Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab

 ,
,
Abstract
1. Introduction
2. Preclinical Data and Mechanisms of Action of Isatuximab
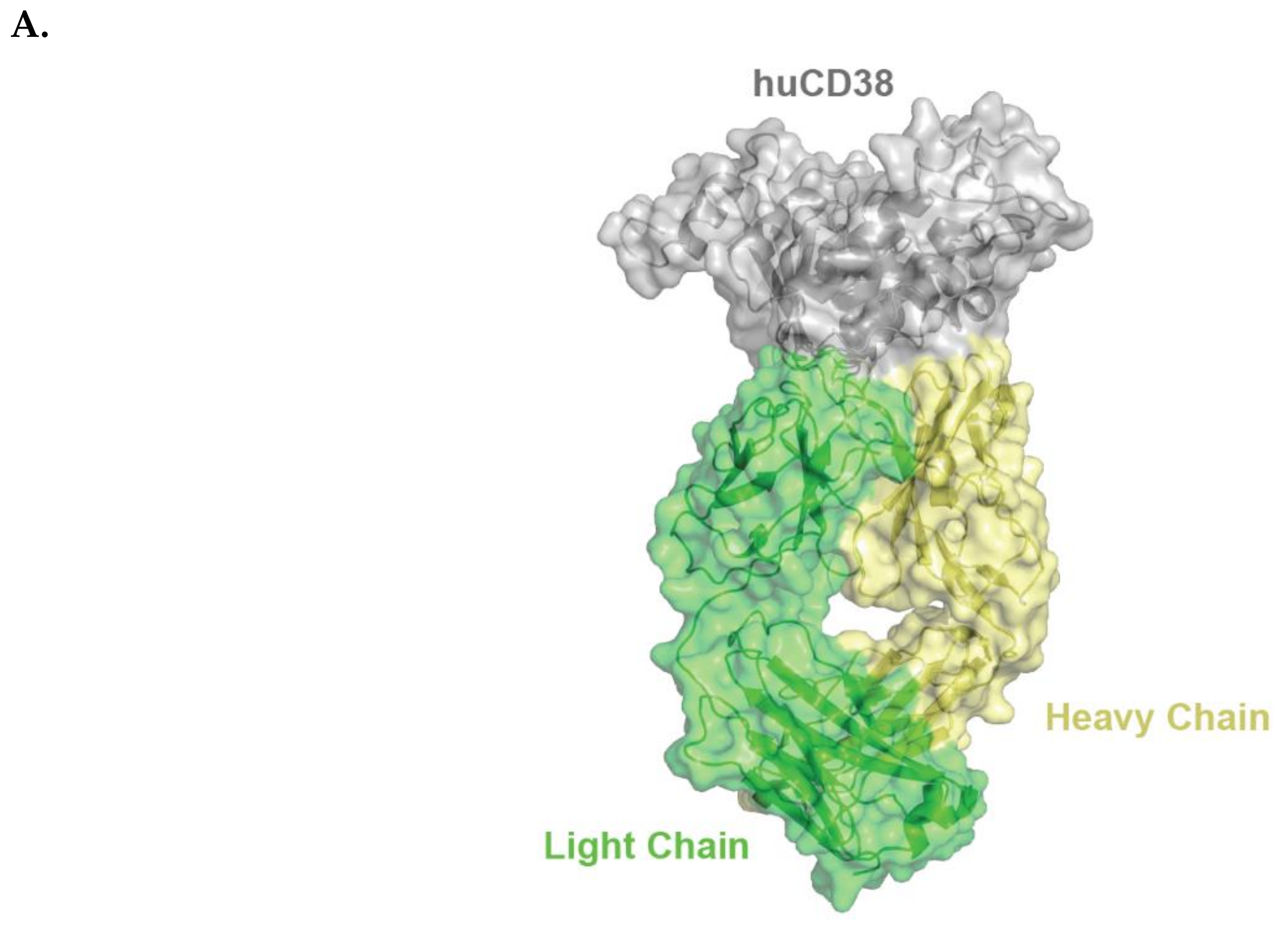
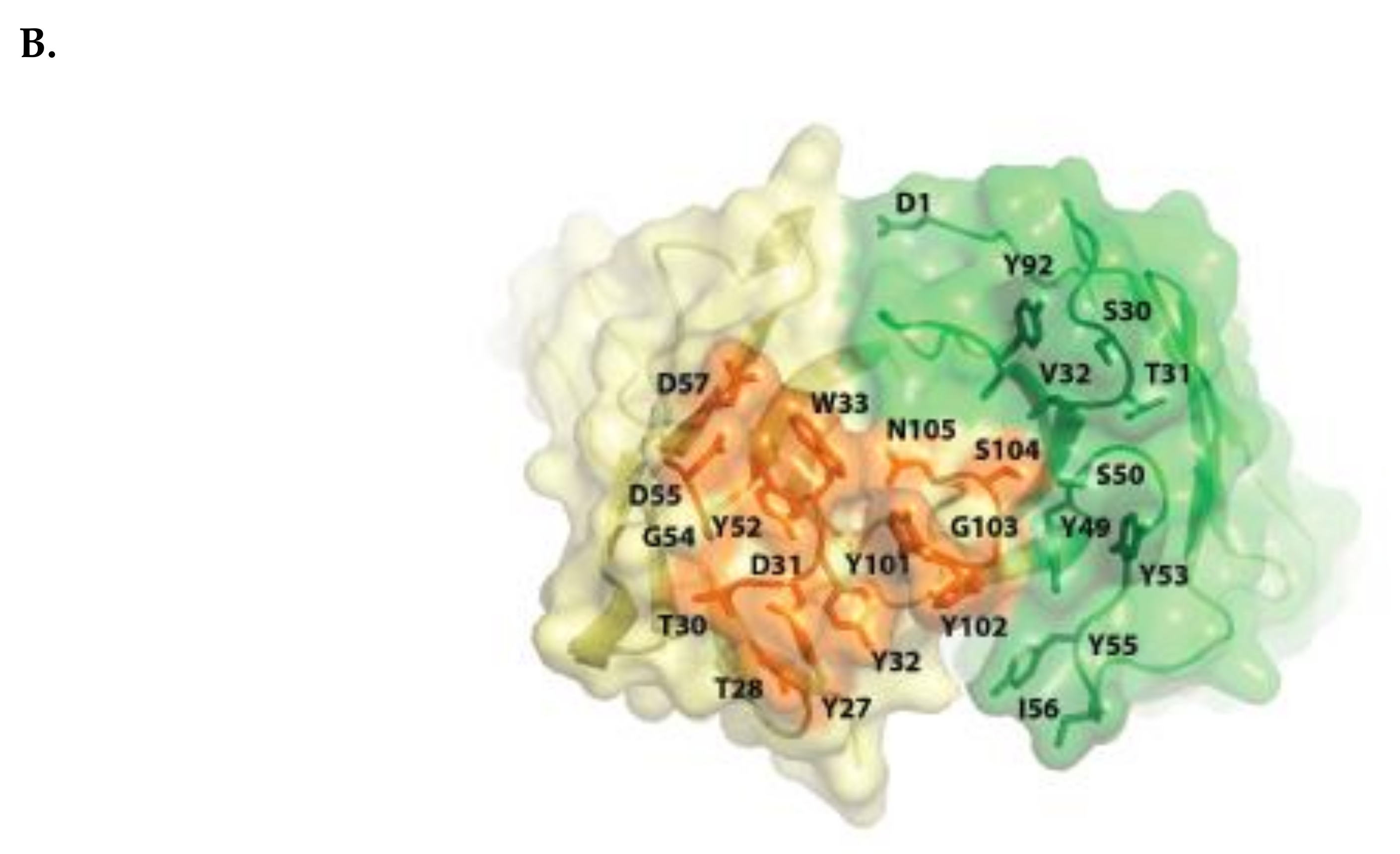
2.1. Isatuximab Mechanism of Binding
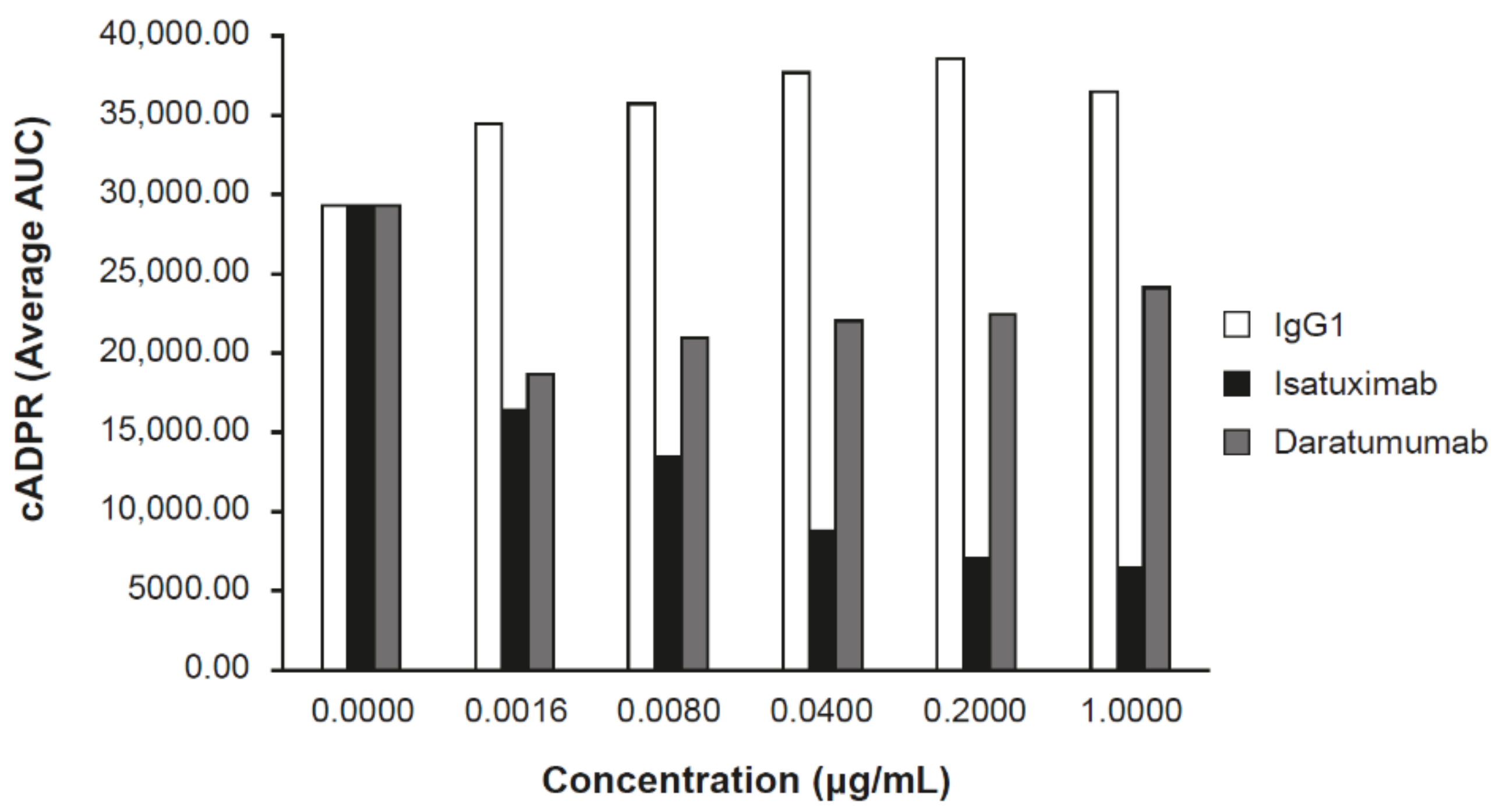
2.2. Isatuximab Uniquely Inhibits CD38 Enzymatic Activity
2.3. Isatuximab Induces Tumor Cell Death Through Effector Functions
2.3.1. ADCC
2.3.2. ADCP
2.3.3. CDC
2.4. Isatuximab Induces Direct Apoptosis
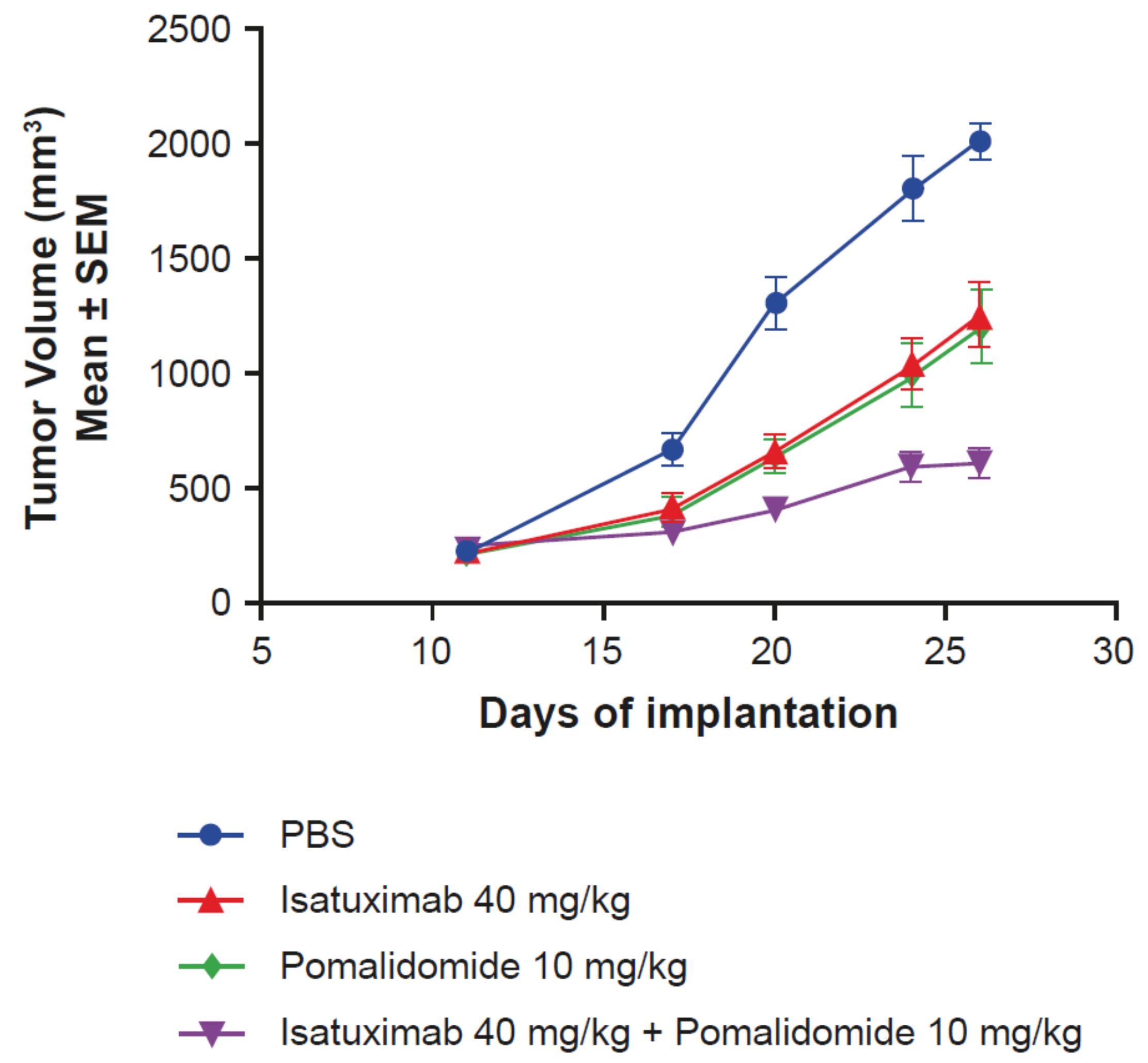
2.5. Activity of Isatuximab in Mouse Tumor Models
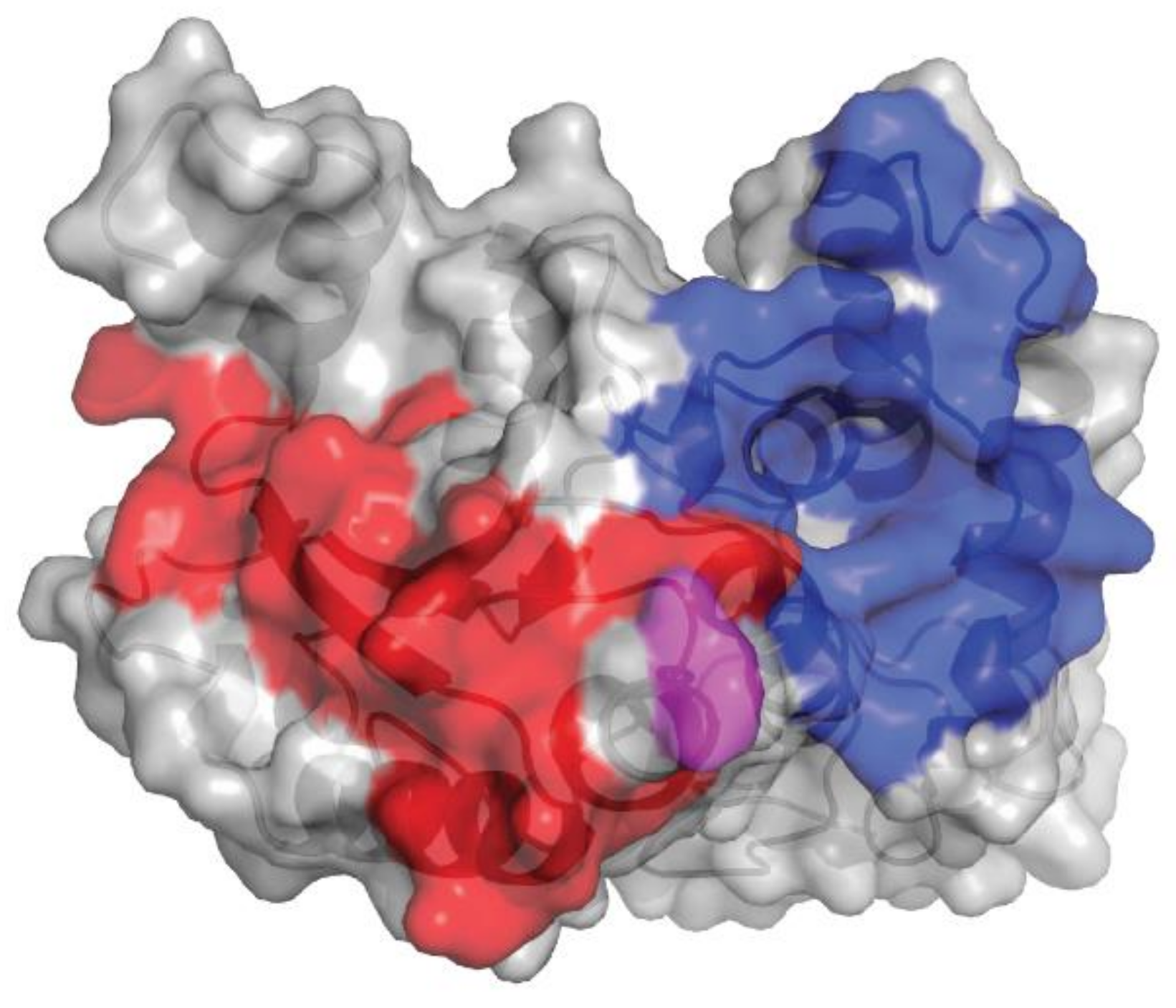
2.6. Isatuximab and Daratumumab Possess Several Mechanistic Differences
3. Targeting CD38 in MM
3.1. Isatuximab Monotherapy Activity in MM
3.2. Combination Therapy for MM with Isatuximab
3.2.1. Isatuximab in Combination with Immunomodulatory Drugs
3.2.2. Isatuximab in Combination with Proteasome Inhibitors
3.3. Isatuximab in Patients with Newly Diagnosed MM
4. Investigation of CD38 Antibodies in Other Therapeutic Areas
4.1. Other Malignancies
4.2. Solid Organ Transplantation
5. Anti-CD38 Agents in Development
5.1. Daratumumab
5.2. MOR202
5.3. TAK-079
6. Future Directions
6.1. Mechanisms of Resistance to CD38 Antibodies
6.2. Predictive Biomarkers for CD38 Antibodies
6.3. New Backbone Regimens with Anti-CD38 Therapies
6.4. Potential of Combining Immunotherapeutic CD38 Approaches in MM
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk. Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft-dependent signaling complex in human B cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Lee, H.C.; Aarhus, R.; Levitt, D. The crystal structure of cyclic ADP-ribose. Nat. Struct. Biol. 1994, 1, 143–144. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar]
- Lee, H.C.; Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Usmani, S.Z. CD38 antibodies in multiple myeloma: Mechanisms of action and modes of resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef]
- Darzalex [Package Insert]; Janssen Biotech, Inc.: Horsham, PA, USA, 2015.
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcgamma receptor-mediated cross-linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Lammerts van Bueren, J.; Jakobs, D.; Kaldenhoven, N.; Roza, M.; Hiddingh, S.; Meesters, J.; Voorhorst, M.; Gresnigt, E.; Wiegman, L.; Ortiz Buijsse, A.; et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood 2014, 124, 3474. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- De Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- Cai, T.; Wetzel, M.C.; Nicolazzi, C.; Vallee, F.; Deckert, J.; Dumontet, C.; Plesa, A.; Kannt, A.; Mikol, V.; Chiron, M.; et al. Preclinical Characterization of SAR650984, a Humanized Anti-CD38 Antibody for the Treatment of Multiple Myeloma. In Proceedings of the 14th International Myeloma Workshop (IMW), Kyoto, Japan, 3–7 April 2013; p. 288. [Google Scholar]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The mechanism of action of the anti-CD38 monoclonal antibody isatuximab in multiple myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zito, A.; Roato, I.; Morandi, F.; Marimpietri, D.; Bolzoni, M.; Toscani, D.; Oldham, R.J.; et al. NAD+-metabolizing ectoenzymes in remodeling tumor-host interactions: The human myeloma model. Cells 2015, 4, 520–537. [Google Scholar] [CrossRef]
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood 2011, 117, 5573–5581. [Google Scholar] [CrossRef]
- Rasche, L.; Kortum, K.M.; Raab, M.S.; Weinhold, N. The impact of tumor heterogeneity on diagnostics and novel therapeutic strategies in multiple myeloma. Int. J. Mol. Sci. 2019, 20, 1248. [Google Scholar] [CrossRef]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A target for immunotherapeutic approaches in multiple myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised international staging system for multiple myeloma: A report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma: Diagnosis and treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global burden of multiple myeloma: A systematic analysis for the Global Burden of Disease study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef]
- Coriu, D.; Dytfeld, D.; Niepel, D.; Spicka, I.; Markuljak, I.; Mihaylov, G.; Ostojic-Kolonic, S.; Fink, L.; Toka, K.; Björklöf, K. Real-world multiple myeloma management practice patterns and outcomes in selected Central and Eastern European countries. Pol. Arch. Intern. Med. 2018, 128, 500–511. [Google Scholar]
- Jagannath, S.; Roy, A.; Kish, J.; Lunacsek, O.; Globe, D.; Eaddy, M.; Kuriakose, E.; Willey, J.; Butler-Bird, S.; Siegel, D. Real-world treatment patterns and associated progression-free survival in relapsed/refractory multiple myeloma among US community oncology practices. Expert Rev. Hematol. 2016, 9, 707–717. [Google Scholar] [CrossRef]
- Yong, K.; Delforge, M.; Driessen, C.; Fink, L.; Flinois, A.; Gonzalez-McQuire, S.; Safaei, R.; Karlin, L.; Mateos, M.; Raab, M.; et al. Multiple myeloma: Patient outcomes in real-world practice. Br. J. Haematol. 2016, 175, 252–264. [Google Scholar] [CrossRef]
- Van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Bringhen, S.; Anttila, P.; Capra, M.; Cavo, M.; Cole, C.E.; Gasparetto, C.J.; Hungria, V.T.M.; Jenner, M.; Vorobyev, V.; et al. Results from a phase II study of isatuximab as a single agent and in combination with dexamethasone in patients with relapsed/refractory multiple myeloma. Blood 2018, 132, 155. [Google Scholar] [CrossRef]
- Martin, T.; Strickland, S.; Glenn, M.; Charpentier, E.; Guillemin, H.; Hsu, K.; Mikhael, J. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.R.; Martin, T.; Vij, R.; Cole, C.; Atanackovic, D.; Zonder, J.A.; Kaufman, J.L.; Mikhael, J.; Bensinger, W.; Dimopoulos, M.A.; et al. Updated data from a phase II dose finding trial of single agent isatuximab (SAR650984, anti-CD38 mAb) in relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2016, 34, 8005. [Google Scholar] [CrossRef]
- Iida, S.; Sunami, K.; Ri, M.; Matsumoto, M.; Shimazaki, C.; Asaoku, H.; Shibayama, H.; Ishizawa, K.; Takamatsu, H.; Ikeda, T.; et al. Phase 1/2 study of isatuximab monotherapy for relapsed and/or refractory multiple myeloma in Japanese patients: PF623. HemaSphere 2019, 3, 265. [Google Scholar] [CrossRef]
- Van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood Cancer J. 2011, 1, e41. [Google Scholar] [CrossRef]
- Martin, T.; Baz, R.; Benson, D.M.; Lendvai, N.; Wolf, J.; Munster, P.; Lesokhin, A.M.; Wack, C.; Charpentier, E.; Campana, F.; et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 129, 3294–3303. [Google Scholar] [CrossRef]
- Mikhael, J.; Richardson, P.; Usmani, S.Z.; Raje, N.; Bensinger, W.; Karanes, C.; Campana, F.; Kanagavel, D.; Dubin, F.; Liu, Q.; et al. A Phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood 2019, 134, 123–133. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Raikumar, S.V.; San-Miguel, J.F.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low dose dexamethasone versus pomalidomide and low dose dexamethasone in relapsed and refractory multiple myeloma: A randomized, multicenter, open-label, phase 3 study. Lancet 2019, in press. [Google Scholar] [CrossRef]
- Richardson, P.G.; Attal, M.; Campana, F.; Le-Guennec, S.; Hui, A.M.; Risse, M.L.; Corzo, K.; Anderson, K.C. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Future Oncol. 2018, 14, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Attal, M.; Rajkumar, S.V.; Miguel, J.S.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. A phase III randomized, open label, multicenter study comparing isatuximab, pomalidomide, and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8004. [Google Scholar]
- Chari, A.; Richter, J.R.; Shah, N.; Wong, S.W.K.; Jagannath, S.; Cho, H.J.; Biran, N.; Wolf, J.; Parekh, S.S.; Munster, P.N.; et al. Phase I-b study of isatuximab + carfilzomib in relapsed and refractory multiple myeloma (RRMM). J. Clin. Oncol. 2018, 36, 8014. [Google Scholar] [CrossRef]
- Martin, T.G.; Mannis, G.N.; Chari, A.; Munster, P.; Campana, F.; Hui, A.-M.; Wolf, J.L. Phase Ib study of isatuximab and carfilzomib in relapse and refractory multiple myeloma. Blood 2016, 128, 2111. [Google Scholar] [CrossRef]
- Ocio, E.M.; Bringhen, S.; Oliva, S.; Rodriguez-Otero, P.; Kanagavel, D.; Oprea, C.; Wei, V.; Doroumian, S.; Martinez-Lopez, J. A phase Ib study of isatuximab in combination with bortezomib, cyclophosphamide, and dexamethasone (VCDI) in patients with newly diagnosed multiple myeloma non-eligible for transplantation. Blood 2017, 130, 3160. [Google Scholar]
- Ocio, E.M.; Rodriguez Otero, P.; Bringhen, S.; Oliva, S.; Nogai, A.; Attal, M.; Moreau, P.; Kanagavel, D.; Fitzmaurice, T.F.; Wu, J.; et al. Preliminary results from a phase I study of isatuximab (ISA) in combination with bortezomib, lenalidomide, dexamethasone (VRd), and in patients with newly diagnosed multiple myeloma (NDMM) non-eligible for transplant. Blood 2018, 132, 595. [Google Scholar] [CrossRef]
- Di Gaetano, R.; Gasparetto, V.; Padoan, A.; Callegari, B.; Candiotto, L.; Sanzari, M.C.; Scapinello, A.; Tagariello, G. Flow cytometry CD4+CD26−CD38+ lymphocyte subset in the microenvironment of Hodgkin lymphoma-affected lymph nodes. Ann. Hematol. 2014, 93, 1319–1326. [Google Scholar] [CrossRef]
- Zaja, F.; Tabanelli, V.; Agostinelli, C.; Calleri, A.; Chiappella, A.; Varettoni, M.; Luigi Zinzani, P.; Volpetti, S.; Sabattini, E.; Fanin, R.; et al. CD38, BCL-2, PD-1, and PD-1L expression in nodal peripheral T-cell lymphoma: Possible biomarkers for novel targeted therapies? Am. J. Hematol. 2017, 92, E1–E2. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bogels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015, 7, 311–321. [Google Scholar] [CrossRef]
- Matas-Céspedes, A.; Vidal-Crespo, A.; Rodriguez, V.; Roue, G.; Campo, E.; Colomer, D.; van Bueren, J.L.; Bakker, J.M.; Wiestner, A.; Parren, P.W.H.I.; et al. Daratumumab, a novel human anti-CD38 monoclonal antibody for the treatment of chronic lymphocytic leukemia and B-cell non–Hodgkin lymphoma. Blood 2012, 120, 3935. [Google Scholar] [CrossRef]
- Mustafa, N.; Nee, H.F.A.; Lee, X.T.J.; Jin, W.; Yu, Y.; Chen, Y.; Yang, J.; Chng, W.J. Daratumumab efficiently targets NK/T cell lymphoma with high CD38 expression. Blood 2017, 130, 2814. [Google Scholar]
- Paulus, A.; Akhtar, S.; Bashir, Y.; Paulus, S.M.; Yousaf, H.; Roy, V.; Ailawadhi, S.; Ansell, S.; Witzig, T.E.; Chanan-Khan, A.A. Drug resistance alters CD38 expression and in vitro response to daratumumab in Waldenstrom macroglobulinemia cells. Blood 2016, 128, 3018. [Google Scholar] [CrossRef]
- Salles, G.; Gopal, A.K.; Minnema, M.C.; Wakamiya, K.; Feng, H.; Schecter, J.M.; Wang, M. Phase 2 study of daratumumab in relapsed/refractory mantle-cell lymphoma, diffuse large B-cell lymphoma, and follicular lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-S.; Eom, H.-S.; Yeh, S.-P.; Cho, S.-G.; Heo, D.S.; Kim, J.S.; Yao, M.; Zhu, J.; Gao, G.; Zhang, L.; et al. Daratumumab monotherapy for patients with relapsed or refractory (R/R) natural killer/T-cell lymphoma (NKTCL), nasal type: An open-label, single-arm, multicenter phase 2 study. Blood 2018, 132, 1617. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Chen, L.M.; Byers, L.A.; Ullrich, S.; Wistuba, I.I.; Qin, X.F.; Gibbons, D.L. CD38 as a novel immune checkpoint and a mechanism of resistance to the blockade of the PD-1/PD-L1 axis. J. Clin. Oncol. 2017, 35, 79. [Google Scholar] [CrossRef]
- Flores-Montero, J.; de Tute, R.; Paiva, B.; Perez, J.J.; Bottcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry B Clin. Cytom. 2016, 90, 61–72. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Rostaing, L.P.; Malvezzi, P. HLA-incompatible kidney transplantation—Worth the risk? N. Engl. J. Med. 2016, 374, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Wiebe, N.; Knoll, G.; Bello, A.; Browne, S.; Jadhav, D.; Klarenbach, S.; Gill, J. Systematic review: Kidney transplantation compared with dialysis in clinically relevant outcomes. Am. J. Transplant. 2011, 11, 2093–2109. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, C.; Loupy, A.; Hill, G.S.; Andrade, J.; Nochy, D.; Antoine, C.; Gautreau, C.; Charron, D.; Glotz, D.; Suberbielle-Boissel, C. Preexisting donor-specific HLA antibodies predict outcome in kidney transplantation. J. Am. Soc. Nephrol. 2010, 21, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Terasaki, P.I. Significance of the positive crossmatch test in kidney transplantation. N. Engl. J. Med. 1969, 280, 735–739. [Google Scholar] [CrossRef]
- Everly, M.J.; Terasaki, P.I. The state of therapy for removal of alloantibody producing plasma cells in transplantation. Semin. Immunol. 2012, 24, 143–147. [Google Scholar] [CrossRef]
- Lemy, A.; Toungouz, M.; Abramowicz, D. Bortezomib: A new player in pre- and post-transplant desensitization? Nephrol. Dial. Transplant. 2010, 25, 3480–3489. [Google Scholar] [CrossRef]
- Kwun, J.; Matignon, M.; Manook, M.; Guendouz, S.; Audard, V.; Kheav, D.; Poullot, E.; Gautreau, C.; Ezekian, B.; Bodez, D.; et al. Daratumumab in sensitized kidney transplantation: Potentials and limitations of experimental and clinical use. J. Am. Soc. Nephrol. 2019, 30, 1206–1219. [Google Scholar] [CrossRef]
- Jordan, S.C.; Vescio, R.; Toyoda, M.; Ammerman, N.; Huang, E.; Peng, A.; Sethi, S.; Najjar, R.; Lim, K.; Vo, A. Daratumumab for treatment of antibody-mediated rejection in a kidney transplant recipient. Am. J. Transplant. 2019, 19, 1062. [Google Scholar]
- Casneuf, T.; Xu, X.; Adams, H.; Axel, A.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef]
- Janssen Submits Application to U.S. FDA Seeking Approval of New DARZALEX® (Daratumumab) Subcutaneous Formulation. Available online: https://www.janssen.com/janssen-submits-application-us-fda-seeking-approval-new-darzalex-daratumumab-subcutaneous (accessed on 18 November 2019).
- Mateos, M.V.; Nahi, H.; Legiec, W.; Grosicki, S.; Vorobyev, V.; Spicka, I.; Hungria, V.; Korenkova, S.; Bahlis, N.J.; Flogegard, M.; et al. Efficacy and safety of the randomized, open-label, non-inferiority, phase 3 study of subcutaneous (SC) versus intravenous (IV) daratumumab (DARA) administration in patients (pts) with relapsed or refractory multiple myeloma (RRMM): COLUMBA. J. Clin. Oncol. 2019, 37, 8005. [Google Scholar]
- Boxhammer, R.; Weirather, J.; Steidl, S.; Endell, J. MOR202, a human anti-CD38 monoclonal antibody, mediates potent tumoricidal activity in vivo and shows synergistic efficacy in combination with different antineoplastic compounds. Blood 2015, 126, 3015. [Google Scholar] [CrossRef]
- Endell, J.; Boxhammer, R.; Wurzenberger, C.; Ness, D.; Steidl, S. The activity of MOR202, a fully human anti-CD38 antibody, is complemented by ADCP and is synergistically enhanced by lenalidomide in vitro and in vivo. Blood 2012, 120, 4018. [Google Scholar] [CrossRef]
- Endell, J.; Samuelsson, C.; Boxhammer, R.; Strauss, S.; Steidl, S. Effect of MOR202, a human CD38 antibody, in combination with lenalidomide and bortezomib, on bone lysis and tumor load in a physiologic model of myeloma. J. Clin. Oncol. 2011, 29, 8078. [Google Scholar] [CrossRef]
- Raab, M.S.; Goldschmidt, H.; Agis, H.; Blau, I.; Einsele, H.; Engelhardt, M.M.; Ferstl, B.; Gramatzki, M.; Röllig, C.; Weisel, K.; et al. A phase I/IIa study of the human anti-CD38 antibody MOR202 (MOR03087) in relapsed or refractory multiple myeloma (rrMM). J. Clin. Oncol. 2015, 33, 8574. [Google Scholar] [CrossRef]
- Raab, M.S.; Chatterjee, M.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Engelhardt, M.M.; Ferstl, B.; Gramatzki, M.; Röllig, C.; et al. MOR202 alone and in combination with pomalidomide or lenalidomide in relapsed or refractory multiple myeloma: Data from clinically relevant cohorts from a phase I/IIa study. J. Clin. Oncol. 2016, 34, 8012. [Google Scholar] [CrossRef]
- Takeda R&D Investor Day 2018. Available online: https://www.takeda.com/siteassets/system/investors/report/quarterlyannouncements/fy2018/0_full_deck_boston_e.pdf (accessed on 27 September 2019).
- Smithson, G.; Zalevsky, J.; Korver, W.; Roepcke, S.; Dahl, M.; Zhao, L.; Yuan, J.; McLean, L.; Elias, K. TAK-079 is a high affinity monoclonal antibody that effectively mediates CD38+ cell depletion. J. Immunol. 2017, 198, 224–320. [Google Scholar]
- Fedyk, E.R.; Berg, D.; Smithson, G.; Estevam, J.; Mclean, L.; Allikmets, K.; Palumbo, A. A single administration of the cytolytic CD38 antibody TAK-079 to healthy subjects: Tolerability, pharmacokinetics and pharmacodynamics. Blood 2018, 132, 3249. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Song, Z.; Yang, G.; Wang, A.; Greco, R.; Theilhaber, J.; Shehu, E.; Ajona, D.; Paiva, B.; Zhu, C.; Wiederschain, D.; et al. Abstract 2966: Isatuximab-induced multiple myeloma cell killing through effector functions is dependent on CD38 expression and complement inhibitors. Cancer Res. 2019, 79, 2966. [Google Scholar]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Sonneveld, P.; Hungria, V.T.M.; Nooka, A.K.; Estell, J.; Barreto, W.G.; Corradini, P.; Min, C.-K.; Medvedova, E.; Weisel, K.; et al. Efficacy and safety of daratumumab, bortezomib, and dexamethasone (D-Vd) versus bortezomib and dexamethasone (Vd) in first relapse patients: Two-year update of Castor. Blood 2018, 132, 3270. [Google Scholar] [CrossRef]
- Weisel, K.C.; San Miguel, J.; Cook, G.; Leiba, M.; Suzuki, K.; Kumar, S.; Cavo, M.; Avet-Loiseau, H.; Quach, H.; Hungria, V.; et al. Efficacy of daratumumab in combination with lenalidomide plus dexamethasone (DRd) or bortezomib plus dexamethasone (DVd) in relapsed or refractory multiple myeloma (RRMM) based on cytogenetic risk status. J. Clin. Oncol. 2017, 35, 8006. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Bene, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Casneuf, T.; Lysaght, A.; LeFave, C.; Bald, J.; Weiss, B.; van de Donk, N.W.C.J.; Lokhorst, H.M.; Ahmadi, T.; Sasser, A.K. Serum proteomic analysis of multiple myeloma subjects treated with daratumumab monotherapy. Blood 2015, 126, 1837. [Google Scholar] [CrossRef]
- Atanackovic, D.; Yousef, S.; Shorter, C.; Tantravahi, S.K.; Steinbach, M.; Iglesias, F.; Sborov, D.; Radhakrishnan, S.V.; Chiron, M.; Miles, R.; et al. In vivo vaccination effect in multiple myeloma patients treated with the monoclonal antibody isatuximab. Leukemia 2019, 1–5. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Ashcroft, J.; Szabo, Z.; Garderet, L. Navigating the treatment landscape in multiple myeloma: which combinations to use and when? Ann. Hematol. 2019, 98, 1–18. [Google Scholar] [CrossRef]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Invest. 2015, 125, 2904. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejia, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef]
- Yousef, S.; Marvin, J.; Steinbach, M.; Langemo, A.; Kovacsovics, T.; Binder, M.; Kroger, N.; Luetkens, T.; Atanackovic, D. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J. 2015, 5, e285. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Puryear, J.; Schinke, C.D.; van Rhee, F.; Zangari, M.; Mathur, P.; Mohan, M.; Susanibar, S.; Jo Kamimoto, J.; Hoque, S.; et al. Nivolumab for treatment of advanced, refractory, high-risk multiple myeloma. Blood 2017, 130, 1858. [Google Scholar]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N.W.C.J. Immunotherapy in myeloma: How far have we come? Ther. Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef]
- Abramson, H. Monoclonal antibodies for the treatment of multiple myeloma: An update. Int. J. Mol. Sci. 2018, 19, 3924. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.J.; Noort, W.A.; van Kessel, B.; de Jong-Korlaar, R.; Bakker, J.; van Bueren, J.J.L.; Parren, P.W.H.I.; Lokhorst, H.M.; van de Donk, N.W.C.J.; et al. Preclinical evidence for the therapeutic potential of CD38-targeted immuno-chemotherapy in multiple myeloma patients refractory to lenalidomide and bortezomib. Clin. Cancer Res. 2015, 21, 2802–2810. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Maximum NK-Mediated Lysis, % | EC50 Values, pM (ng/mL) |
|---|---|---|
| LP-1 | 37 | 13 (2.02) |
| MOLP-8 | 28 | 1 (0.16) |
| NCI-H929 | 27 | 50 (7.61) |
| Clinical Trial | Phase | Patients | Primary Endpoints |
|---|---|---|---|
| NCT02960555 a | 2 | Smoldering myeloma | Determine the rate of response according to the IMWGC of isatuximab as monotherapy |
| NCT01084252 | 1/2 | RRMM | Evaluate DLTs and the ORR of isatuximab as monotherapy |
| NCT02812706 | 1/2 | RRMM | Evaluate DLTs and the ORR of isatuximab in Japanese patients |
| NCT03733717 | 1 | RRMM | Evaluate the pharmacokinetics, safety, and tolerability of isatuximab in Chinese patients |
| NCT02514668 | 1 | RRMM | Evaluate safety, tolerability, and the ORR of isatuximab in patients previously treated with daratumumab |
| Clinical Trial | Phase | Patients | Treatment |
|---|---|---|---|
| NCT03319667 (IMROZ) | 3 | NDMM ineligible for transplant | Isatuximab + VRd vs VRd |
| NCT03275285 (IKEMA) | 3 | RRMM, 1–3 prior lines of therapy | Isatuximab + Kd vs Kd |
| NCT03617731a (GMMG HD7) | 3 | NDMM eligible for transplant | Isatuximab + VRd, then maintenance with R |
| NCT03104842b | 2 | NDMM with high-risk cytogenetic profile | Isatuximab + KRd |
| NCT03194867 | 1/2 | RRMM, ≥3 prior lines of therapy | Isatuximab + cemiplimab |
| NCT04083898c | 1/2 | Penta-refractory MM | Isatuximab + BPr |
| NCT01749969 | 1b | RRMM | Isatuximab + Rd |
| NCT02332850d | 1 | RRMM, ≥2 prior lines of therapy | Isatuximab + K |
| NCT02513186 | 1 | NDMM ineligible for transplant | Isatuximab + VCd; Isatuximab + VRd |
| NCT04045795 | 1 | RRMM | Isatuximab IV + Pd; Isatuximab SC + Pd |
| Clinical Trial | Phase | Malignancy | Treatment |
|---|---|---|---|
| NCT03860844 | 2 | RR ALL and AML | Isatuximab + standard chemotherapies |
| NCT03499808 a | 2 | RR systemic light-chain AL | Isatuximab |
| NCT03769181 | 1/2 | RR cHL, DLBCL, and PTCL | Isatuximab + cemiplimab (anti-PD-1) |
| NCT03367819 | 1/2 | mCRPC and NSCLC | Isatuximab + cemiplimab (anti-PD-1) |
| NCT03637764 | 1/2 | Unresectable HCC, platinum-refractory recurrent/metastatic SCCHN, platinum-resistant/refractory EOC and recurrent GBM | Isatuximab + atezolizumab (anti-PD-L1) |
| NCT03555149 (Morpheus-CRC) b | 1/2 | mCRC that became refractory to first- and second-line standard therapies | Atezolizumab (anti-PD-L1) combined with other immunotherapies, including isatuximab |
| NCT03869190 (MORPHEUS mUC) b | 1/2 | Locally advanced or metastatic UC that has progressed during or following a platinum-containing regimen | Atezolizumab (anti-PD-L1) combined with other immunotherapies, including isatuximab |
| Agent | Company | Modality | Highest Phase |
|---|---|---|---|
| Daratumumab-rHuPH20 (Dara-SC) | Janssen/Genmab | mAb | BLA |
| Isatuximab | Sanofi | mAb | BLA |
| MOR202/TJ202 (MOR03087) | I-Mab/MorphoSys | mAb | 3 |
| TAK-079 (SC) | Takeda | mAb | 1/2 |
| CAR-T/TCR-T | Shenzhen BinDeBio | Cell therapy | 1/2 |
| Multi-CAR-T | Shenzhen Geno Immune | Cell therapy | 1/2 |
| TAK-573 | Takeda | Immunocytokine | 1/2 |
| SAR442085 | Sanofi | Fc-engineered | 1 |
| TAK-169 | Takeda | ETB | 1 |
| T-007 | Sorrento Therapeutics | Cell therapy | 1 |
| AMG 424 | Amgen | TCE | 1 |
| GBR 1342 | Glenmark | TCE | 1 |
| Isatuximab (SC) | Sanofi | mAb | 1 |
| HexaBody-CD38 | Janssen/Genmab | Fc engineered | Preclinical |
| CD38-ARM (KP1196, KP1237) | Kleo/PeptiDream | ARM | Preclinical |
| TSK011010/CID103 | CASI Pharmaceuticals | mAb | Preclinical |
| STI-5171 | CASI Pharmaceuticals | mAb | Preclinical |
| Anti-CD38/IGF-1 R bsAb scFV | I’rom Group/GeneTry | bsAb | Preclinical |
| Anti-CD38 SIFbody | Momenta Pharmaceuticals | Fc engineered | Preclinical |
| CAR38-MILs | WindMIL | Cell therapy | Preclinical |
| CD38 DART | Sorrento Therapeutics | Cell therapy | Preclinical |
| Actinium-225 dara | Actinium Pharmaceuticals | Radionuclide | Preclinical |
| STI-6129 | Sorrento Therapeutics | ADC | Preclinical |
| Anti-CD38/anti-CD3 | IGM Biosciences | TCE | Preclinical |
| CD38 TCE | Sorrento Therapeutics | TCE | Preclinical |
| Y-150 | Wuhan YZY | TCE | Preclinical |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, T.G.; Corzo, K.; Chiron, M.; van de Velde, H.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells 2019, 8, 1522. https://doi.org/10.3390/cells8121522
Martin TG, Corzo K, Chiron M, van de Velde H, Abbadessa G, Campana F, Solanki M, Meng R, Lee H, Wiederschain D, et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells. 2019; 8(12):1522. https://doi.org/10.3390/cells8121522
Chicago/Turabian StyleMartin, Thomas G., Kathryn Corzo, Marielle Chiron, Helgi van de Velde, Giovanni Abbadessa, Frank Campana, Malini Solanki, Robin Meng, Helen Lee, Dmitri Wiederschain, and et al. 2019. "Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab" Cells 8, no. 12: 1522. https://doi.org/10.3390/cells8121522
APA StyleMartin, T. G., Corzo, K., Chiron, M., van de Velde, H., Abbadessa, G., Campana, F., Solanki, M., Meng, R., Lee, H., Wiederschain, D., Zhu, C., Rak, A., & Anderson, K. C. (2019). Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells, 8(12), 1522. https://doi.org/10.3390/cells8121522