínez-Chantar, M. L. (2019). Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis. Cells, 8(12), 1575. https://doi.org/10.3390/cells8121575