Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells




Abstract
:1. Introduction
2. Materials and Methods
2.1. MCF7 Stable shRNA IGF1R/INSR Cell Lines
2.2. T47D IGF1R/INSR siRNA Silencing
2.3. Protein Analysis and Immunoblotting
2.4. Real-Time Quantitative Polymerase Chain Reactions (RT-QPCR)
2.5. Transient Co-Transfections
2.6. Co-Immunoprecipitation (Co-IP) Assays
2.7. Proliferation Assays
2.8. Migration Assays
2.9. DNA Affinity Chromatography
2.10. Statistical Analyses
3. Results
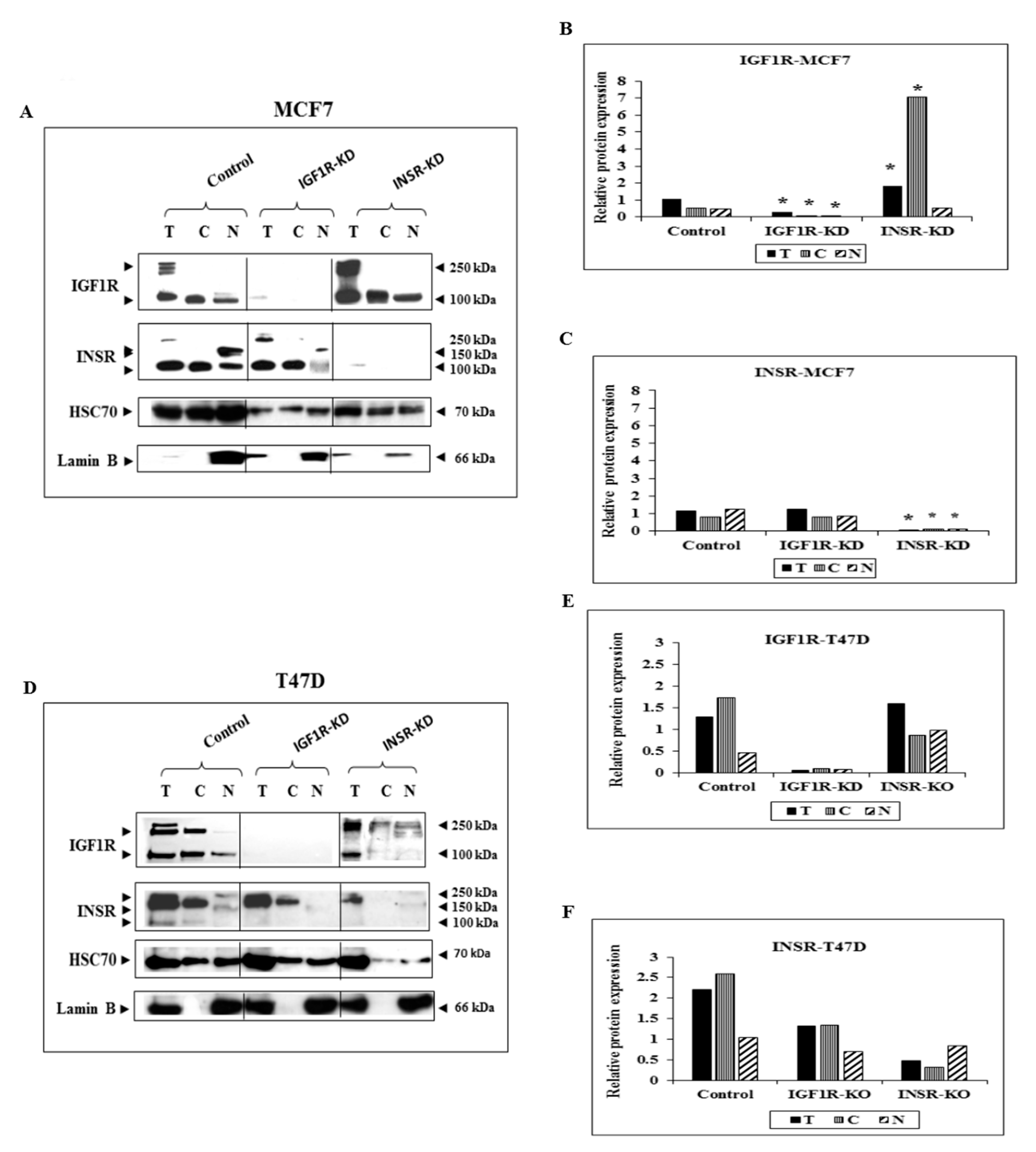
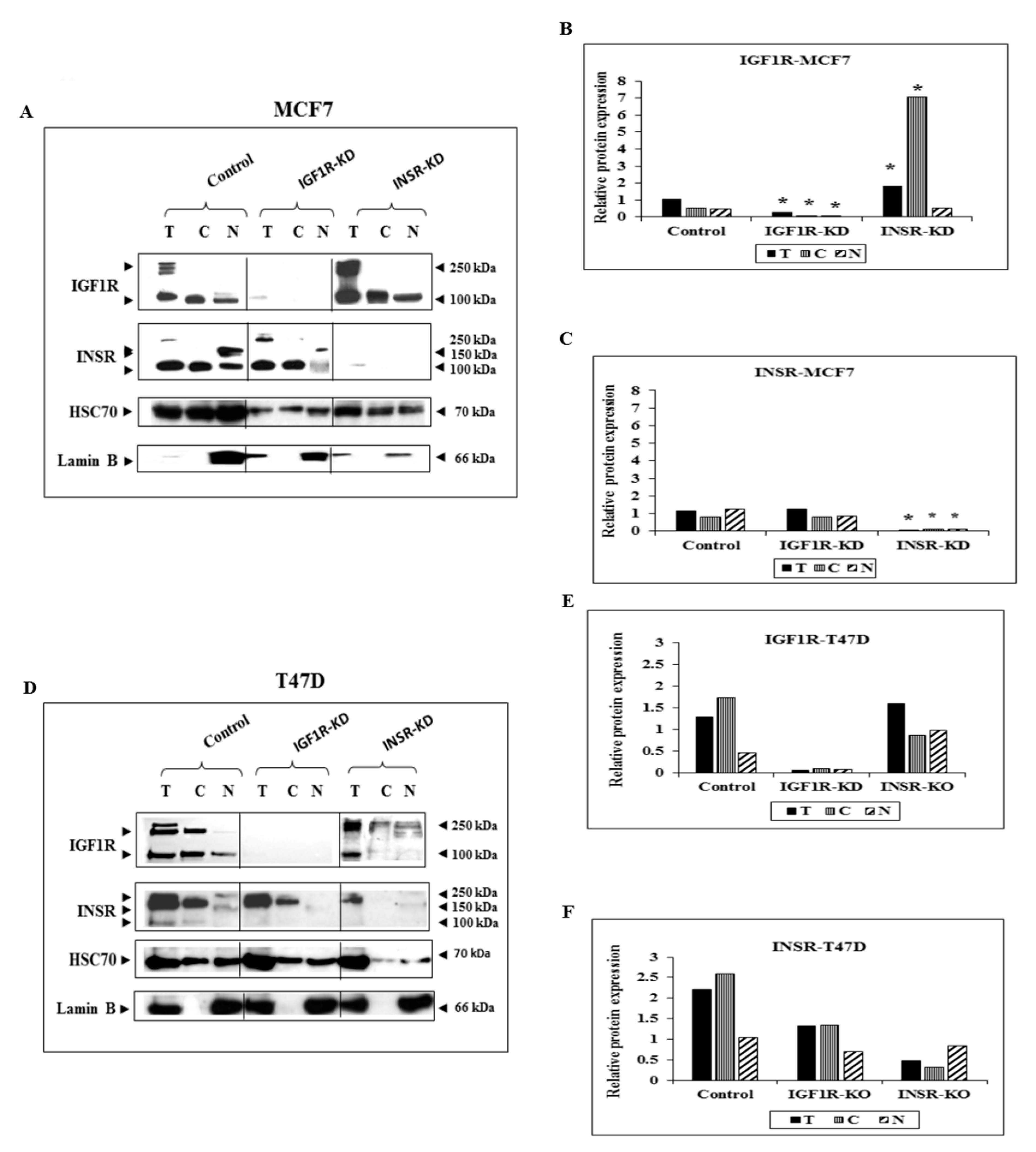
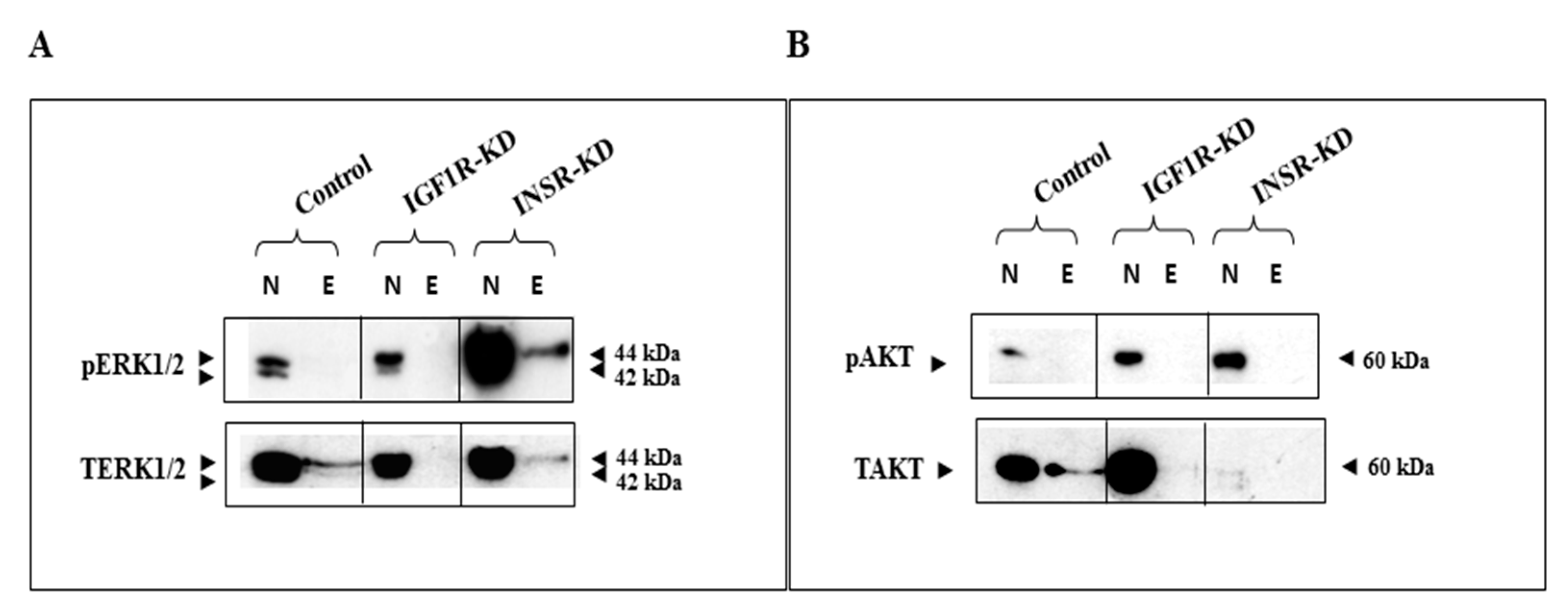
3.1. Subcellular Analysis of IGF1R and INSR Expression in Breast Cancer-Derived Cell Lines with Disrupted IGF1R/INSR
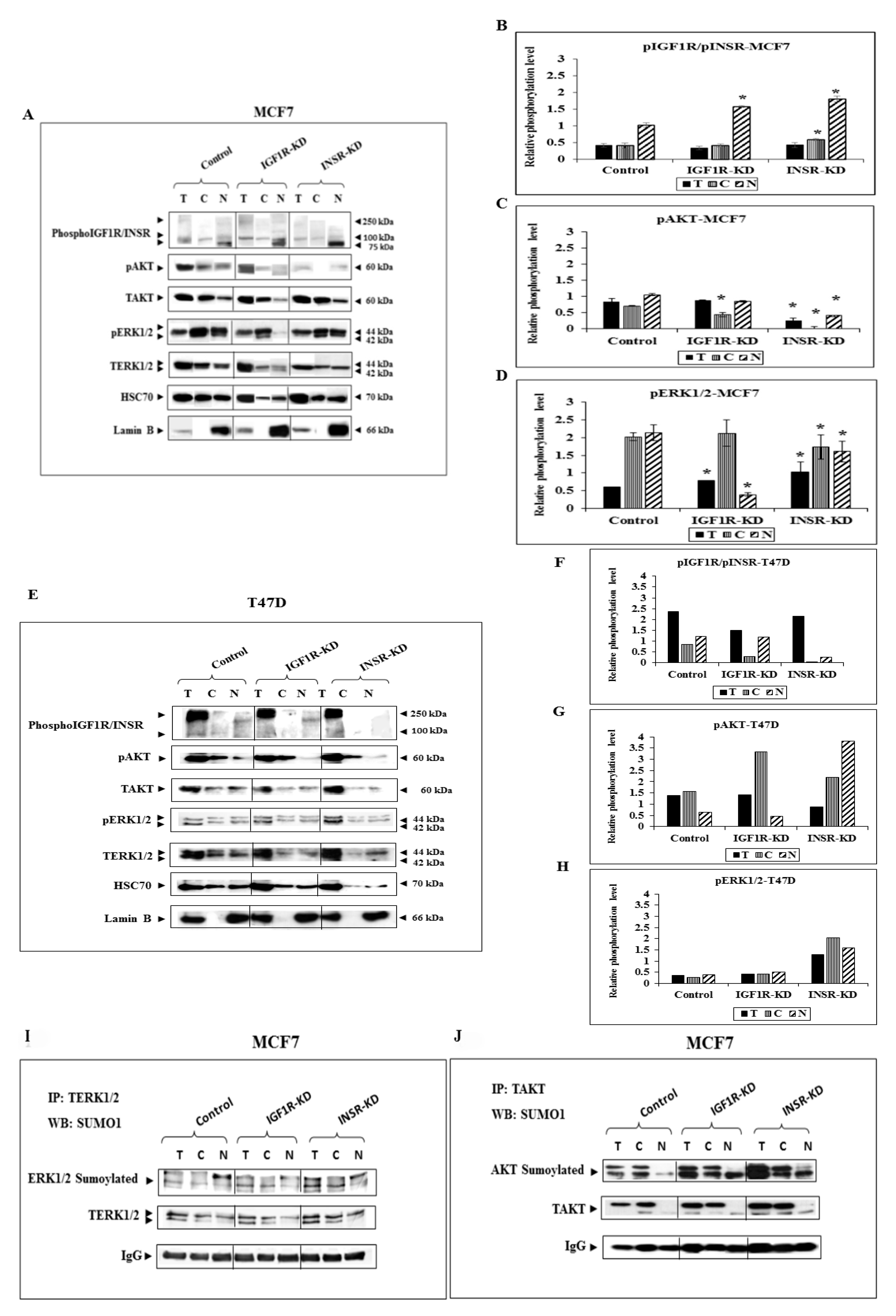
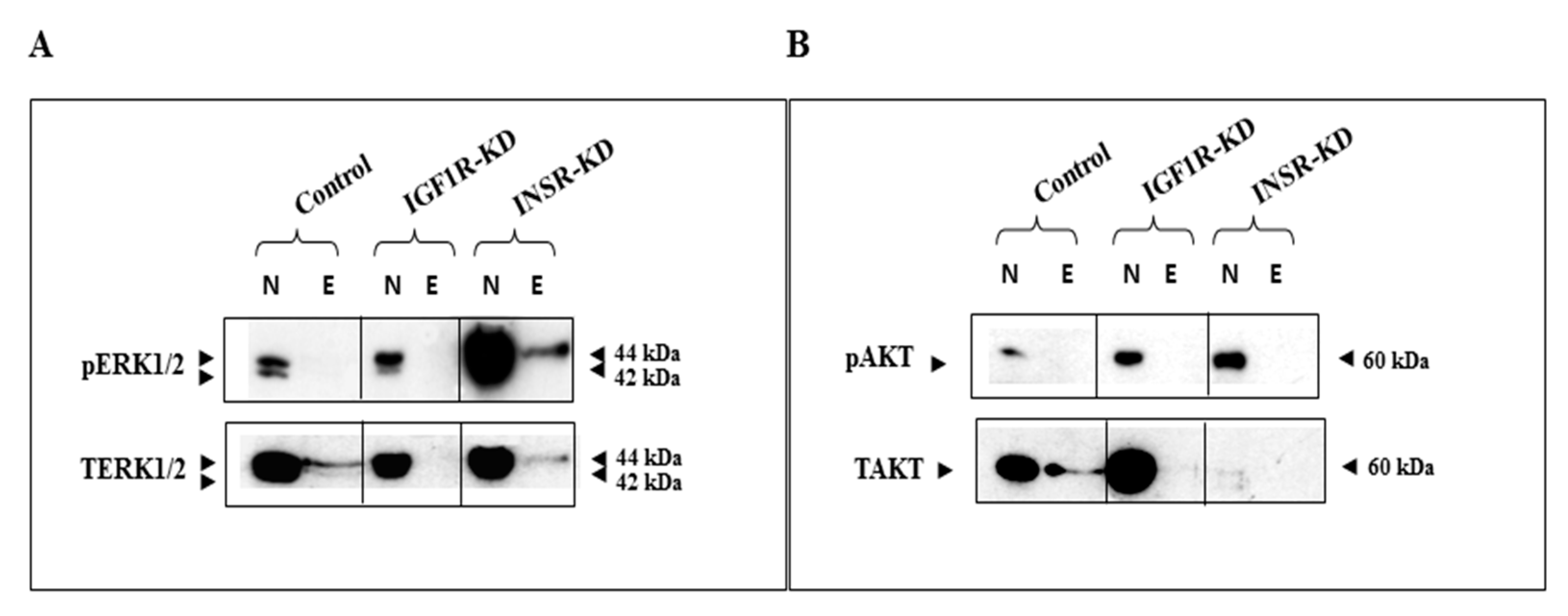
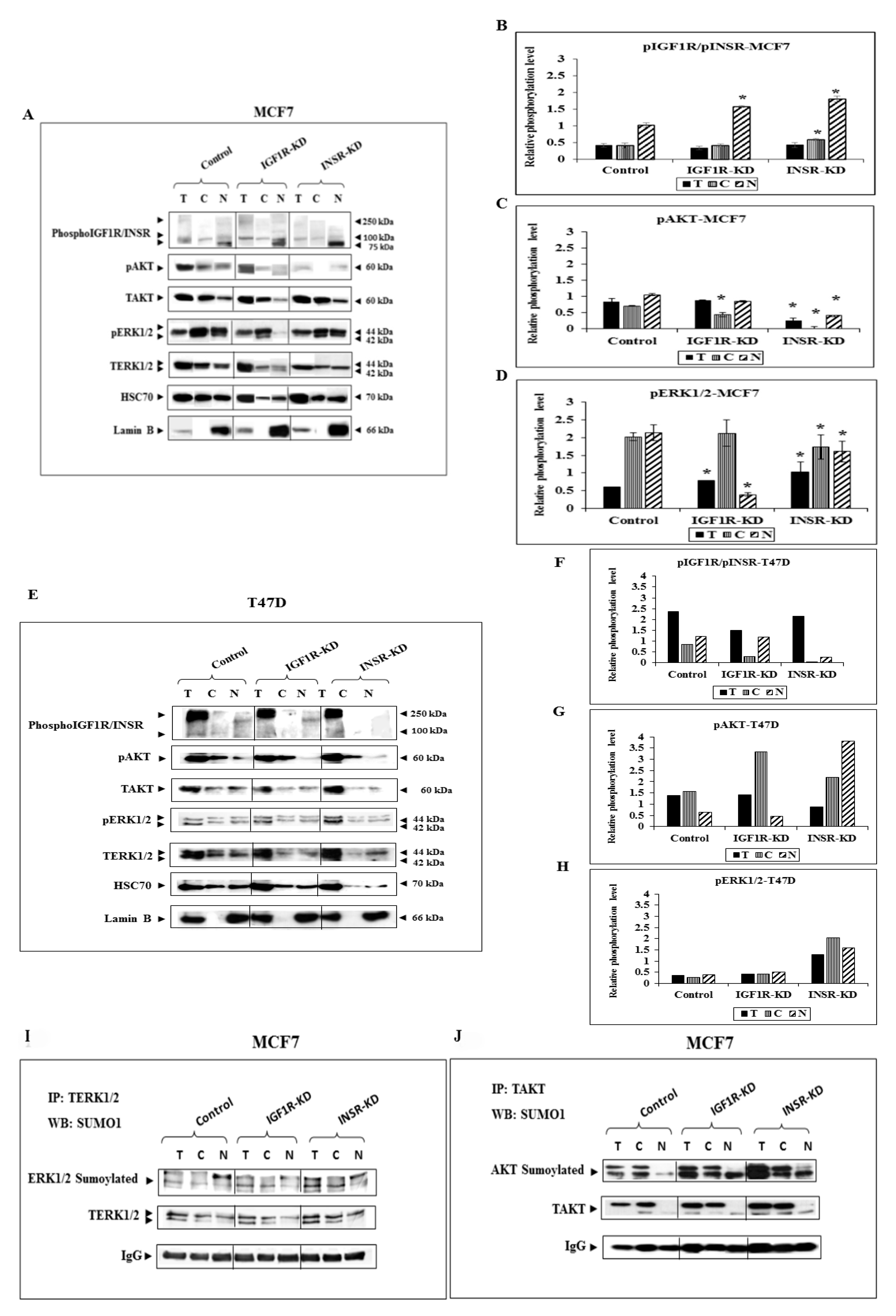
3.2. Effect of IGF1R/INSR Silencing on ERK and AKT Expression and Activation
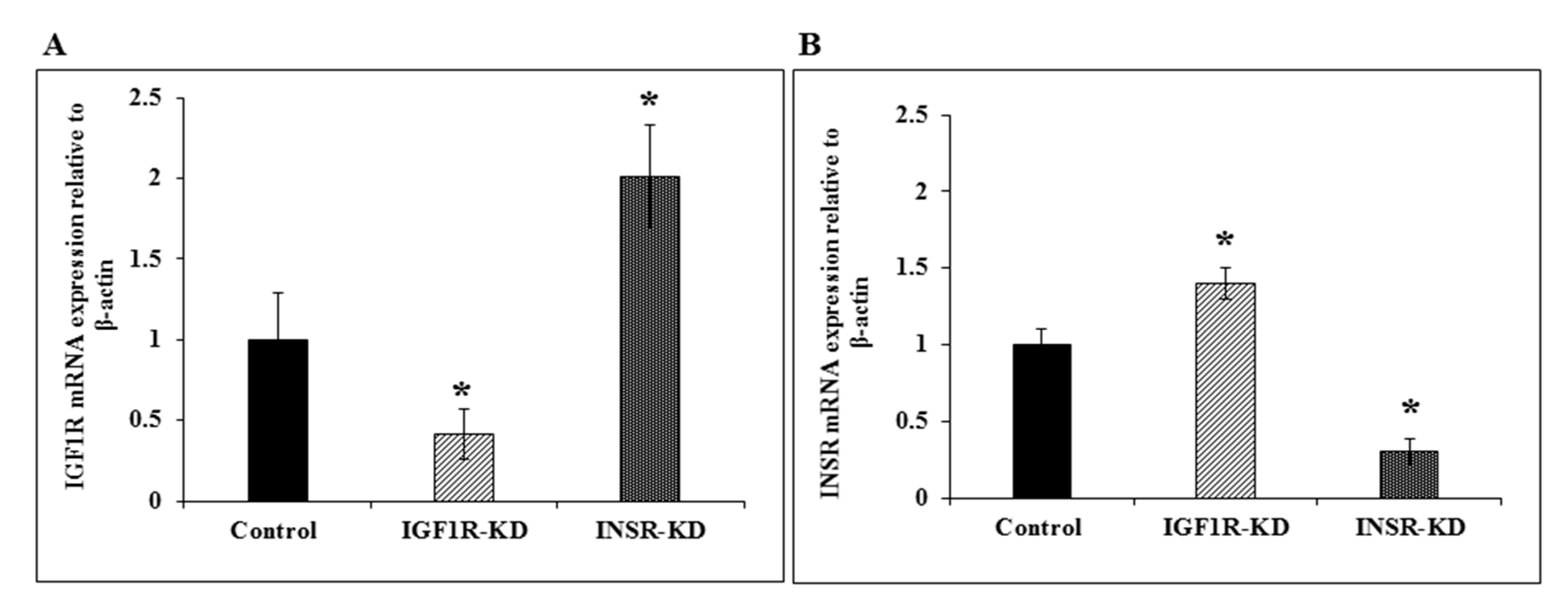
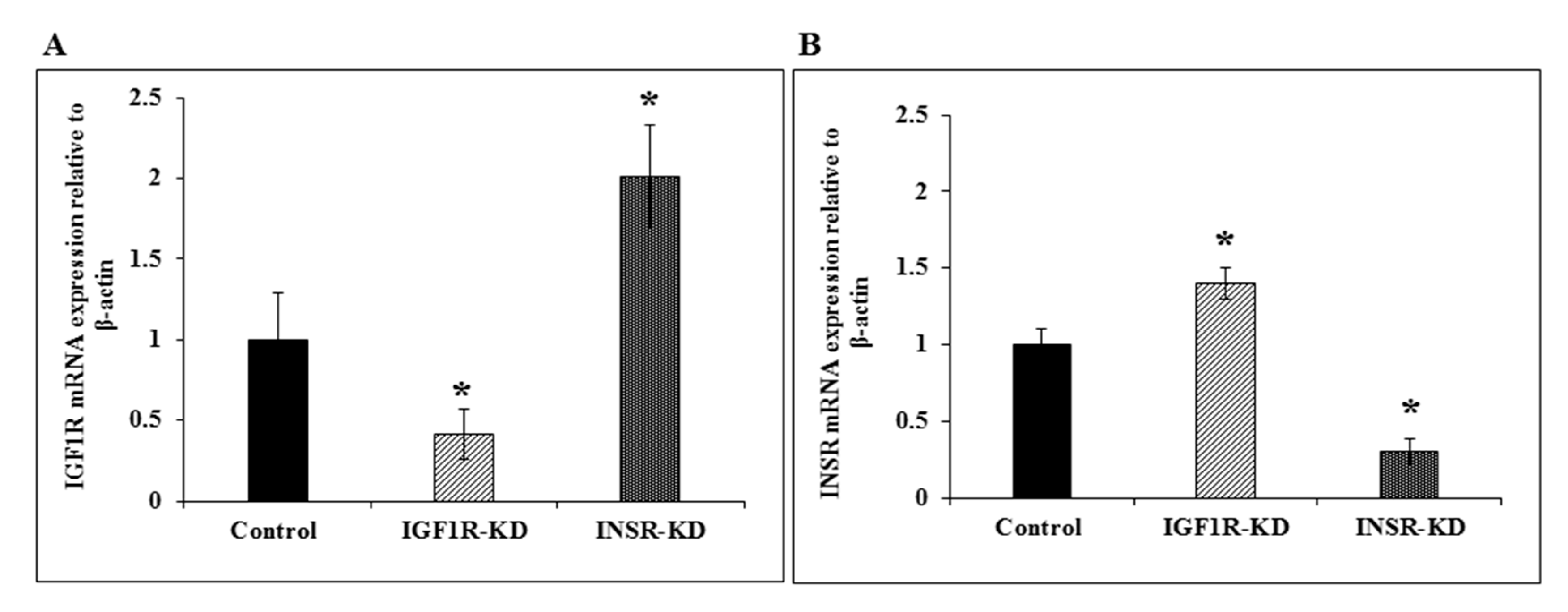
3.3. RT-QPCR of IGF1R and INSR mRNA Levels
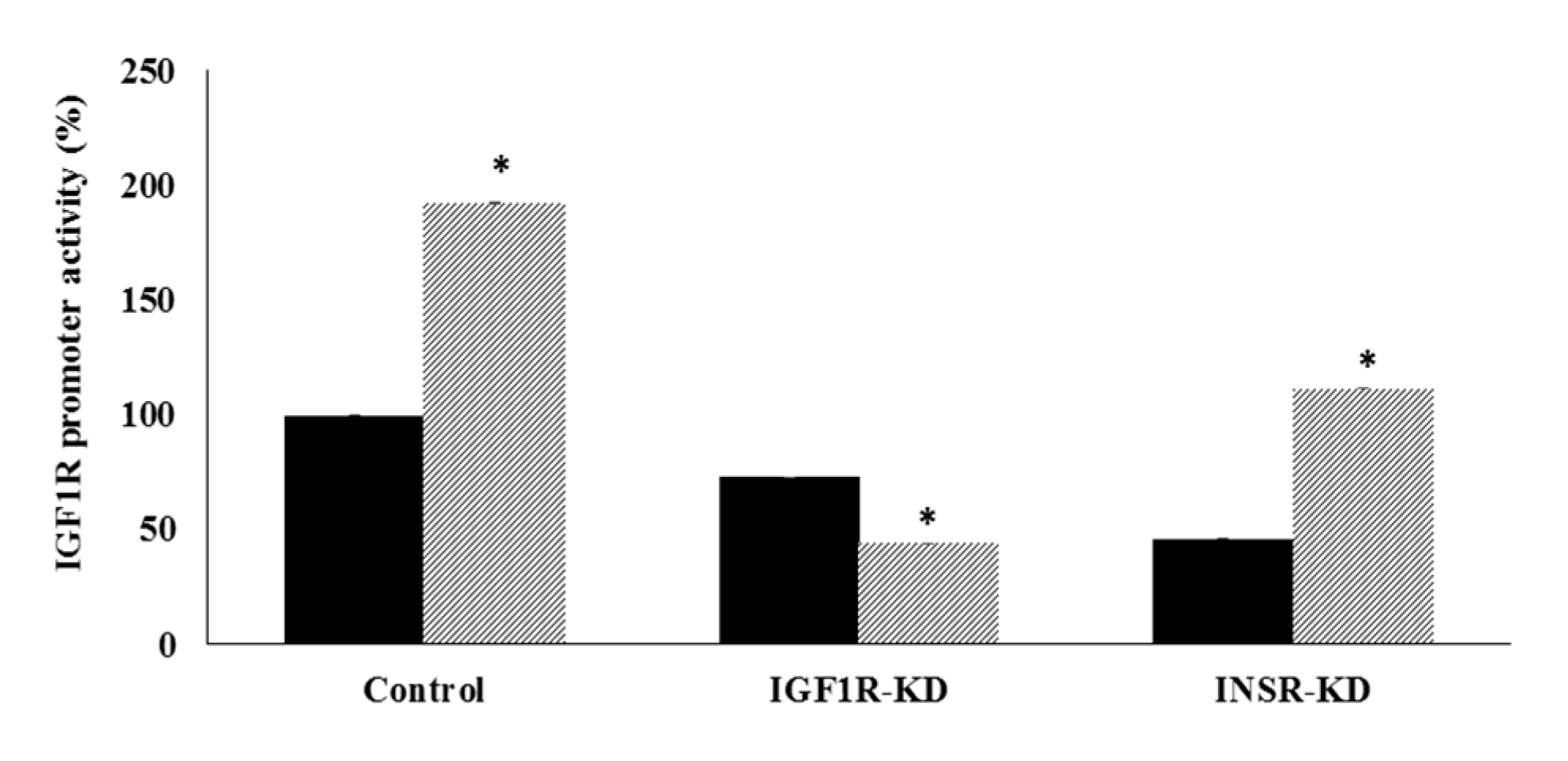
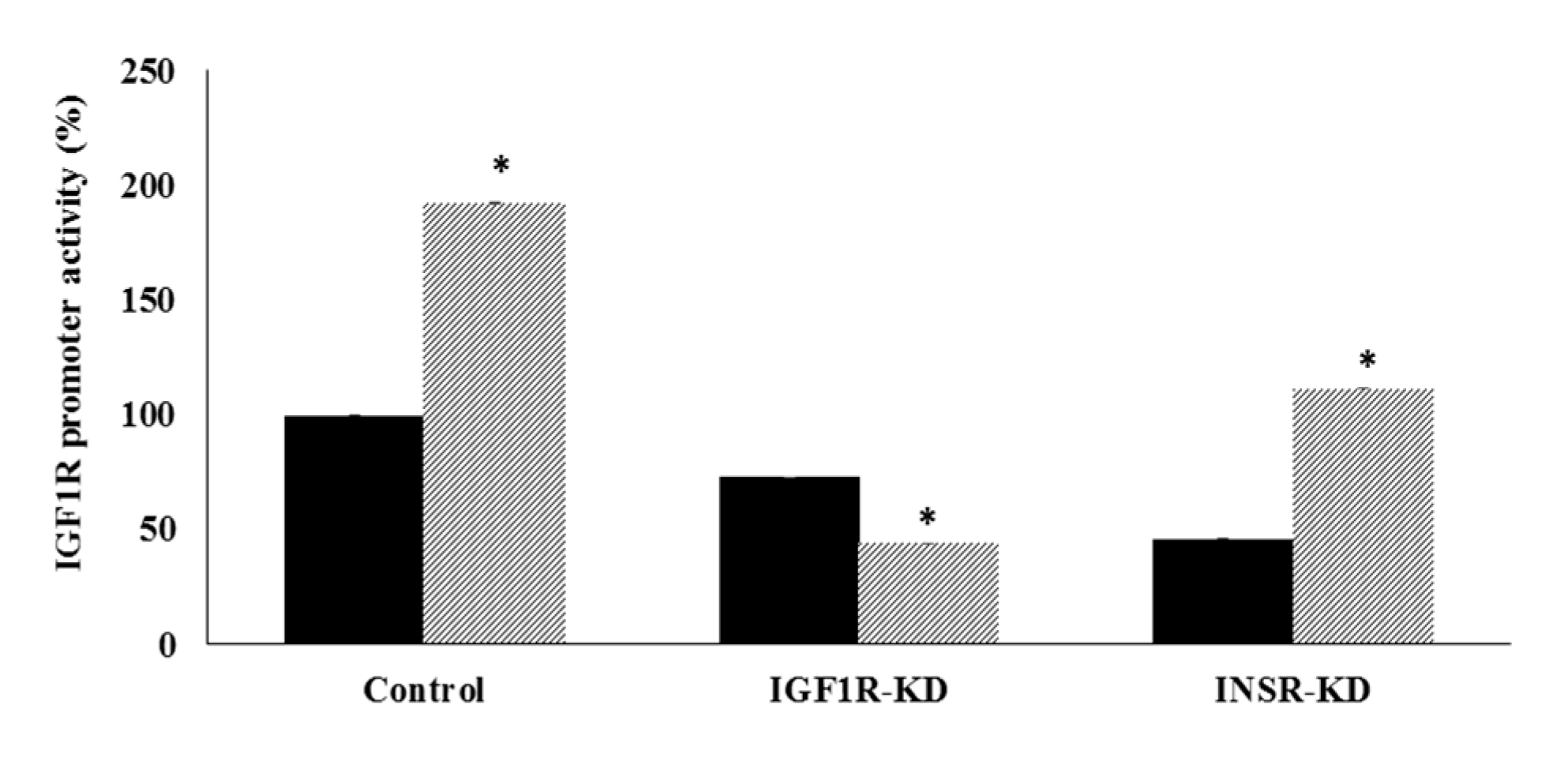
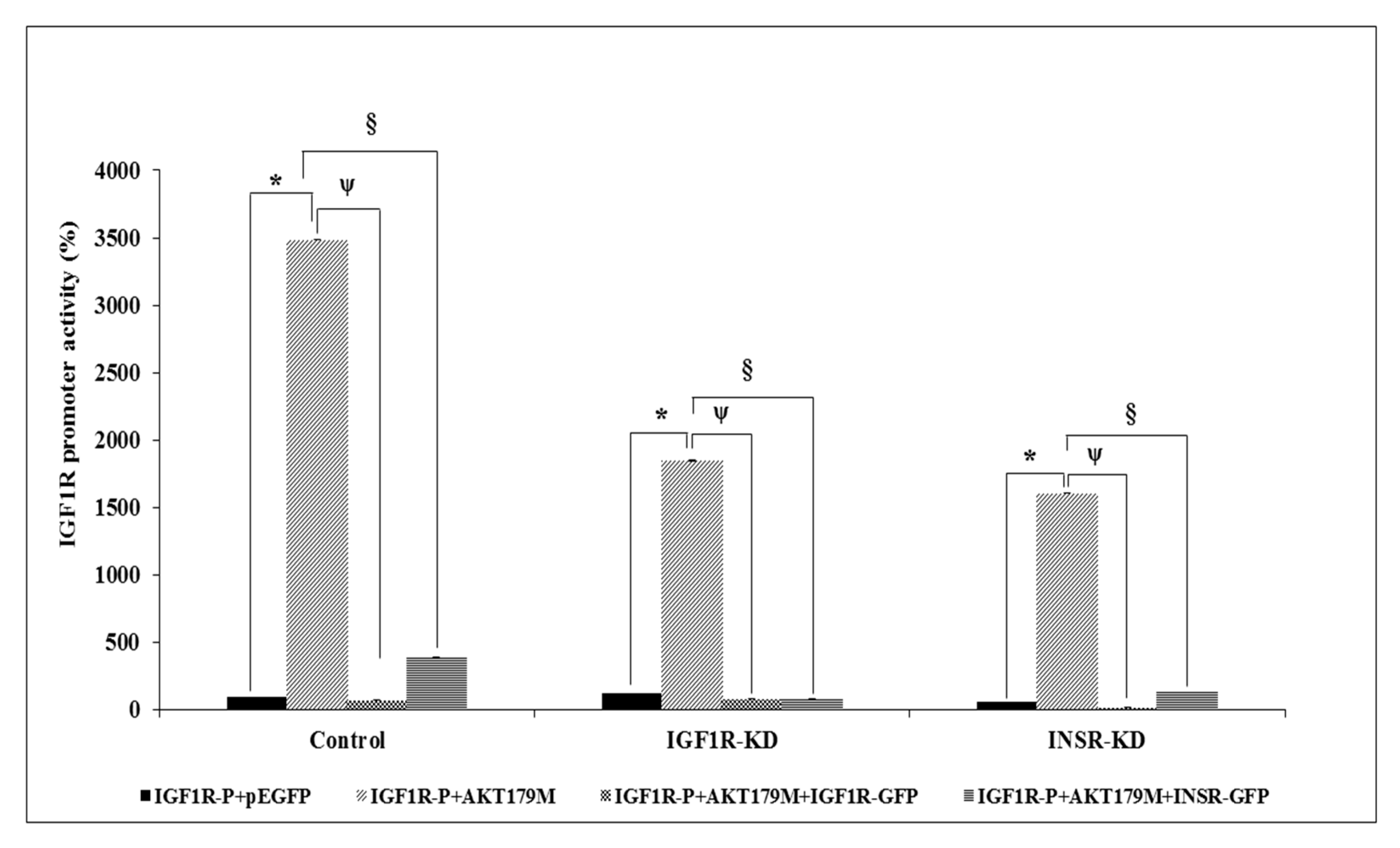
3.4. Effect of IGF1R/INSR Silencing on IGF1R Promoter Activity
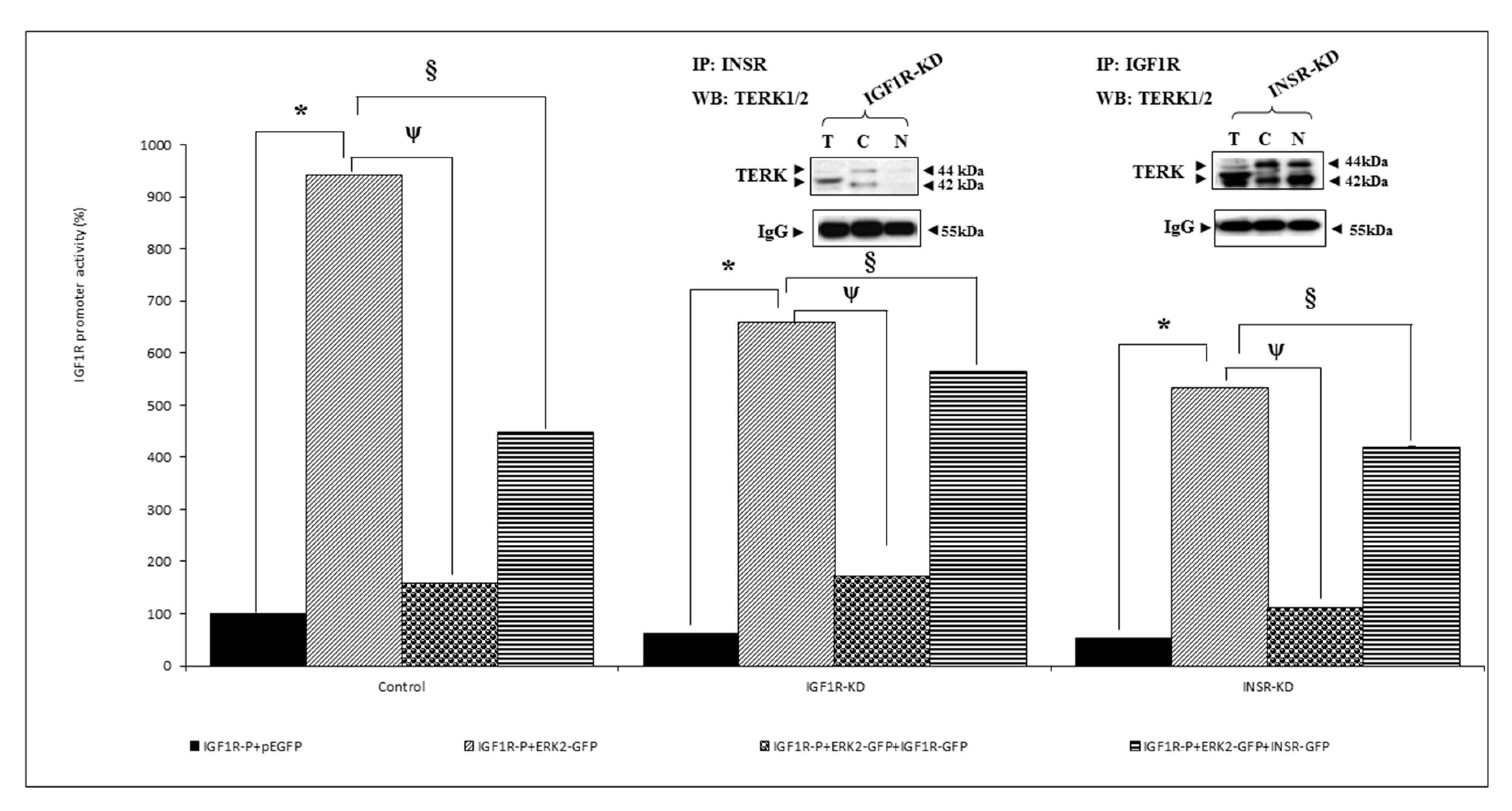
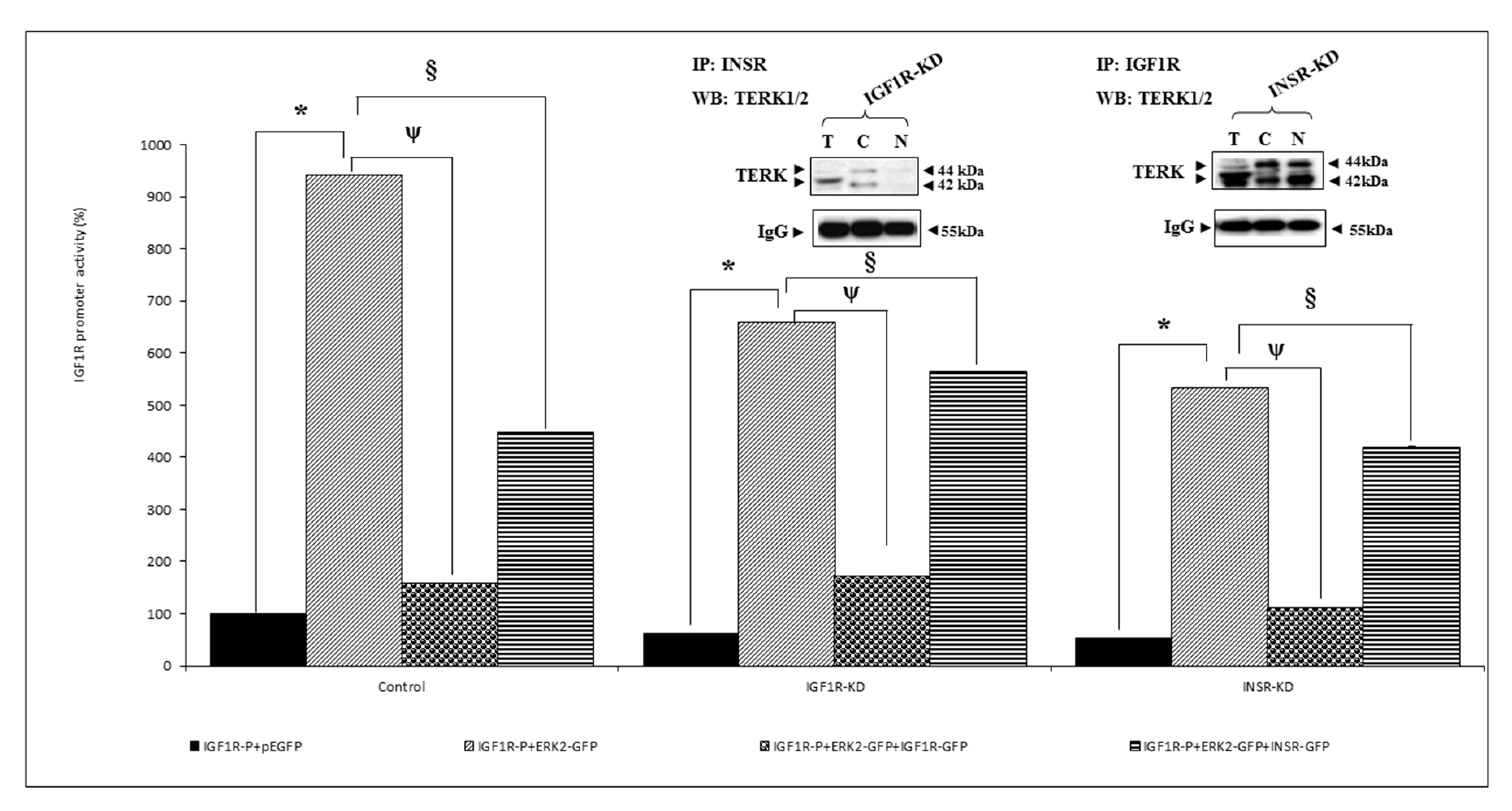
3.5. Effect of ERK on IGF1R Promoter Activity
3.6. Analysis of Physical Interactions between IGF1R and ERK1/2
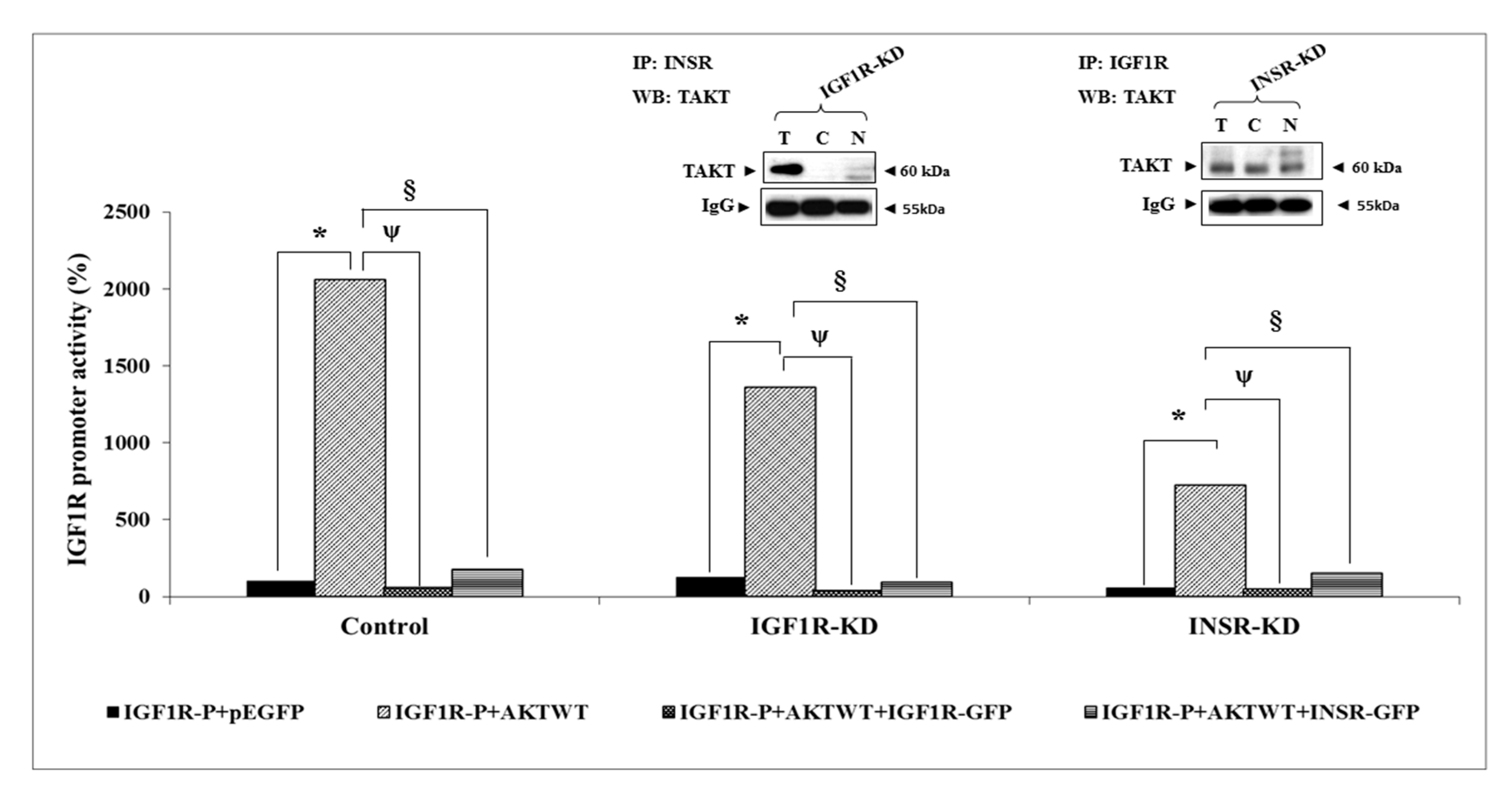
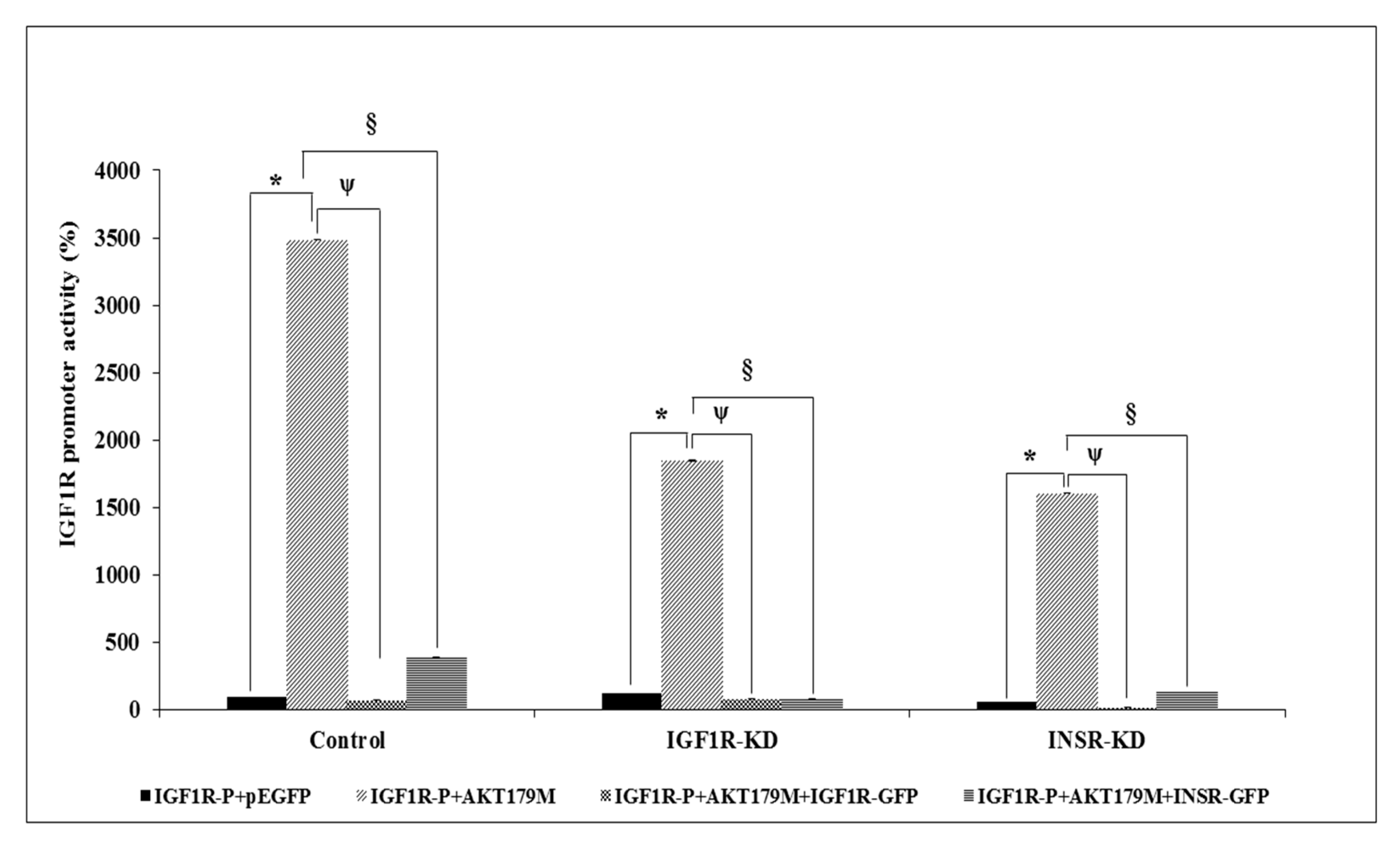
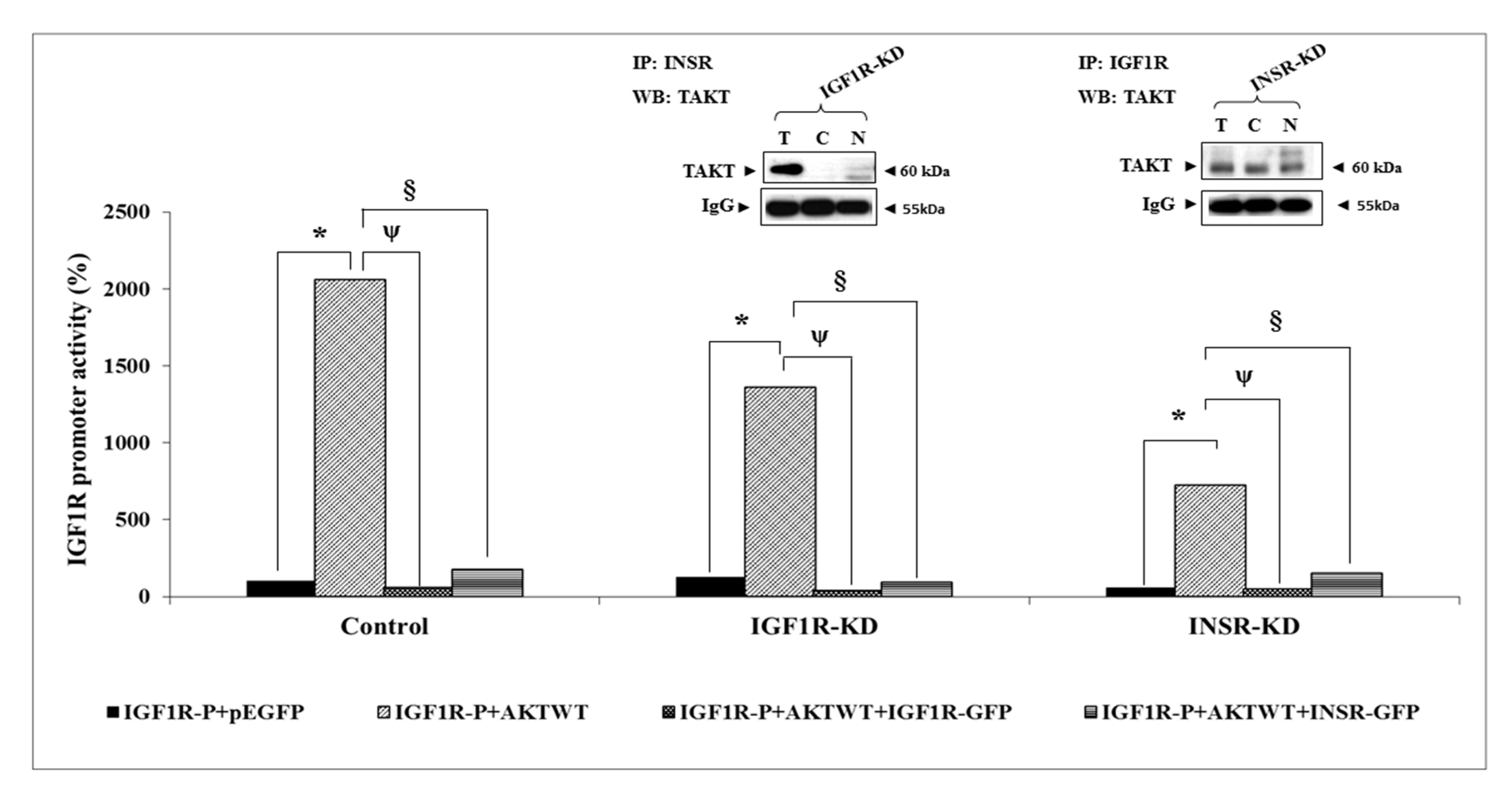
3.7. Effect of AKT on IGF1R Promoter Activity
3.8. Binding of ERK1/2 and AKT to the Proximal IGF1R Promoter
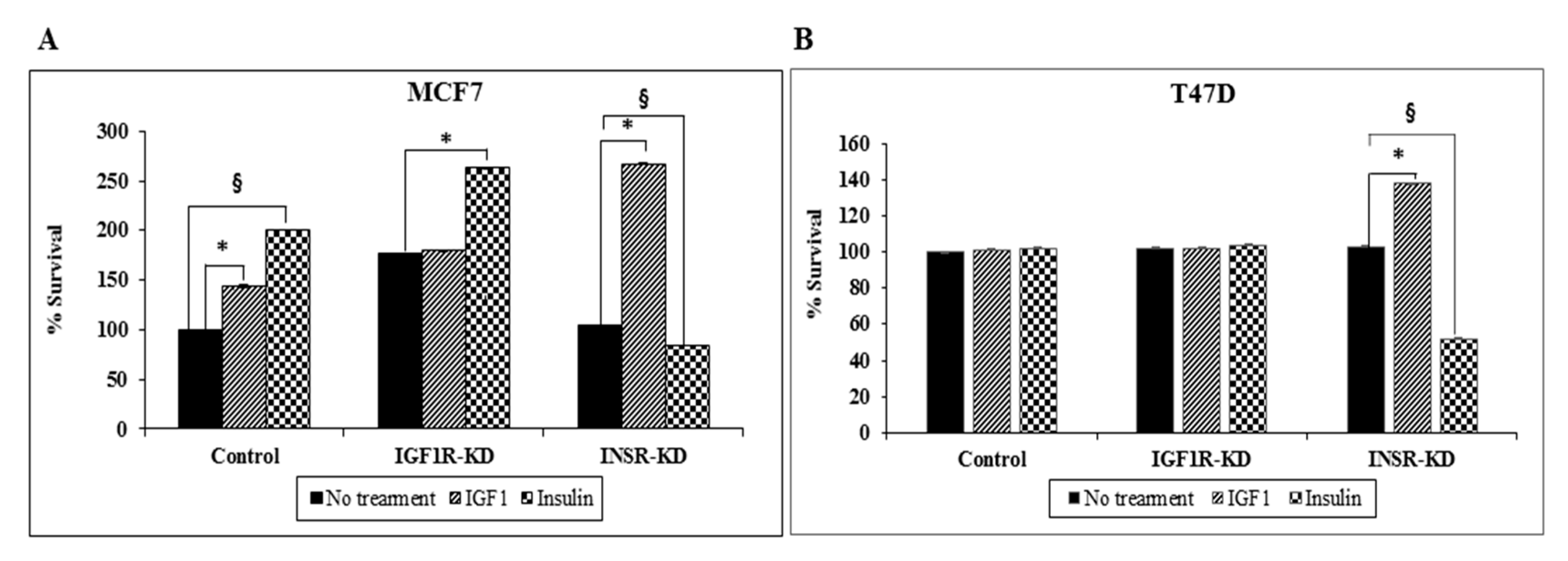
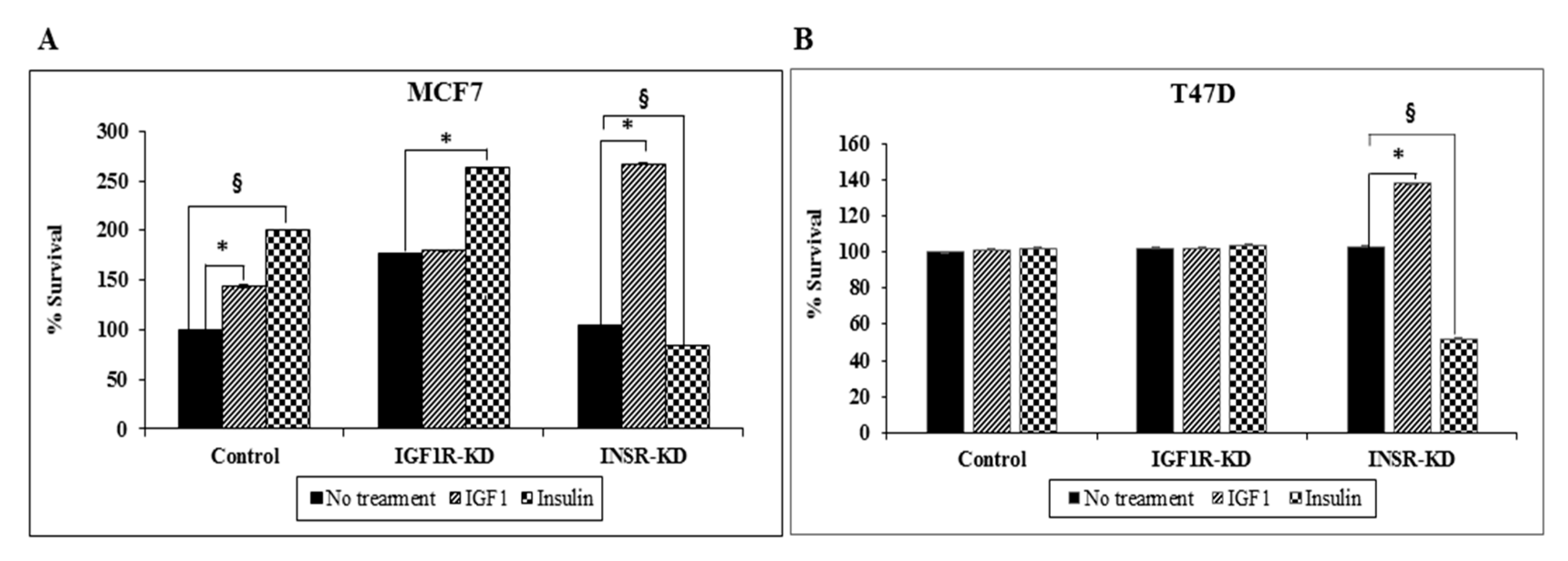
3.9. Effect of IGF1R/INSR Silencing on Cell Proliferation
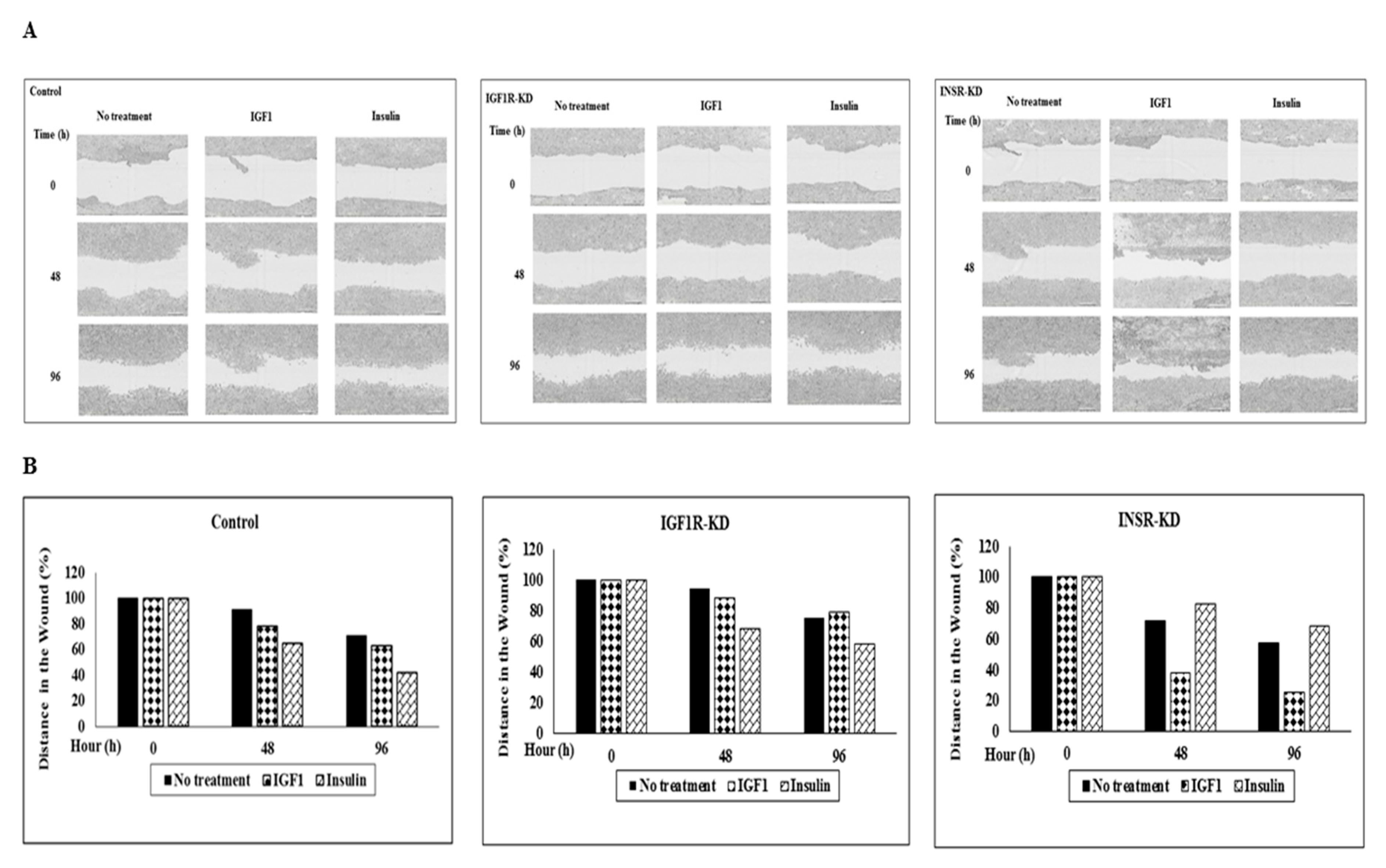
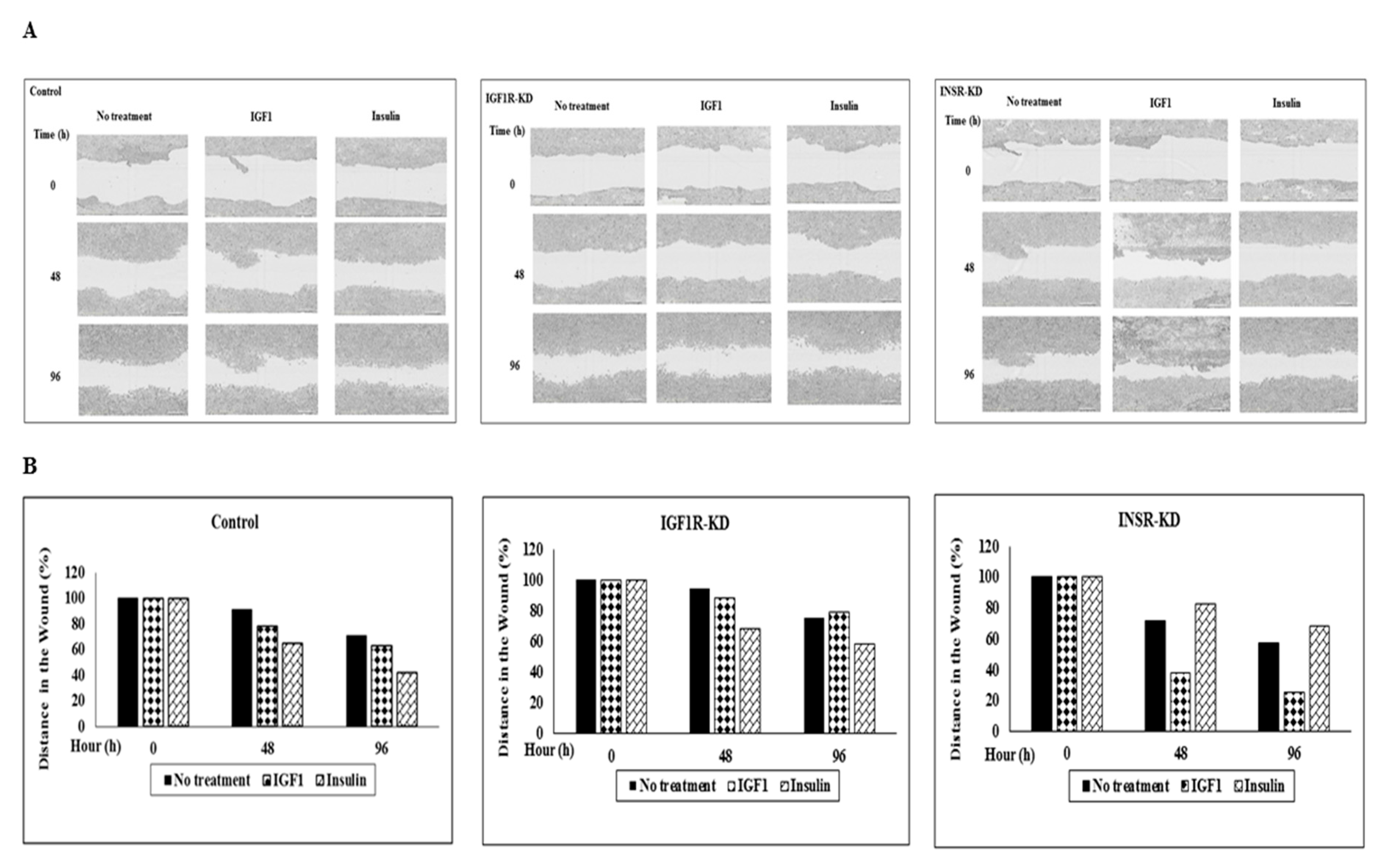
3.10. Effect of IGF1R/INSR Silencing on Cell Migration
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.; Boone, D.; Lee, A. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Bruchim, I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol. 2012, 13, e537–e544. [Google Scholar] [CrossRef]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R. The insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef]
- Rostoker, R.; Abelson, S.; Bitton-Worms, K.; Genkin, I.; Ben-Shmuel, S.; Dakwar, M.; Orr, Z.S.; Caspi, A.; Tzukerman, M.; LeRoith, D. Highly specific role of the insulin receptor in breast cancer progression. Endocr. Relat. Cancer 2015, 22, 145–157. [Google Scholar] [CrossRef]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Target Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Pasmanik-Chor, M.; Yeheskel, A.; Edry, L.; Shomron, N.; Warman, N.; Wertheimer, E.; Maor, S.; Shochat, L.; Werner, H. Insulin-like growth factor-I receptor (IGF-IR) translocates to nucleus and autoregulates IGF-IR gene expression in breast cance cells. J. Biol. Chem. 2012, 287, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Solomon-Zemler, R.; Sarfstein, R.; Werner, H. Nuclear insulin-like growth factor-1 receptor (IGF1R) displays proliferative and regulatory activities in non-malignant cells. PLoS ONE 2017, 12, e0185164. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Lin, Y.; Badin, M.; Vasilcanu, D.; Strömberg, T.; Jernberg-Wiklund, H.; Sehat, B.; Larsson, O. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem. Biophys. Res. Commun. 2011, 404, 667–671. [Google Scholar] [CrossRef]
- Aleksic, T.; Gray, N.E.; Wu, X.; Rieunier, G.; Osher, E.; Mills, J.; Verrill, C.; Bryant, R.J.; Han, C.; Hutchinson, K.; et al. Nuclear IGF-1R interacts with regulatory regions of chromatin to promote RNA polymerase II recruitment and gene expression associated with advanced tumor stage. Cancer Res. 2018, 78, 3497–3509. [Google Scholar] [CrossRef]
- Warsito, D.; Sjöström, S.; Andersson, S.; Larsson, O.; Sehat, B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012, 13, 244–250. [Google Scholar] [CrossRef]
- Sarfstein, R.; Werner, H. Nuclear insulin and insulin-like growth factor-1 receptors: A novel paradigm in signal transduction. Endocrinology 2013, 154, 1672–1679. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The role of the insulin-like growth factor-1 system in breast cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 enhances IGF1 response in Y537S and D538G ESR1 mutant breast cancer cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef]
- Chan, J.Y.; Hackel, B.J.; Yee, D. Targeting insulin receptor in breast cancer using small engineered protein scaffolds. Mol. Cancer Ther. 2017, 16, 1324–1334. [Google Scholar] [CrossRef]
- Bruchim, I.; Werner, H. Targeting IGF-1 signaling pathways in gynecologic malignancies. Expert Opin. Ther. Targets 2013, 17, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NP J. Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Chalbos, D.; Vignon, F.; Keydar, I.; Rochefort, H. Estrogens stimulate cell proliferation and induce secretory proteins in a human breast cancer cell line (T47D). J. Clin. Endocrinol. Metab. 1982, 55, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Vidnovic, N.; Ellisen, L.W.; Leong, C.-O. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer 2009, 101, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Werner, H.; Bach, M.A.; Stannard, B.; Roberts, C.T., Jr.; LeRoith, D. Structural and functional analysis of the insulin-like growth factor I receptor gene promoter. Mol. Endocrinol. 1992, 6, 1545–1558. [Google Scholar]
- Werner, H.; Rauscher, F.J., III; Sukhatme, V.P.; Drummond, I.A.; Roberts, C.T., Jr.; LeRoith, D. Transcriptional repression of the insulin-like growth factor I receptor (IGF-I-R) gene by the tumor suppressor WT1 involves binding to sequences both upstream and downstream of the IGF-I-R gene transcription start site. J. Biol. Chem. 1994, 269, 12577–12582. [Google Scholar]
- Sarfstein, R.; Belfiore, A.; Werner, H. Identification of insulin-like growth factor-I receptor gene promoter-binding proteins in estrogen receptor (ER)-positive and ER-depleted breast cancer cells. Cancers 2010, 2, 233–261. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Yuen, J.S.P.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type I insulin-like growth factor-I receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Worrall, C.; Takahashi, S.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell. Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Picci, P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr. Opin. Oncol. 2008, 20, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F. IGF and insulin receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 381–406. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Vella, V.; Nicolosi, M.L.; Giuliano, M.; Morrione, A.; Malaguarnera, R.; Belfiore, A. Insulin receptor isoform A modulates metabolic reprogramming of breast cancer cells in response to IGF2 and insulin stimulation. Cells 2019, 8, 1017. [Google Scholar] [CrossRef] [Green Version]
- Titone, R.; Zhu, M.; Robertson, D.M. Insulin mediates de novo nuclear accumulation of the IGF-1/insulin hybrid receptor in corneal epithelial cells. Sci. Rep. 2018, 8, 4378. [Google Scholar] [CrossRef] [Green Version]
- Crudden, C.; Song, D.; Cismas, S.; Trocme, E.; Pasca, S.; Calin, G.A.; Girnita, A.; Girnita, L. Below the surface: IGF-1R therapeutic targeting and its endocytic journey. Cells 2019, 8, 1223. [Google Scholar] [CrossRef] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouysségur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999, 18, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT-a major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic targeting of the IGF axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Product Size |
|---|---|---|
| IGF1R-F | 5′-GTGGAGACAGGGGCTTTTATT-3′ | 122 bp |
| IGF1R-R | 5′-CTCCAGCCTCCTTAGATCACA-3′ | |
| INSR-F | 5′-AGTGTGGAGACATCTGTCCG-3′ | 121 bp |
| INSR-R | 5′-GTCGGGCAAACTTTCTGGCA-3′ | |
| βActin-F | 5′-CCTGGCACCCAGCACAAT-3′ | 144 bp |
| βActin-R | 5′-GGGCCGGACTCGTCATACT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarfstein, R.; Nagaraj, K.; LeRoith, D.; Werner, H. Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells. Cells 2019, 8, 1499. https://doi.org/10.3390/cells8121499
Sarfstein R, Nagaraj K, LeRoith D, Werner H. Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells. Cells. 2019; 8(12):1499. https://doi.org/10.3390/cells8121499
Chicago/Turabian StyleSarfstein, Rive, Karthik Nagaraj, Derek LeRoith, and Haim Werner. 2019. "Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells" Cells 8, no. 12: 1499. https://doi.org/10.3390/cells8121499
APA StyleSarfstein, R., Nagaraj, K., LeRoith, D., & Werner, H. (2019). Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells. Cells, 8(12), 1499. https://doi.org/10.3390/cells8121499