Mitochondrial MiRNA in Cardiovascular Function and Disease



Abstract
1. Introduction
2. MitomiR Regulation of Mitochondrial Functions
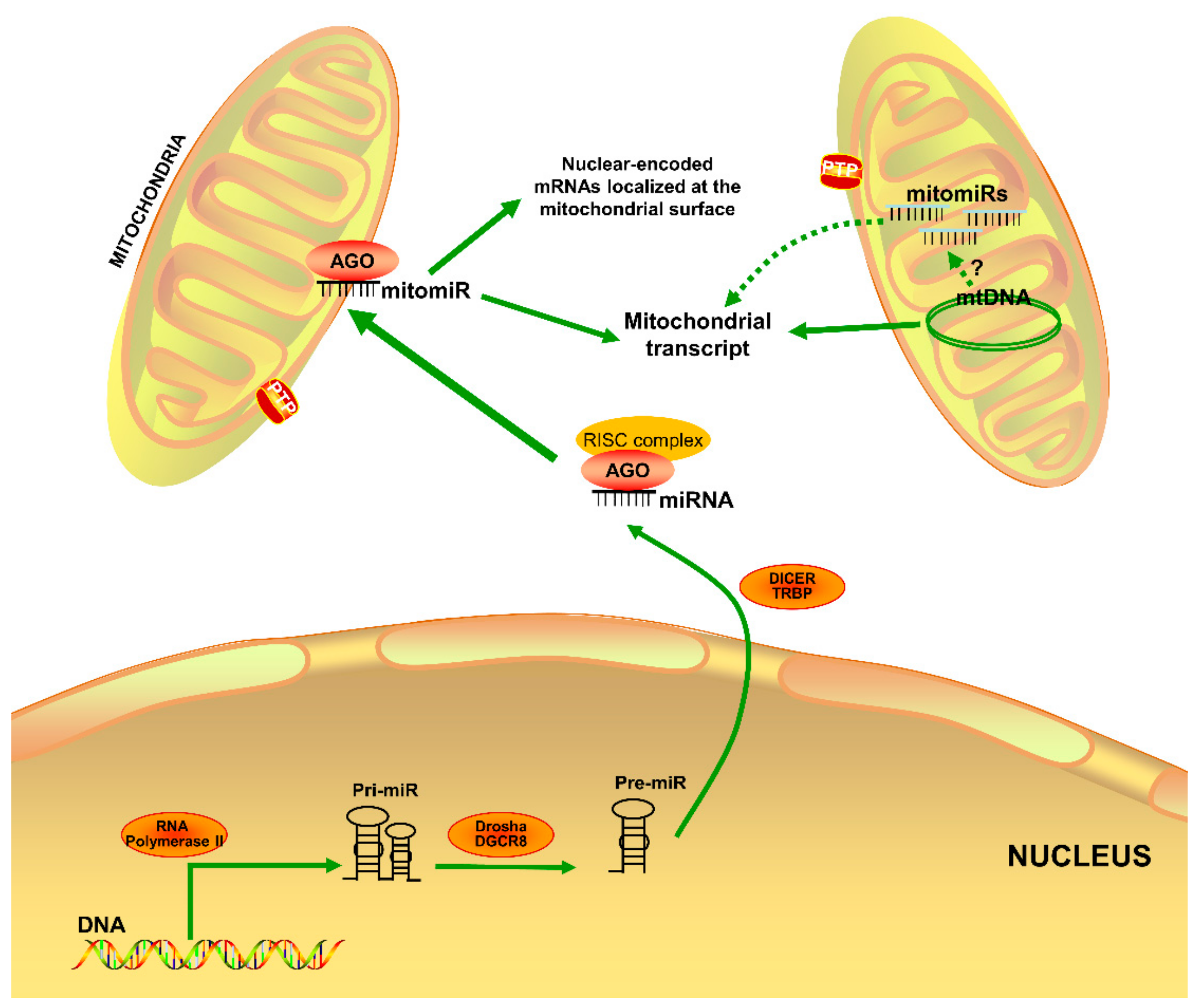
2.1. MitomiR Biogenesis
2.2. MitomiRs and Mitochondrial Energy Metabolism
2.3. MitomiRs and Mitochondrial Fission/Fusion
3. MitomiR Function in Cardiac Health and Diseases
3.1. Heart Failure
3.2. Ischemic Heart Disease
3.3. Cardiac Hypertrophy
3.4. Diabetic Heart
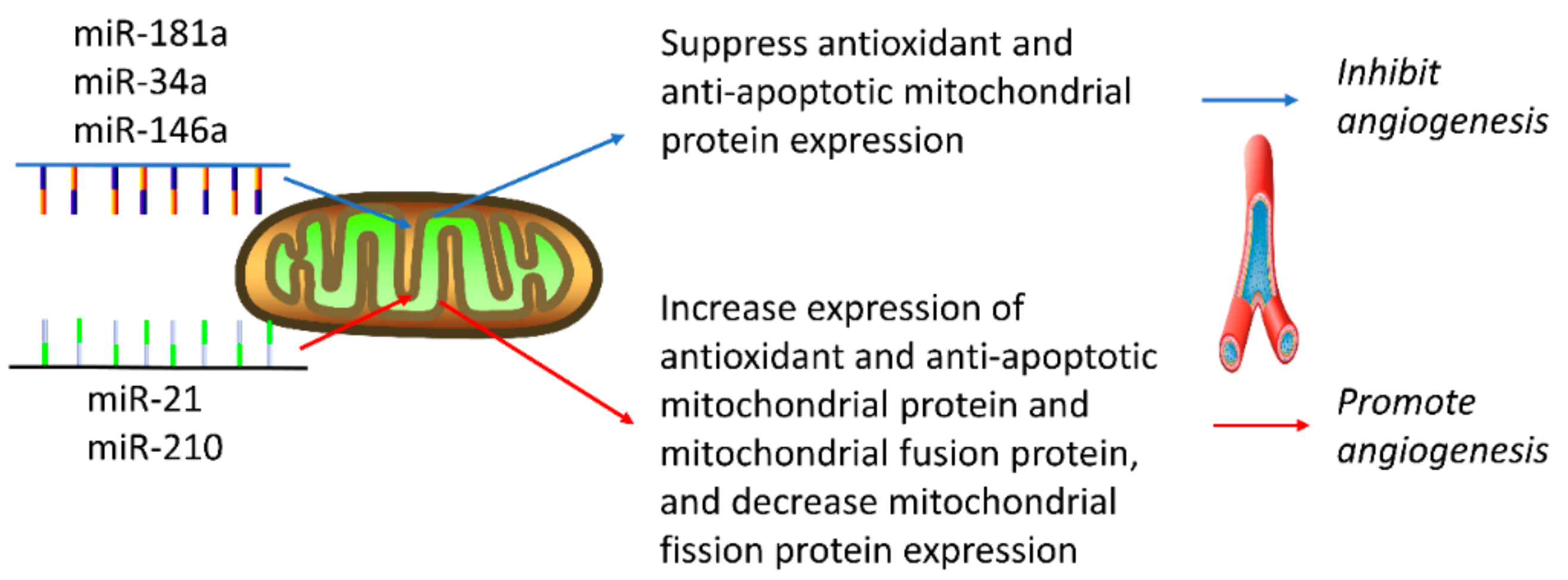
4. MitomiR Regulation of Angiogenesis
4.1. MiRNAs that Inhibit Angiogenesis
4.2. MiRNAs that Promote Angiogenesis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arter. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Vandekeere, S.; Dewerchin, M.; Carmeliet, P. Angiogenesis Revisited: An Overlooked Role of Endothelial Cell Metabolism in Vessel Sprouting. Microcirculation 2015, 22, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, W. Mitochondrial dynamics in the heart as a novel therapeutic target for cardioprotection. Chonnam Med. J. 2013, 49, 101–107. [Google Scholar] [CrossRef]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta. Pharm. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef]
- Reichard, A.; Asosingh, K. The role of mitochondria in angiogenesis. Mol. Biol. Rep. 2019, 46, 1393–1400. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kaminska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef]
- Fu, G.; Brkic, J.; Hayder, H.; Peng, C. MicroRNAs in Human Placental Development and Pregnancy Complications. Int. J. Mol. Sci. 2013, 14, 5519–5544. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Wilfred, B.R.; Wang, W.X.; Nelson, P.T. Energizing miRNA research: A review of the role of miRNAs in lipid metabolism, with a prediction that miR-103/107 regulates human metabolic pathways. Mol. Genet. Metab. 2007, 91, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Weidhaas, J.B.; Slack, F.J. MicroRNAs as potential cancer therapeutics. Oncogene 2008, 27, S52–S57. [Google Scholar] [CrossRef] [PubMed]
- Fasanaro, P.; Greco, S.; Ivan, M.; Capogrossi, M.C.; Martelli, F. microRNA: Emerging therapeutic targets in acute ischemic diseases. Pharmacol. Ther. 2010, 125, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Badalian-Very, G. Clinical applications of microRNAs. F1000Research 2013, 2, 136. [Google Scholar] [CrossRef]
- Shi, L.; Liao, J.; Liu, B.; Zeng, F.; Zhang, L. Mechanisms and therapeutic potential of microRNAs in hypertension. Drug Discov. Today 2015, 20, 1188–1204. [Google Scholar] [CrossRef]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today 2015, 20, 799–811. [Google Scholar] [CrossRef]
- Li, B.; Meng, X.; Zhang, L. microRNAs and cardiac stem cells in heart development and disease. Drug Discov. Today 2019, 24, 233–240. [Google Scholar] [CrossRef]
- Bian, Z.; Li, L.M.; Tang, R.; Hou, D.X.; Chen, X.; Zhang, C.Y.; Zen, K. Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Res. 2010, 20, 1076–1078. [Google Scholar] [CrossRef]
- Barrey, E.; Saint-Auret, G.; Bonnamy, B.; Damas, D.; Boyer, O.; Gidrol, X. Pre-microRNA and mature microRNA in human mitochondria. PLoS ONE 2011, 6, e20220. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Res. 2019, 79, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, C.; Zhou, X.; Han, X.; Li, J.; Wang, Z.; Shang, H.; Liu, Y.; Cao, H. Mitochondria Associated MicroRNA Expression Profiling of Heart Failure. Biomed Res. Int. 2017, 2017, 4042509. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Hathaway, Q.A.; Hollander, J.M. Role of microRNA in metabolic shift during heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H33–H45. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Ma, Z.; Huo, Y.; Xiao, Z.; Li, Y.; Wang, Y. Dynamic mechanisms for pre-miRNA binding and export by Exportin-5. RNA 2011, 17, 1511–1528. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Ipsaro, J.J.; Joshua-Tor, L. From guide to target: Molecular insights into eukaryotic RNA-interference machinery. Nat. Struct. Mol. Biol. 2015, 22, 20–28. [Google Scholar] [CrossRef]
- Srinivasan, H.; Das, S. Mitochondrial miRNA (MitomiR): A new player in cardiovascular health. Can. J. Physiol. Pharm. 2015, 93, 855–861. [Google Scholar] [CrossRef]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, S.E.; Salehi, S.; Cortes, M.; Vilca-Melendez, H.; Menon, K.; Srinivasan, P.; Prachalias, A.; Jassem, W.; Heaton, N. An in silico argument for mitochondrial microRNA as a determinant of primary non function in liver transplantation. Sci. Rep. 2018, 8, 3105. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Liu, C.; Ye, Z. Deep sequencing of small RNA repertoires in mice reveals metabolic disorders-associated hepatic miRNAs. PLoS ONE 2013, 8, e80774. [Google Scholar] [CrossRef]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Colleoni, F.; Padmanabhan, N.; Yung, H.W.; Watson, E.D.; Cetin, I.; Tissot van Patot, M.C.; Burton, G.J.; Murray, A.J. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: A role for miRNA-210 and protein synthesis inhibition. PLoS ONE 2013, 8, e55194. [Google Scholar] [CrossRef]
- Aschrafi, A.; Schwechter, A.D.; Mameza, M.G.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J. Neurosci. 2008, 28, 12581–12590. [Google Scholar] [CrossRef]
- Yan, K.; An, T.; Zhai, M.; Huang, Y.; Wang, Q.; Wang, Y.; Zhang, R.; Wang, T.; Liu, J.; Zhang, Y.; et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. 2019, 10, 500. [Google Scholar] [CrossRef]
- Jaquenod De Giusti, C.; Roman, B.; Das, S. The Influence of MicroRNAs on Mitochondrial Calcium. Front. Physiol. 2018, 9, 1291. [Google Scholar] [CrossRef]
- McCarron, J.G.; Wilson, C.; Sandison, M.E.; Olson, M.L.; Girkin, J.M.; Saunter, C.; Chalmers, S. From structure to function: Mitochondrial morphology, motion and shaping in vascular smooth muscle. J. Vasc. Res. 2013, 50, 357–371. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Wang, K.; Long, B.; Jiao, J.Q.; Wang, J.X.; Liu, J.P.; Li, Q.; Li, P.F. miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 2012, 3, 781. [Google Scholar] [CrossRef] [PubMed]
- Duroux-Richard, I.; Roubert, C.; Ammari, M.; Presumey, J.; Grun, J.R.; Haupl, T.; Grutzkau, A.; Lecellier, C.H.; Boitez, V.; Codogno, P.; et al. miR-125b controls monocyte adaptation to inflammation through mitochondrial metabolism and dynamics. Blood 2016, 128, 3125–3136. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Rippo, M.R.; Olivieri, F.; Monsurro, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef]
- Das, S.; Kohr, M.; Dunkerly-Eyring, B.; Lee, D.I.; Bedja, D.; Kent, O.A.; Leung, A.K.; Henao-Mejia, J.; Flavell, R.A.; Steenbergen, C. Divergent Effects of miR-181 Family Members on Myocardial Function Through Protective Cytosolic and Detrimental Mitochondrial microRNA Targets. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Wang, L.; Huang, H.; Fan, Y.; Kong, B.; Hu, H.; Hu, K.; Guo, J.; Mei, Y.; Liu, W.L. Effects of downregulation of microRNA-181a on H2O2-induced H9c2 cell apoptosis via the mitochondrial apoptotic pathway. Oxid. Med. Cell Longev. 2014, 2014, 960362. [Google Scholar] [CrossRef]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Ono, K.; Iwanaga, Y.; Horie, T.; Nagao, K.; Takemura, G.; Kinoshita, M.; Kuwabara, Y.; Mori, R.T.; Hasegawa, K.; et al. MicroRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. J. Biol. Chem. 2010, 285, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.; Sun, C.; Guo, J.; Zhao, J.; Du, Z. BDNF-mediates Down-regulation of MicroRNA-195 Inhibits Ischemic Cardiac Apoptosis in Rats. Int. J. Biol. Sci. 2016, 12, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef]
- Bienertova-Vasku, J.; Sana, J.; Slaby, O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013, 336, 1–7. [Google Scholar] [CrossRef]
- Puissegur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef]
- Heggermont, W.A.; Papageorgiou, A.P.; Quaegebeur, A.; Deckx, S.; Carai, P.; Verhesen, W.; Eelen, G.; Schoors, S.; van Leeuwen, R.; Alekseev, S.; et al. Inhibition of MicroRNA-146a and Overexpression of Its Target Dihydrolipoyl Succinyltransferase Protect Against Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction. Circulation 2017, 136, 747–761. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Castellano, L.; Mills, A.; Dabrowska, A.F.; Sikkel, M.B.; Pellegrino, L.; Jacob, J.; Frampton, A.E.; Krell, J.; Coombes, R.C.; et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015, 6, e1754. [Google Scholar] [CrossRef]
- Zhao, Y.; Ponnusamy, M.; Liu, C.; Tian, J.; Dong, Y.; Gao, J.; Wang, C.; Zhang, Y.; Zhang, L.; Wang, K.; et al. MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 2871–2881. [Google Scholar] [CrossRef]
- Shepherd, D.L.; Hathaway, Q.A.; Pinti, M.V.; Nichols, C.E.; Durr, A.J.; Sreekumar, S.; Hughes, K.M.; Stine, S.M.; Martinez, I.; Hollander, J.M. Exploring the mitochondrial microRNA import pathway through Polynucleotide Phosphorylase (PNPase). J. Mol. Cell Cardiol. 2017, 110, 15–25. [Google Scholar] [CrossRef]
- Jagannathan, R.; Thapa, D.; Nichols, C.E.; Shepherd, D.L.; Stricker, J.C.; Croston, T.L.; Baseler, W.A.; Lewis, S.E.; Martinez, I.; Hollander, J.M. Translational Regulation of the Mitochondrial Genome Following Redistribution of Mitochondrial MicroRNA in the Diabetic Heart. Circ. Cardiovasc. Genet. 2015, 8, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dai, B.; Fan, J.; Chen, C.; Nie, X.; Yin, Z.; Zhao, Y.; Zhang, X.; Wang, D.W. The Different Roles of miRNA-92a-2-5p and let-7b-5p in Mitochondrial Translation in db/db Mice. Mol. Ther. Nucleic Acids 2019, 17, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Naito, Y.; Mizushima, K.; Takanami, Y.; Kawai, Y.; Ichikawa, H.; Yoshikawa, T. The microRNA miR-696 regulates PGC-1{alpha} in mouse skeletal muscle in response to physical activity. Am. J. Physiol. Metab. 2010, 298, E799–E806. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhang, X.J.; Feng, C.; Sun, T.; Wang, K.; Wang, Y.; Zhou, L.Y.; Li, P.F. MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 2015, 6, e1677. [Google Scholar] [CrossRef]
- Siengdee, P.; Trakooljul, N.; Murani, E.; Schwerin, M.; Wimmers, K.; Ponsuksili, S. MicroRNAs Regulate Cellular ATP Levels by Targeting Mitochondrial Energy Metabolism Genes during C2C12 Myoblast Differentiation. PLoS ONE 2015, 10, e0127850. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Mobius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid. Med. Cell Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Fiedler, J.; Thum, T. MicroRNAs in myocardial infarction. Arter. Thromb. Vasc. Biol. 2013, 33, 201–205. [Google Scholar] [CrossRef]
- Corn, P.G. Hypoxic regulation of miR-210: Shrinking targets expand HIF-1’s influence. Cancer Biol. Ther. 2008, 7, 265–267. [Google Scholar] [CrossRef]
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210—Micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Devlin, C.; Greco, S.; Martelli, F.; Ivan, M. miR-210: More than a silent player in hypoxia. IUBMB Life 2011, 63, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell Biol. 2011, 31, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016, 89, 202–212. [Google Scholar] [CrossRef]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2018, 300, 41–50. [Google Scholar] [CrossRef]
- Li, B.; Dasgupta, C.; Huang, L.; Meng, X.; Zhang, L. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol. Immunol. 2019. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 Modulates the Cellular Energy Metabolism Shift During H2O2-Induced Oxidative Stress by Repressing ISCU in H9c2 Cardiomyocytes. Cell Physiol. Biochem. 2017, 43, 383–394. [Google Scholar] [CrossRef]
- Martinez, S.R.; Ma, Q.; Dasgupta, C.; Meng, X.; Zhang, L. MicroRNA-210 suppresses glucocorticoid receptor expression in response to hypoxia in fetal rat cardiomyocytes. Oncotarget 2017, 8, 80249–80264. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1519–H1530. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Shao, S.; Dong, H.; Bian, X.; Yang, X.; Dong, S. MicroRNA-214 protects cardiac myocytes against H2O2-induced injury. J. Cell Biochem. 2014, 115, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Demkes, C.J.; van Rooij, E. MicroRNA-146a as a Regulator of Cardiac Energy Metabolism. Circulation 2017, 136, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Lopaschuk, G.D.; McCormack, J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 1997, 34, 25–33. [Google Scholar] [CrossRef]
- Duncan, J.G. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim. Biophys. Acta 2011, 1813, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Tahergorabi, Z.; Khazaei, M. A review on angiogenesis and its assays. Iran J. Basic. Med. Sci. 2012, 15, 1110–1126. [Google Scholar]
- Sun, L.L.; Li, W.D.; Lei, F.R.; Li, X.Q. The regulatory role of microRNAs in angiogenesis-related diseases. J. Cell Mol. Med. 2018, 22, 4568–4587. [Google Scholar] [CrossRef]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensa, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging 2018, 10, 2855–2873. [Google Scholar] [CrossRef]
- Li, L.; Yuan, L.; Luo, J.; Gao, J.; Guo, J.; Xie, X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin. Exp. Med. 2013, 13, 109–117. [Google Scholar] [CrossRef]
- Susnow, N.; Zeng, L.; Margineantu, D.; Hockenbery, D.M. Bcl-2 family proteins as regulators of oxidative stress. Semin. Cancer Biol. 2009, 19, 42–49. [Google Scholar] [CrossRef]
- Autret, A.; Martin, S.J. Bcl-2 family proteins and mitochondrial fission/fusion dynamics. Cell Mol. Life Sci. 2010, 67, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tahiri, H.; Cai, C.; Gu, M.; Gagnon, C.; Hardy, P. microRNA-181a inhibits ocular neovascularization by interfering with vascular endothelial growth factor expression. Cardiovasc. Ther. 2018, 36, e12329. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Huang, X. miR-210: Fine-tuning the hypoxic response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar] [CrossRef]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm Circ. 2018, 8. [Google Scholar] [CrossRef]
- Melnik, B.C. MiR-21: An environmental driver of malignant melanoma? J. Transl Med. 2015, 13, 202. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Li, J.; Ma, C.; Chen, S.; Lei, W.; Yang, Y.; Liu, S.; Bihl, J.; Chen, C. Loading MiR-210 in Endothelial Progenitor Cells Derived Exosomes Boosts Their Beneficial Effects on Hypoxia/Reoxygeneation-Injured Human Endothelial Cells via Protecting Mitochondrial Function. Cell Physiol. Biochem. 2018, 46, 664–675. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| miRNA | Cell Type/Tissue | Targeting Mitochondria | Pathology | Reference |
|---|---|---|---|---|
| miR-181c and miR-378 | Human and rat heart | fatty acid metabolism, electron transport activity, and energy metabolism pathways | Heart failure | [33,58] |
| miR-696, miR-532, miR-690, miR-345-3p | Human heart | fatty acid biosynthesis, energy metabolism, and oxidative stress pathways | Heart failure | [33] |
| miR-762 | Neonatal rat cardiac myocytes, pig and mouse heart | energy metabolism pathways (pyruvate dehydrogenase kinase 4, serum/glucocorticoid-regulated kinase 1), and mitochondrial fusion regulators | Ischemic heart disease | [61,62,63] |
| miR-210 | Fetal rat cardiomyocytes, and mouse embryonic fibroblasts | energy metabolism and oxidative stress pathways (mitochondrial iron-sulfur cluster homologue (ISCU1/2)) | Ischemic heart disease | [64,65,66] |
| miR-146a | Human and mouse heart, and neonatal rat cardiomyocytes | energy metabolism and oxidative stress pathways (dihydrolipoamide succinyltransferase (DLST) | Cardiac hypertrophy | [67] |
| miR-1 | Mouse heart | mitochondrial calcium signaling (the mitochondrial calcium uniporter (MCU)) | Cardiac hypertrophy | [64,65,66] |
| miR-30 | Neonatal rat cardiac cells and rat heart | mitochondrial apoptosis signaling (Bcl-2 and Bnip3L/Nix) and the mitochondrial fission regulator dynamin-related protein 1 (Drp1) | Cardiac hypertroph | [55,68] |
| miR-485-5p | Neonatal rat cardiomyocytes and mouse heart | mitochondrial fusion-fission regulators (mitochondrial anchored protein ligase and mitochondrial fusion protein2 (Mfn2)) | Cardiac hypertrophy | [69] |
| miRNA-378 | Mouse heart and HL-1 cardiomyocyte | ATP synthase membrane subunit 6 | Diabetic heart | [70,71] |
| miR-92a | Neonatal rat cardiac myocytes, and mouse heart | mitochondrial gene cytochrome-b | Diabetic heart | [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, R.; Hu, X.-Q.; Zhang, L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells 2019, 8, 1475. https://doi.org/10.3390/cells8121475
Song R, Hu X-Q, Zhang L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells. 2019; 8(12):1475. https://doi.org/10.3390/cells8121475
Chicago/Turabian StyleSong, Rui, Xiang-Qun Hu, and Lubo Zhang. 2019. "Mitochondrial MiRNA in Cardiovascular Function and Disease" Cells 8, no. 12: 1475. https://doi.org/10.3390/cells8121475
APA StyleSong, R., Hu, X.-Q., & Zhang, L. (2019). Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells, 8(12), 1475. https://doi.org/10.3390/cells8121475