Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model



Abstract
1. Introduction
2. Materials and Methods
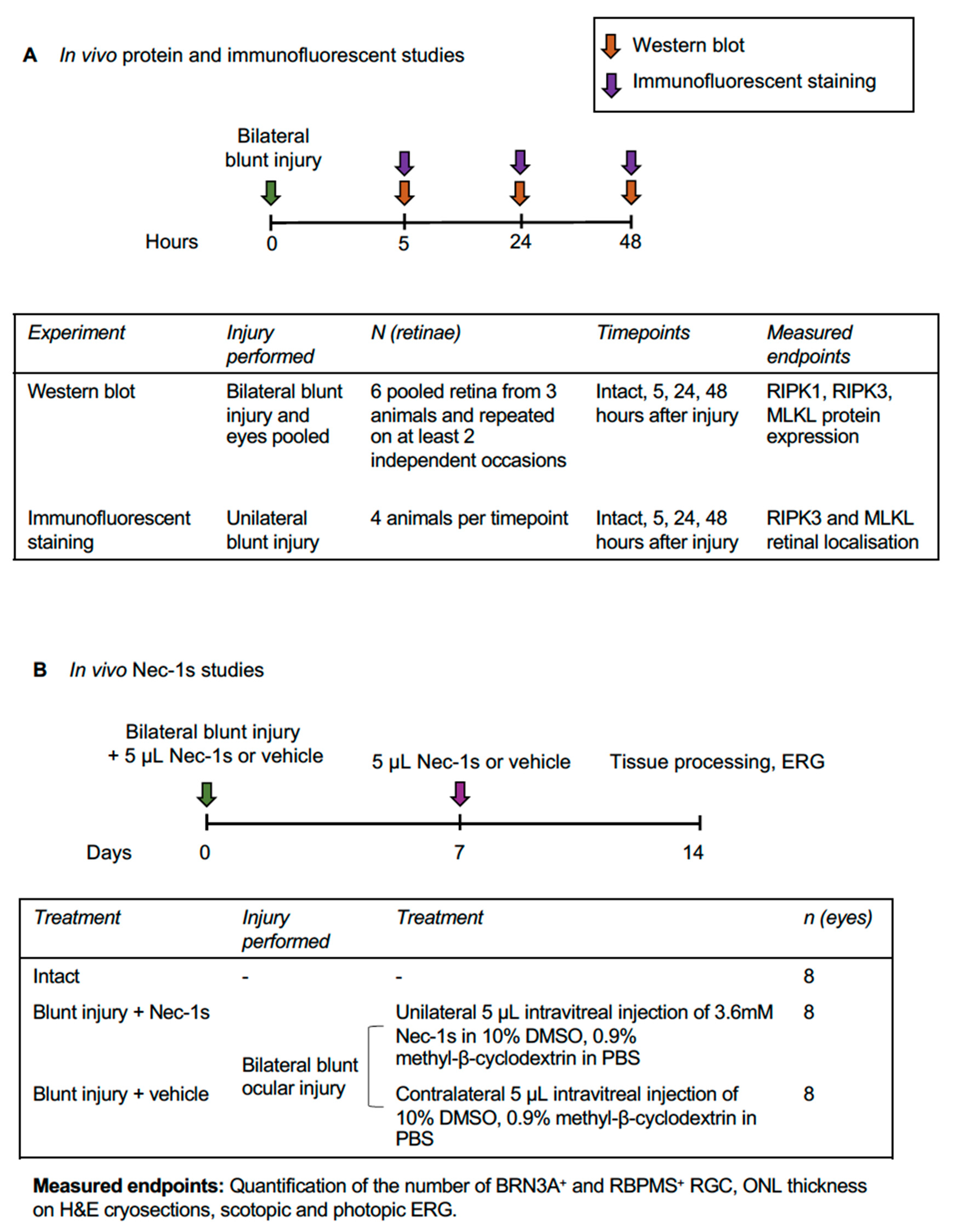
2.1. Experimental Design
2.2. Animal Care and Procedures
2.3. Western Blot
2.4. Intravitreal Injection of Nec-1s
2.5. Tissue Preparation for Immunohistochemistry
2.6. Immunohistochemistry Protocol
2.7. Electroretinography (ERG)
2.8. Assessment of ONL Thickness
2.9. Assessment of RGC Survival
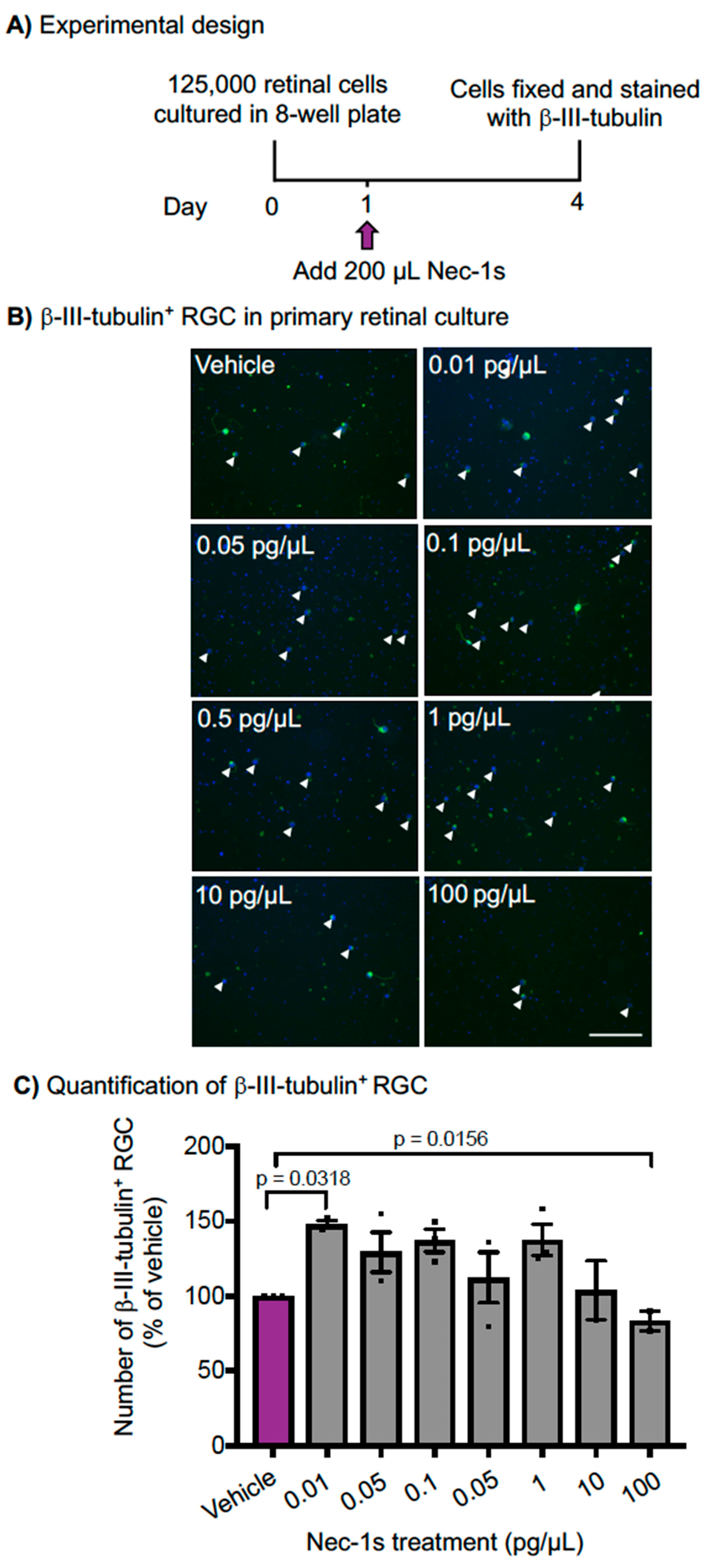
2.10. Primary RGC Culture and Immunocytochemistry
2.11. Statistics
3. Results
3.1. MLKL Protein Expression was Higher 5 h after Blunt Ocular Injury Compared with Intact Eyes, but RIPK1 and RIPK3 Expression Did Not Change
3.2. RIPK3 is Localized to BRN3A+ RGC whilst MLKL is Localized to OPL, ONL, INL and RBPMS+ RGC
3.3. Nec-1s is Neuroprotective In Vitro
3.4. Nec-1s Preserved BRN3A+ and RBPMS+ RGC at the Center of the Blunt Injury Site Compared to Vehicle Treated Eyes but did not Preserve Photoreceptors
3.5. Nec-1s Treatment did not Preserve ERG Amplitudes After Blunt Ocular Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| BRN3A | Brain-Specific Homeobox/POU Domain Protein 3A |
| BSA | Bovine Serum Albumin |
| DAPI | 4′,6’-Diamidino-2-Phenylindole |
| DMSO | Dimethyl Sulfoxide |
| Dpi | Days Post Injury |
| ERG | Electroretinogram |
| GCL | Ganglion Cell Layer |
| GLM | Generalized Linear Models |
| H&E | Hematoxylin and Eosin |
| IDO | Indoleamine-2,3-Dioxygenase |
| IHC | Immunohistochemistry |
| INL | Inner Nuclear Layer |
| MLKL | Mixed Lineage Kinase Domain Like Pseudokinase |
| Nec-1 | Necrostatin-1 |
| Nec-1s | Necrostatin-1s |
| OCTc | Optimal Cutting Temperature Compound |
| ONC | Optic Nerve Crush |
| ON | Optic Nerve |
| ONH | Optic Nerve Head |
| ONL | Outer Nuclear Layer |
| OPL | Outer Plexiform Layer |
| PBS | Phosphate Buffered Saline |
| PFA | Paraformaldehyde |
| PVDF | Polyvinylidene Fluoride |
| RBPMS | Retinal Binding Protein with Multiple Splicing |
| RGC | Retinal Ganglion Cells |
| RT | Room Temperature |
| RIPK1 | Receptor Interacting Kinase 1 |
| RIPK3 | Receptor Interacting Kinase 3 |
| SDS | Sodium Dodecyl Sulphate |
| SEM | Standard Error of the Mean |
| siRNA | Small Interfering RNA |
| TON | Traumatic Optic Neuropathy |
| TNF | Tumor Necrosis Factor |
| WB | Western Blot |
References
- Blanch, R.J.; Bindra, M.S.; Jacks, A.S.; Scott, R.A.H. Ophthalmic injuries in British Armed Forces in Iraq and Afghanistan. Eye 2011, 25, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Klein, B.E.K.; Klein, R. The prevalence and 5-year incidence of ocular trauma—The Beaver Dam Eye Study. Ophthalmology 2000, 107, 2196–2202. [Google Scholar] [CrossRef]
- Levin, L.A.; Beck, R.W.; Joseph, M.P.; Seiff, S.; Kraker, R. The treatment of traumatic optic neuropathy: The International Optic Nerve Trauma Study. Ophthalmology 1999, 106, 1268–1277. [Google Scholar] [CrossRef]
- Chaon, B.C.; Lee, M.S. Is there treatment for traumatic optic neuropathy? Curr. Opin. Ophthalmol. 2015, 26, 445–449. [Google Scholar] [CrossRef]
- Blanch, R.J.; Scott, R.A.H. Primary blast injury of the eye. J. R. Army Med. Corps 2008, 154, 76. [Google Scholar]
- Sarkies, N. Traumatic optic neuropathy. Eye 2004, 18, 1122–1125. [Google Scholar] [CrossRef]
- Guy, W.M.; Soparkar, C.N.; Alford, E.L.; Patrinely, J.R.; Sami, M.S.; Parke, R.B. Traumatic optic neuropathy and second optic nerve injuries. JAMA Ophthalmol. 2014, 132, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Sipperley, J.O.; Quigley, H.A.; Gass, D.M. Traumatic retinopathy in primates. The explanation of commotio retinae. Arch. Ophthalmol. 1978, 96, 2267–2273. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G. Steroids for traumatic optic neuropathy. Cochrane Database Syst. Rev. 2013, 6, CD006032. [Google Scholar] [CrossRef]
- Lee, V.; Ford, R.L.; Xing, W.; Bunce, C.; Foot, B. Surveillance of traumatic optic neuropathy in the UK. Eye 2010, 24, 240–250. [Google Scholar] [CrossRef]
- Weichel, E.D.; Colyer, M.H.; Ludlow, S.E.; Bower, K.S.; Eiseman, A.S. Combat Ocular Trauma Visual Outcomes during Operations Iraqi and Enduring Freedom. Ophthalmology 2008, 115, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Blanch, R.J.; Good, P.A.; Shah, P.; Bishop, J.R.B.; Logan, A.; Scott, R.A.H. Visual outcomes after blunt ocular trauma. Ophthalmology 2013, 120, 1588–1591. [Google Scholar] [CrossRef] [PubMed]
- Bricker-Anthony, C.; Hines-Beard, J.; Rex, T.S. Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4853–4862. [Google Scholar] [CrossRef] [PubMed]
- Hines-Beard, J.; Marchetta, J.; Gordon, S.; Chaum, E.; Geisert, E.E.; Rex, T.S. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp. Eye Res. 2012, 99, 63–70. [Google Scholar] [CrossRef]
- Blanch, R.J.; Ahmed, Z.; Sik, A.; Snead, D.R.J.; Good, P.A.; O’Neill, J.; Berry, M.; Scott, R.A.H.; Logan, A. Neuroretinal cell death in a murine model of closed globe injury: Pathological and functional characterization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7220–7226. [Google Scholar] [CrossRef]
- Berkelaar, M.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [CrossRef]
- Villegas-Perez, M.P.; Vidal-Sanz, M.; Rasminsky, M.; Bray, G.M.; Aguayo, A.J. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J. Neurobiol. 1993, 24, 23–36. [Google Scholar] [CrossRef]
- Berry, M.; Carlile, J.; Hunter, A. Peripheral nerve explants grafted into the vitreous body of the eye promote the regeneration of retinal ganglion cell axons severed in the optic nerve. J. Neurocytol. 1996, 25, 147–170. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Gizycka, A.; Chorostowska-Wynimko, J. Programmed necrosis and necroptosis–molecular mechanisms. Postepy. Hig. Med. Dosw. 2015, 69, 1353–1363. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef]
- Shen, H.; Liu, C.; Zhang, D.; Yao, X.; Zhang, K.; Li, H.; Chen, G. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 2017, 8, 2641. [Google Scholar] [CrossRef]
- Yang, S.H.; Lee, D.K.; Shin, J.; Lee, S.; Baek, S.; Kim, J.; Jung, H.; Hah, J.M.; Kim, Y. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol. Med. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Tao, Y.; Zhang, S.; Wang, J.; Feng, X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 2014, 266, 91–101. [Google Scholar] [CrossRef]
- You, Z.; Savitz, S.I.; Yang, J.; Degterev, A.; Yuan, J.; Cuny, G.D.; Moskowitz, M.A.; Whalen, M.J. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2008, 28, 1564–1573. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Degterev, A.; Ivanov, D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp. Eye Res. 2014, 123, 1–7. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, 437. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.N.; Thompson, A.M.; McCance, E.; Berry, M.; Logan, A.; Blanch, R.J.; Ahmed, Z. Caspase-2 mediates site-specific retinal ganglion cell death after blunt ocular injury. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4453–4462. [Google Scholar] [CrossRef] [PubMed]
- Blanch, R.J.; Ahmed, Z.; Thompson, A.R.; Akpan, N.; Snead, D.R.J.; Berry, M.; Troy, C.M.; Scott, R.A.H.; Logan, A. Caspase-9 mediates photoreceptor death after blunt ocular trauma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6350–6357. [Google Scholar] [CrossRef][Green Version]
- Ahmed, Z.; Suggate, E.L.; Brown, E.R.; Dent, R.G.; Armstrong, S.J.; Barrett, L.B.; Berry, M.; Logan, A. Schwann cell-derived factor-induced modulation of the NgR/p75NTR/EGFR axis disinhibits axon growth through CNS myelin in vivo and in vitro. Brain 2006, 129, 1517–1533. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Mead, B.; Tomarev, S. Evaluating retinal ganglion cell loss and dysfunction. Exp. Eye Res. 2016, 151, 96–106. [Google Scholar] [CrossRef]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Nieto-Lopez, L.; Canovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a Marker of Retinal Ganglion Cells: Qualitative and Quantitative Time Course Studies in Naive and Optic Nerve-Injured Retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef]
- Kwong, J.M.K.; Quan, A.; Kyung, H.; Piri, N.; Caprioli, J. Quantitative Analysis of Retinal Ganglion Cell Survival with Rbpms Immunolabeling in Animal Models of Optic Neuropathies. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9694–9702. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Muller, L.P.D.; Brecha, N.C. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J. Comp. Neurol. 2014, 522, 1411–1443. [Google Scholar] [CrossRef]
- Thompson, A.; Berry, M.; Logan, A.; Ahmed, Z. Activation of the BMP4/Smad1 Pathway Promotes Retinal Ganglion Cell Survival and Axon Regeneration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Warren, P.J.; O’Neill, J.; de Cogan, F.; Spivak, I.; Ashush, H.; Kalinski, H.; Ahmed, Z.; Berry, M.; Feinstein, E.; Scott, R.A.; et al. siRNA-Mediated Knockdown of the mTOR Inhibitor RTP801 Promotes Retinal Ganglion Cell Survival and Axon Elongation by Direct and Indirect Mechanisms. Investig. Ophthalmol. Vis. Sci. 2016, 57, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Logan, A.; Berry, M.; Leadbeater, W.; Scheven, B.A. Intravitreally transplanted dental pulp stem cells promote neuroprotection and axon regeneration of retinal ganglion cells after optic nerve injury. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7544–7556. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Davies, K.A.; Tanzer, M.C.; Griffin, M.D.W.; Mok, Y.F.; Young, S.N.; Qin, R.; Petrie, E.J.; Czabotar, P.E.; Silke, J.; Murphy, J.M. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 2018, 25, 1567–1580. [Google Scholar] [CrossRef]
- Trichonas, G.; Murakami, Y.; Thanos, A.; Morizane, Y.; Kayama, M.; Debouck, C.M.; Hisatomi, T.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc. Natl. Acad. Sci. USA 2010, 107, 21695–21700. [Google Scholar] [CrossRef]
- Murakami, Y.; Miller, J.W.; Vavvas, D.G. RIP Kinase-Mediated Necrosis as an Alternative Mechanism of Photoreceptor Death. Oncotarget 2011, 2, 497–509. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, 961–969. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J.; et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef]
- Thomas, C.N.; Berry, M.; Logan, A.; Blanch, R.J.; Ahmed, Z. Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discov. 2017, 3. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Vigneswara, V.; Berry, M.; Logan, A.; Ahmed, Z. Caspase-2 is upregulated after sciatic nerve transection and its inhibition protects dorsal root ganglion neurons from apoptosis after serum withdrawal. PLoS ONE 2013, 8, 57861. [Google Scholar] [CrossRef]
- Logan, A.; Ahmed, Z.; Baird, A.; Gonzalez, A.M.; Berry, M. Neurotrophic factor synergy is required for neuronal survival and disinhibited axon regeneration after CNS injury. Brain 2006, 129, 490–502. [Google Scholar] [CrossRef]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, 124. [Google Scholar] [CrossRef]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef]
- Notman, R.; Noro, M.; O’Malley, B.; Anwar, J. Molecular basis for dimethylsulfoxide (DMSO) action on lipid membranes. J. Am. Chem. Soc. 2006, 128, 13982–13983. [Google Scholar] [CrossRef]
- Ahmed, Z.; Kalinski, H.; Berry, M.; Almasieh, M.; Ashush, H.; Slager, N.; Brafman, A.; Spivak, I.; Prasad, N.; Mett, I.; et al. Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis. 2011, 2, e173. [Google Scholar] [CrossRef]
- Do, Y.J.; Sul, J.W.; Jang, K.H.; Kang, N.S.; Kim, Y.H.; Kim, Y.G.; Kim, E. A novel RIPK1 inhibitor that prevents retinal degeneration in a rat glaucoma model. Exp. Cell Res. 2017, 359, 30–38. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Degterev, A.; David, J.; Rosenbaum, P.S.; Roth, S.; Grotta, J.C.; Cuny, G.D.; Yuan, J.; Savitz, S.I. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 2010, 88, 1569–1576. [Google Scholar] [CrossRef]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef]
- Politi, K.; Przedborski, S. Axonal Degeneration: RIPK1 Multitasking in ALS. Curr. Biol. 2016, 26, 932–934. [Google Scholar] [CrossRef][Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Cougnoux, A.; Cluzeau, C.; Mitra, S.; Li, R.; Williams, I.; Burkert, K.; Xu, X.; Wassif, C.A.; Zheng, W.; Porter, F.D. Necroptosis in Niemann-Pick disease, type C1: A potential therapeutic target. Cell Death Dis. 2016, 7, e2147. [Google Scholar] [CrossRef]
- Zamaraev, A.V.; Kopeina, G.S.; Buchbinder, J.H.; Zhivotovsky, B.; Lavrik, I.N. Caspase-2 is a negative regulator of necroptosis. Int. J. Biochem. Cell Biol. 2018, 102, 101–108. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chang, T.W.; Hsieh, W.H.; Hung, M.C.; Lin, I.H.; Lai, S.C.; Tzeng, Y.J. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discov. 2016, 2, 16065. [Google Scholar] [CrossRef]
- Mead, B.; Thompson, A.; Scheven, B.A.; Logan, A.; Berry, M.; Leadbeater, W. Comparative evaluation of methods for estimating retinal ganglion cell loss in retinal sections and wholemounts. PLoS ONE 2014, 9, e110612. [Google Scholar] [CrossRef]
- Sanchez-Migallon, M.C.; Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Brain derived neurotrophic factor maintains Brn3a expression in axotomized rat retinal ganglion cells. Exp. Eye Res. 2011, 92, 260–267. [Google Scholar] [CrossRef]
- Sanchez-Migallon, M.C.; Valiente-Soriano, F.J.; Nadal-Nicolas, F.M.; Vidal-Sanz, M.; Agudo-Barriuso, M. Apoptotic Retinal Ganglion Cell Death After Optic Nerve Transection or Crush in Mice: Delayed RGC Loss with BDNF or a Caspase 3 Inhibitor. Investig. Ophthalmol. Vis. Sci. 2016, 57, 81–93. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Giani, A.; Kataoka, K.; Morizane, Y.; Kayama, M.; Thanos, A.; Nakatake, S.; Notomi, S.; et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014, 21, 270–277. [Google Scholar] [CrossRef]
- Viringipurampeer, I.A.; Metcalfe, A.L.; Bashar, A.E.; Sivak, O.; Yanai, A.; Mohammadi, Z.; Moritz, O.L.; Gregory-Evans, C.Y.; Gregory-Evans, K. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum. Mol. Genet. 2016, 25, 1501–1516. [Google Scholar] [CrossRef]
- Viswanathan, S.; Frishman, L.J.; Robson, J.G.; Harwerth, R.S.; Smith, E.L., 3rd. The photopic negative response of the macaque electroretinogram: Reduction by experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1124–1136. [Google Scholar]
- Li, B.; Barnes, G.E.; Holt, W.F. The decline of the photopic negative response (PhNR) in the rat after optic nerve transection. Doc. Ophthalmol. 2005, 111, 23–31. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen (Origin) | Dilutions | Company | Catalogue Number |
| RIPK1 (mouse) | 1:200 (WB) | BD Pharmigen | 551041 |
| RIPK3 (B2) (mouse) | 1:200 (IHC) | Santa Cruz | SC374639 |
| RIPK3 (rabbit) | 1:1000 (WB) | Abcam | AB56164 |
| MLKL (rat) | 1:5000 (WB), 1:1000 (IHC) | Millipore | MABC604 |
| BRN3A (C-20) (goat) | 1:200 (IHC) | Santa Cruz | SC-31984 |
| RBPMS (rabbit) | 1:400 (IHC) | Millipore | ABN1362 |
| β-actin (mouse) | 1: 10,000 (WB) | Sigma | A5441 |
| βIII-tubulin (mouse) | 1:200 (ICC) | Sigma | T8660 |
| Secondary Antibodies (Origin) | Dilutions | Company | Catalogue Number |
| Anti-goat Alexa Fluor 594 (goat) | 1:400 (IHC) | Invitrogen | A11058 |
| Anti-rabbit Alexa Fluor 488 (donkey) | 1:400 (IHC) | Invitrogen | A21206 |
| Anti-rabbit Alexa Fluor 594 (donkey) | 1:400 (IHC) | Invitrogen | A21207 |
| Anti-rat Alexa Fluor 488 (goat) | 0.736111 | Invitrogen | A11006 |
| Anti-rabbit HRP-linked (goat) | 1:1000 (WB) | Cell Signalling Technologies | 7074S |
| Anti-rat HRP-linked (goat) | 1:5000 (WB) | Cell Signalling Technologies | 7077S |
| Anti-mouse HRP-linked (horse) | 1:1000 (WB) | Cell Signalling Technologies | 7076S |
| A | Mean Number of BRN3A+ RGC per mm of Retina (95% CI) | |||
| Treatment | 0 µm | 600 µm | 1200 µm | 1800 µm |
| Intact | 56.95 | 59.87 | 57.48 | 63.10 |
| (52.7–61.2) | (56.2–63.5) | (51.7–63.3) | (42.2–74.0) | |
| Blunt + Nec-1s | 44.2 | 44.7 | 49.3 | 54.7 |
| (39.1–50.0) | (35.9–55.6) | (36.9–65.9) | (39.5–75.9) | |
| Blunt + vehicle | 36.0 | 41.0 | 48.1 | 56.8 |
| (27.0–48.2) | (24.8–67.8) | (25.4–91.3) | (27.4–118) | |
| p value for comparison | p = 0.017 | n/a | n/a | n/a |
| B | Mean Number of RBPMS+ RGC per mm of Retina (95% CI) | |||
| Treatment | 0 µm | 600 µm | 1200 µm | 1800 µm |
| Intact | 62.68 | 64.70 | 63.84 | 72.16 |
| (58.2–67.2) | (60.7–68.7) | (58.0–69.7) | (61.3–83.6) | |
| Blunt + Nec-1s | 45.2 | 46.9 | 51.8 | 58.9 |
| (40.4–50.5)0 | (37.9–57.9) | (39.6–67.8) | (44.7–77.6) | |
| Blunt + vehicle | 38.0 | 43.6 | 50.5 | 60.9 |
| (29.3–49.1) | (27.4–69.3) | (29.0–87.9) | (33.0–112.6) | |
| p value for comparison | p = 0.02 | n/a | n/a | n/a |
| C | Mean Number of Pixels of ONL Thickness (95% CI) | |||
| Treatment | 0 µm | 600 µm | 1200 µm | 1800 µm |
| Intact | 98.12 | 101.72 | 120.66 | 144.34 |
| (94.4–101.9) | (94.1–109.4) | (104.9–136.4) | (125.6–163.1) | |
| Blunt + Nec-1s | 52.91 | 54.36 | 54.53 | 82.31 |
| (45.1–60.7) | (45.5–63.3) | (47.6–61.5) | (76.8–87.8) | |
| Blunt + vehicle | 46.47 | 47.90 | 52.38 | 76.79 |
| (36.6–56.3) | (42.0–53.7) | (42.5–62.3) | (69.0–93.5) | |
| p value for comparison | p = 0.229 | p = 0.292 | p = 0.625 | p = 0.143 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, C.N.; Thompson, A.M.; Ahmed, Z.; Blanch, R.J. Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model. Cells 2019, 8, 1517. https://doi.org/10.3390/cells8121517
Thomas CN, Thompson AM, Ahmed Z, Blanch RJ. Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model. Cells. 2019; 8(12):1517. https://doi.org/10.3390/cells8121517
Chicago/Turabian StyleThomas, Chloe N., Adam M. Thompson, Zubair Ahmed, and Richard J. Blanch. 2019. "Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model" Cells 8, no. 12: 1517. https://doi.org/10.3390/cells8121517
APA StyleThomas, C. N., Thompson, A. M., Ahmed, Z., & Blanch, R. J. (2019). Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model. Cells, 8(12), 1517. https://doi.org/10.3390/cells8121517