Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study

 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Results and Discussion
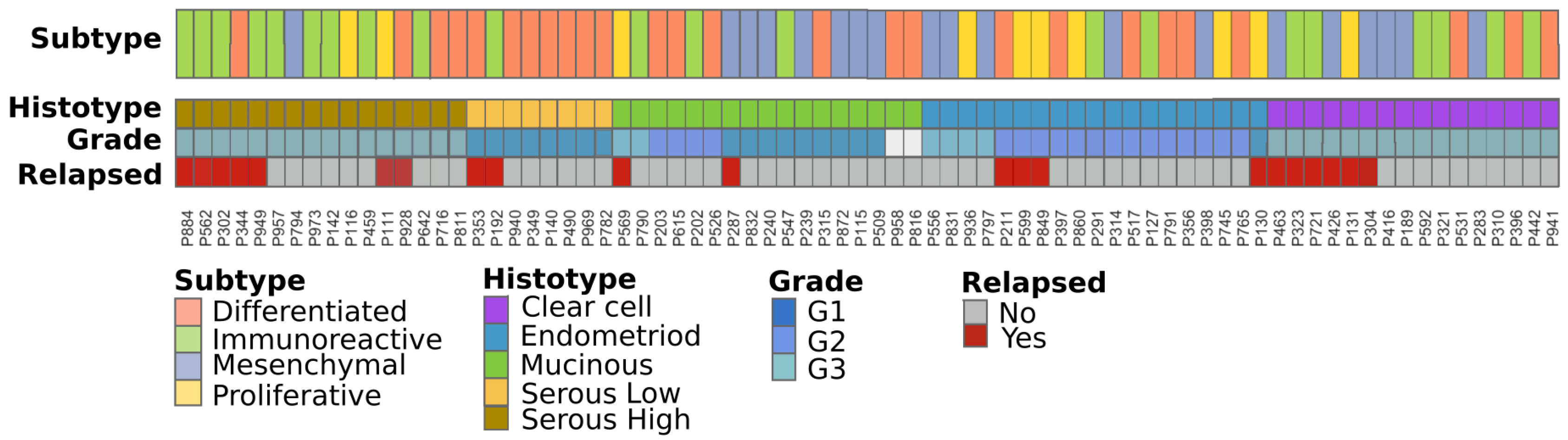
2.1. Patient Cohorts and Sample Collection
2.2. Stage I Classification Using Advanced Ovarian Cancer Expression Subtypes
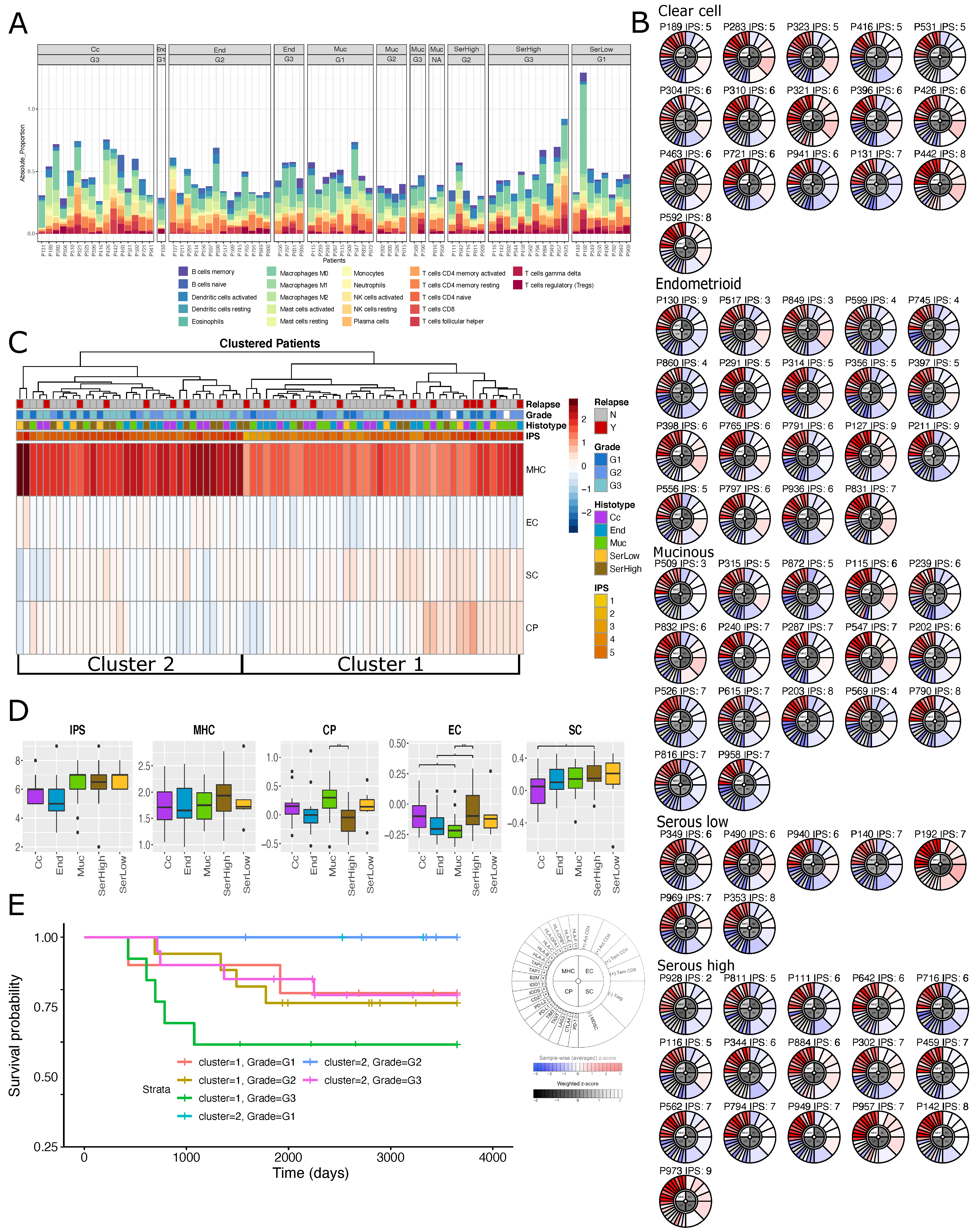
2.3. Immuno-Phenotype of Stage I EOC Patients
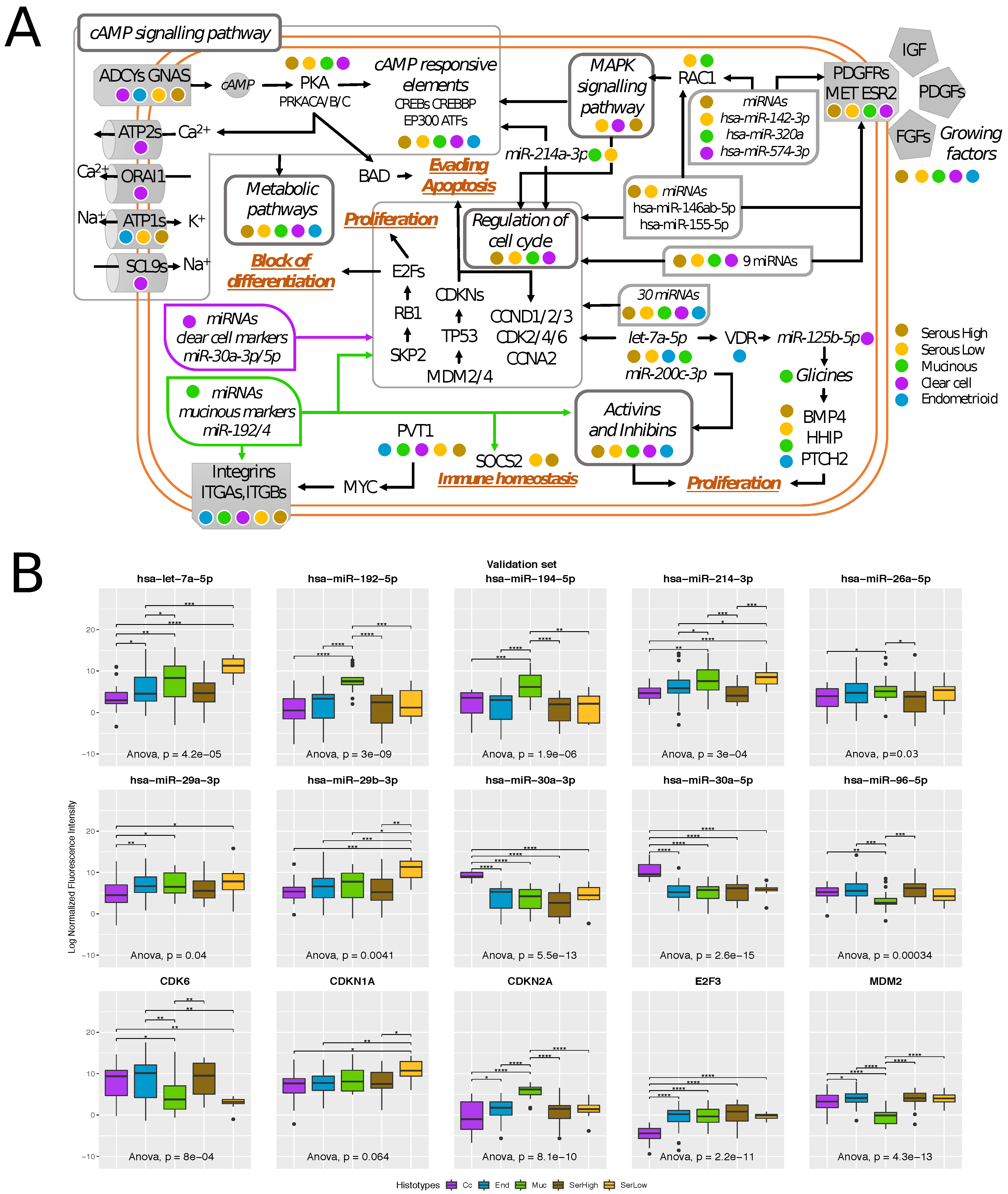
2.4. Transcriptional Alterations of Ovarian Cancer Stage I
2.5. Histotype-Specific Transcripts Validation
3. Materials and Methods
3.1. Patient Cohorts
3.2. Expression Analyses
3.3. Ovarian Cancer Molecular Signature Identification
3.4. Immuno-Phenotype Analyses
3.5. Network Analysis
3.6. qRT-PCR Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EOC | Epithelial Ovarian Cancer |
| FIGO | International Federation of Gynecological and Obstetrics |
| IPS | Immunophenoscore |
| IPG | Immunophenogram |
| KM | Kaplan–Meier |
| MUC | Mucinous |
| SER HIGH | High Grade Serous |
| SER LOW | Low Grade Serous |
| CC | Clear Cell |
| END | Endometrioid |
| DIF | Differentiated subtype |
| MES | Mesenchymal subtype |
| MHC | Major Histocompatibility Complex |
| CTL | Cytotoxic T Lymphocyte |
| HLA | Human Leukocyte Antigen |
| GO | Gene Ontology |
| IMR | Immunoreactive subtype |
| PRO | Proliferative subtype |
References
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Hanley, G.E.; McAlpine, J.N.; Miller, D.; Huntsman, D.; Schrader, K.A.; Blake Gilks, C.; Mitchell, G. A population-based analysis of germline BRCA1 and BRCA2 testing among ovarian cancer patients in an era of histotype-specific approaches to ovarian cancer prevention. BMC Cancer 2018, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.E.M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumor–immune cell interactions. Nat. Rev. Genet. 2016, 17, 441. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: An empirical gene set approach exploiting pathway topology. Nucleic Acids Res. 2012, 41, e19. [Google Scholar] [CrossRef] [PubMed]
- Calura, E.; Paracchini, L.; Fruscio, R.; Di Feo, A.; Ravaggi, A.; Peronne, J.; Martini, P.; Sales, G.; Beltrame, L.; Bignotti, E.; et al. A prognostic regulatory pathway in stage I epithelial ovarian cancer: New hints for the poor prognosis assessment. Ann. Oncol. 2016, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Cavalieri, D.; Fruscio, R.; Calura, E.; Garavaglia, D.; Fuso Nerini, I.; Mangioni, C.; Cattoretti, G.; Clivio, L.; Beltrame, L.; et al. Association between miR-200c and the survival of patients with stage I epithelial ovarian cancer: A retrospective study of two independent tumor tissue collections. Lancet Oncol. 2011, 12, 273–285. [Google Scholar] [CrossRef]
- Martini, P.; Paracchini, L.; Caratti, G.; Mello-Grand, M.; Fruscio, R.; Beltrame, L.; Calura, E.; Sales, G.; Ravaggi, A.; Bignotti, E.; et al. lncRNAs as Novel Indicators of Patients’ Prognosis in Stage I Epithelial Ovarian Cancer: A Retrospective and Multicentric Study. Clin. Cancer Res. 2017, 23, 2356–2366. [Google Scholar] [CrossRef]
- Calura, E.; Fruscio, R.; Paracchini, L.; Bignotti, E.; Ravaggi, A.; Martini, P.; Sales, G.; Beltrame, L.; Clivio, L.; Ceppi, L.; et al. MiRNA landscape in stage I epithelial ovarian cancer defines the histotype specificities. Clin. Cancer Res. 2013, 19, 4114–4123. [Google Scholar] [CrossRef]
- Calura, E.; Martini, P.; Sales, G.; Beltrame, L.; Chiorino, G.; D’Incalci, M.; Marchini, S.; Romualdi, C. Wiring miRNAs to pathways: A Topological approach to integrate miRNA and mRNA expression profiles. Nucleic Acids Res. 2014, 42, e96. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Syst. Biol. 2018, 1711, 243–259. [Google Scholar]
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Haanen, J.; Ribas, A.; Schumacher, T. CANCER IMMUNOLOGY The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Nagaraj, A.B.; Razi, A.; Singh, S.; Kamalakaran, S.; Banerjee, N.; Joseph, P.; Mankovich, A.; Mittal, P.; DiFeo, A.; et al. InFlo: A novel systems biology framework identifies cAMP-CREB1 axis as a key modulator of platinum resistance in ovarian cancer. Oncogene 2017, 36, 2472–2482. [Google Scholar] [CrossRef]
- Parolia, A.; Cielik, M.; Chinnaiyan, A.M. Competing for enhancers: PVT1 fine-tunes MYC expression. Cell Res. 2018, 28, 785–786. [Google Scholar] [CrossRef]
- Boudjadi, S.; Carrier, J.C.; Groulx, J.F.; Beaulieu, J.F. Integrins expression is controlled by c-MYC in colorectal cancer cells. Oncogene 2016, 35, 1671–1678. [Google Scholar] [CrossRef]
- Wei, L.; Yin, F.; Zhang, W.; Li, L. ITGA 1 and cell adhesion-mediated drug resistance in ovarian cancer. Int. Clin. Exp. Pathol. 2017, 10, 5522–5529. [Google Scholar]
- Nirschl, C.J.; Suarez-Farinas, M.; Izar, B.; Prakadan, S.; Dannenfelser, R.; Tirosh, I.; Liu, Y.; Zhu, Q.; Devi, K.S.P.; Carroll, S.L.; et al. IFN-Dependent Tissue-Immune Homeostasis Is Co-opted in the Tumor Microenvironment. Cell 2017, 170, 127–141. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Helland, Å.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef]
- Bentink, S.; Haibe-Kains, B.; Risch, T.; Fan, J.B.; Hirsch, M.S.; Holton, K.; Rubio, R.; April, C.; Chen, J.; Wickham-Garcia, E.; et al. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS ONE 2012, 7, e30269. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. JNCI J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453. [Google Scholar] [CrossRef]
- Raivo, K. Pheatmap: Pretty Heatmaps. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 1 December 2019).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. g raphite-a Bioconductor package to convert pathway topology to gene network. BMC Bioinform. 2012, 13, 20. [Google Scholar] [CrossRef]
- Sales, G.; Calura, E.; Romualdi, C. meta Graphite—A new layer of pathway annotation to get metabolite networks. Bioinformatics 2018, 35, 1258–1260. [Google Scholar] [CrossRef]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. miRTarBase: A database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2010, 39, D163–D169. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA–target interactions. Nucleic Acids Res. 2008, 37, D105–D110. [Google Scholar] [CrossRef]
- James, F.R.; Jiminez-Linan, M.; Alsop, J.; Mack, M.; Song, H.; Brenton, J.D.; Pharoah, P.D.P.; Ali, H.R. Association between tumor infiltrating lymphocytes, histotype and clinical outcome in epithelial ovarian cancer. BMC Cancer 2017, 17, 657. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Annotation | Training Set | Validation Set |
|---|---|---|
| HISTOTYPES | ||
| Clear cell | 16 (21%) | 22 (16.6%) |
| Endometroid | 19 (25%) | 55 (41.7%) |
| Mucinous | 17 (22.4%) | 21 (15.9%) |
| Serous high-grade | 16 (21.0%) | 26 (76.4%) |
| Serous low-grade | 8 (10.6%) | 8 (23.5%) |
| GRADES | ||
| G1 | 18 (23.7%) | 32 (32.6%) |
| G2 | 18 (23.7%) | 31 (31.7%) |
| G3 | 38 (50.0%) | 35 (35.7%) |
| NA | 2 (2.6%) | 0 (0%) |
| RELAPSING | ||
| No | 55 (72.4%) | 105 (79.5%) |
| Yes | 21 (27.6%) | 24 (18.2%) |
| NA | 0 (0%) | 3 (2.3%) |
| VITAL STATUS AT THE LAST FOLLOW UP | ||
| Alive | 57 (75%) | 102 (77.3%) |
| Dead of EOC | 15 (19.8%) | 9 (6.8%) |
| Dead of other cause | 2 (2.6%) | 13 (9.9%) |
| Unknown | 0 (0%) | 4 (3%) |
| Awarded | 2 (2.6%) | 4 (3%) |
| FIGO SUBSTAGE | ||
| A | 25 (32.9%) | 27 (20.5%) |
| B | 4 (5.3%) | 6 (4.5%) |
| C | 47 (61.8%) | 52 (39.4%) |
| NA | 0 (0%) | 47 (35.6%) |
| Mean age in years [min-max] | 53 [21–82] | 55 [17–80] |
| Mean follow up in years [min-max] | 9 [1–18] | 6 [0–17] |
| Total number of patients | 76 | 132 |
| Validation Set | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Muc vs SerHigh | Muc vs SerLow | Muc vs End | Muc vs Cc | SerHigh vs End | SerLow vs End | SerHigh vs Cc | SerLow vs Cc | End vs Cc | SerH vs SerL | |||||||||||
| p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | p | adj-p | |
| hsa-let-7a-5p | 0.0012 | 0.0091 | 0.9849 | 0.9863 | 0.0602 | 0.3635 | 0.0016 | 0.0074 | 0.0520 | 0.2758 | 0.1683 | 0.5946 | 0.4018 | 0.6980 | 0.0169 | 0.0805 | 0.1688 | 0.5215 | 0.0221 | 0.2328 |
| hsa-miR-192-5p | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.6842 | 0.9218 | 0.8806 | 0.9527 | 0.1767 | 0.6247 | 0.7796 | 0.8982 | 0.6812 | 0.8535 | 0.3771 | 0.7724 |
| hsa-miR-194-5p | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.8083 | 0.9218 | 0.7085 | 0.8940 | 0.1768 | 0.6247 | 0.9347 | 0.9527 | 0.5082 | 0.7506 | 0.4415 | 0.7784 |
| hsa-miR-214-3p | 0.0000 | 0.0005 | 0.6570 | 0.9118 | 0.0014 | 0.0153 | 0.0000 | 0.0001 | 0.3073 | 0.8143 | 0.0040 | 0.1054 | 0.8778 | 0.9481 | 0.0000 | 0.0001 | 0.2122 | 0.5367 | 0.0002 | 0.0096 |
| hsa-miR-26a-5p | 0.0072 | 0.0346 | 0.9291 | 0.9863 | 0.0790 | 0.4037 | 0.0033 | 0.0135 | 0.3287 | 0.8280 | 0.1551 | 0.5874 | 0.9999 | 0.9999 | 0.0095 | 0.0630 | 0.2958 | 0.6990 | 0.0283 | 0.2328 |
| hsa-miR-29a-3p | 0.1899 | 0.4575 | 0.0512 | 0.3874 | 0.3262 | 0.6668 | 0.0957 | 0.2205 | 0.7376 | 0.9218 | 0.0132 | 0.1754 | 0.7564 | 0.9481 | 0.0025 | 0.0187 | 0.5136 | 0.7506 | 0.0070 | 0.1189 |
| hsa-miR-29b-3p | 0.0044 | 0.0260 | 0.1692 | 0.9118 | 0.0031 | 0.0274 | 0.0003 | 0.0016 | 0.9426 | 0.9796 | 0.0024 | 0.1054 | 0.4083 | 0.6980 | 0.0007 | 0.0097 | 0.4482 | 0.7506 | 0.0035 | 0.0917 |
| hsa-miR-30a-3p | 0.8097 | 0.9331 | 0.5548 | 0.9118 | 0.8213 | 0.9572 | 0.0000 | 0.0000 | 0.9633 | 0.9818 | 0.7056 | 0.8940 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.4771 | 0.7784 |
| hsa-miR-30a-5p | 0.9686 | 0.9881 | 0.5980 | 0.9118 | 0.7880 | 0.9492 | 0.0000 | 0.0000 | 0.7934 | 0.9218 | 0.6702 | 0.8940 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.5494 | 0.8089 |
| hsa-miR-96-5p | 0.4640 | 0.6767 | 0.9863 | 0.9863 | 0.5985 | 0.8572 | 0.0003 | 0.0018 | 0.7916 | 0.9218 | 0.6623 | 0.8940 | 0.0011 | 0.0140 | 0.0020 | 0.0176 | 0.0004 | 0.0058 | 0.5140 | 0.7784 |
| CDK6 | 0.0004 | 0.0032 | 0.7787 | 0.9863 | 0.0124 | 0.0940 | 0.0017 | 0.0074 | 0.4418 | 0.9218 | 0.0986 | 0.4751 | 0.9367 | 0.9547 | 0.0284 | 0.1157 | 0.5240 | 0.7506 | 0.0090 | 0.1189 |
| CDKN1A | 0.0064 | 0.0339 | 0.5615 | 0.9118 | 0.4863 | 0.8054 | 0.0217 | 0.0715 | 0.0093 | 0.1910 | 0.8607 | 0.9527 | 0.2890 | 0.6831 | 0.2496 | 0.4267 | 0.0413 | 0.2739 | 0.1272 | 0.4496 |
| CDKN2A | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.2831 | 0.7896 | 0.7871 | 0.9426 | 0.0129 | 0.0857 | 0.0463 | 0.1504 | 0.0020 | 0.0209 | 0.5659 | 0.8107 |
| E2F3 | 0.3567 | 0.6301 | 0.8805 | 0.9863 | 0.3187 | 0.6668 | 0.0001 | 0.0004 | 0.0788 | 0.3797 | 0.4780 | 0.8940 | 0.0028 | 0.0249 | 0.0118 | 0.0697 | 0.0000 | 0.0003 | 0.6869 | 0.8307 |
| MDM2 | 0.0000 | 0.0000 | 0.0001 | 0.0016 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.5109 | 0.9218 | 0.5543 | 0.8940 | 0.0008 | 0.0140 | 0.1640 | 0.3858 | 0.0138 | 0.1218 | 0.2344 | 0.5730 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calura, E.; Ciciani, M.; Sambugaro, A.; Paracchini, L.; Benvenuto, G.; Milite, S.; Martini, P.; Beltrame, L.; Zane, F.; Fruscio, R.; et al. Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study. Cells 2019, 8, 1554. https://doi.org/10.3390/cells8121554
Calura E, Ciciani M, Sambugaro A, Paracchini L, Benvenuto G, Milite S, Martini P, Beltrame L, Zane F, Fruscio R, et al. Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study. Cells. 2019; 8(12):1554. https://doi.org/10.3390/cells8121554
Chicago/Turabian StyleCalura, Enrica, Matteo Ciciani, Andrea Sambugaro, Lara Paracchini, Giuseppe Benvenuto, Salvatore Milite, Paolo Martini, Luca Beltrame, Flaminia Zane, Robert Fruscio, and et al. 2019. "Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study" Cells 8, no. 12: 1554. https://doi.org/10.3390/cells8121554
APA StyleCalura, E., Ciciani, M., Sambugaro, A., Paracchini, L., Benvenuto, G., Milite, S., Martini, P., Beltrame, L., Zane, F., Fruscio, R., Marchette, M. D., Borella, F., Tognon, G., Ravaggi, A., Katsaros, D., Bignotti, E., Odicino, F., D’Incalci, M., Marchini, S., & Romualdi, C. (2019). Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study. Cells, 8(12), 1554. https://doi.org/10.3390/cells8121554