The Role of MicroRNAs upon Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease


Abstract
1. Introduction
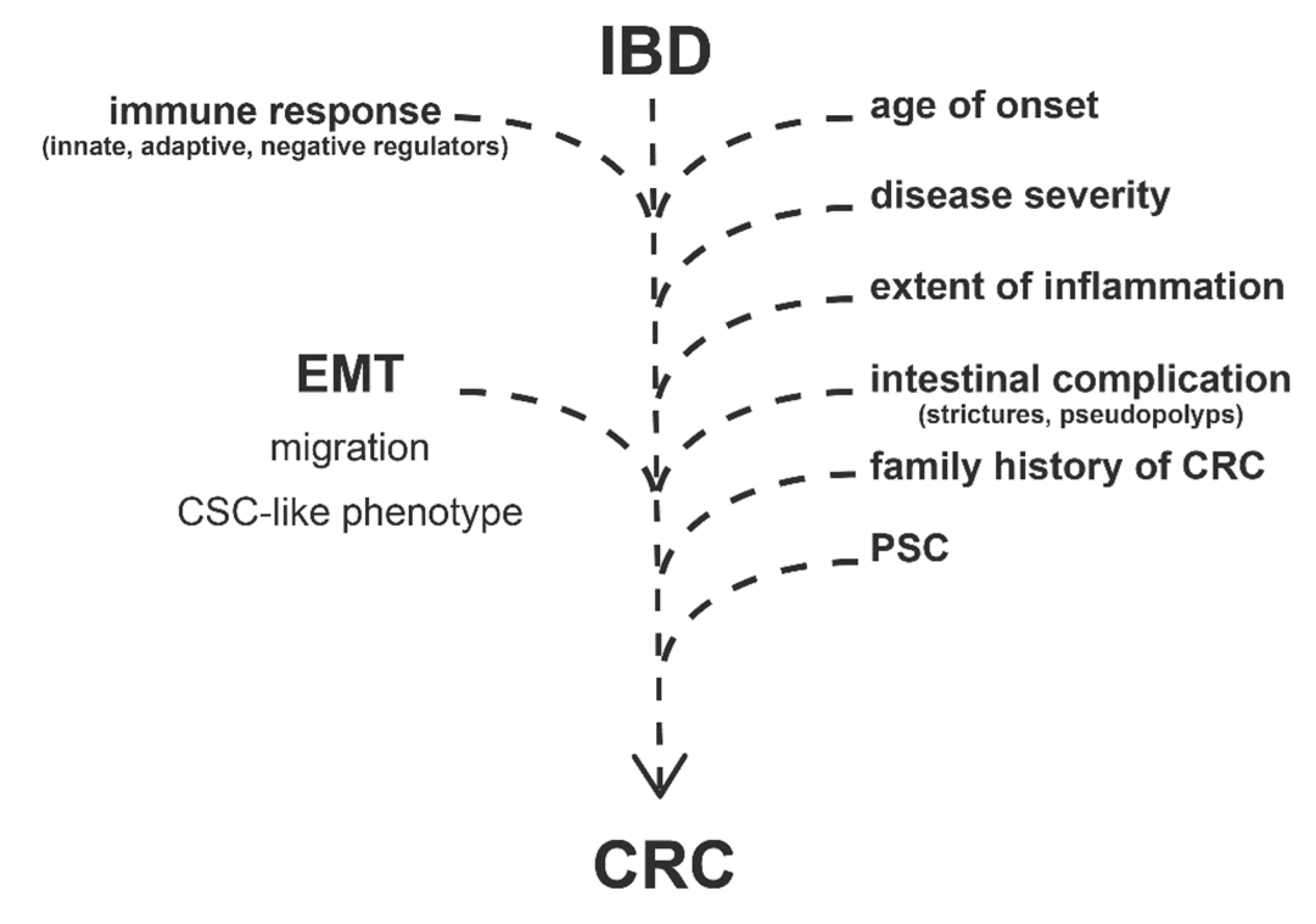
2. Risk Factors of CRC in IBD
3. Inflammation, EMT, and Tumorigenesis
4. Molecular Mechanism of Epithelial-to-Mesenchymal Transition
4.1. Disengagement from the Bondage of Junctions
4.2. Control of EMT by Transcription Factors
4.3. Influence of Signaling Pathways
4.4. Role of EMT in IBD
5. MicroRNAs
5.1. Biogenesis and Function of MicroRNAs
5.2. Role of miRNAs in IBD
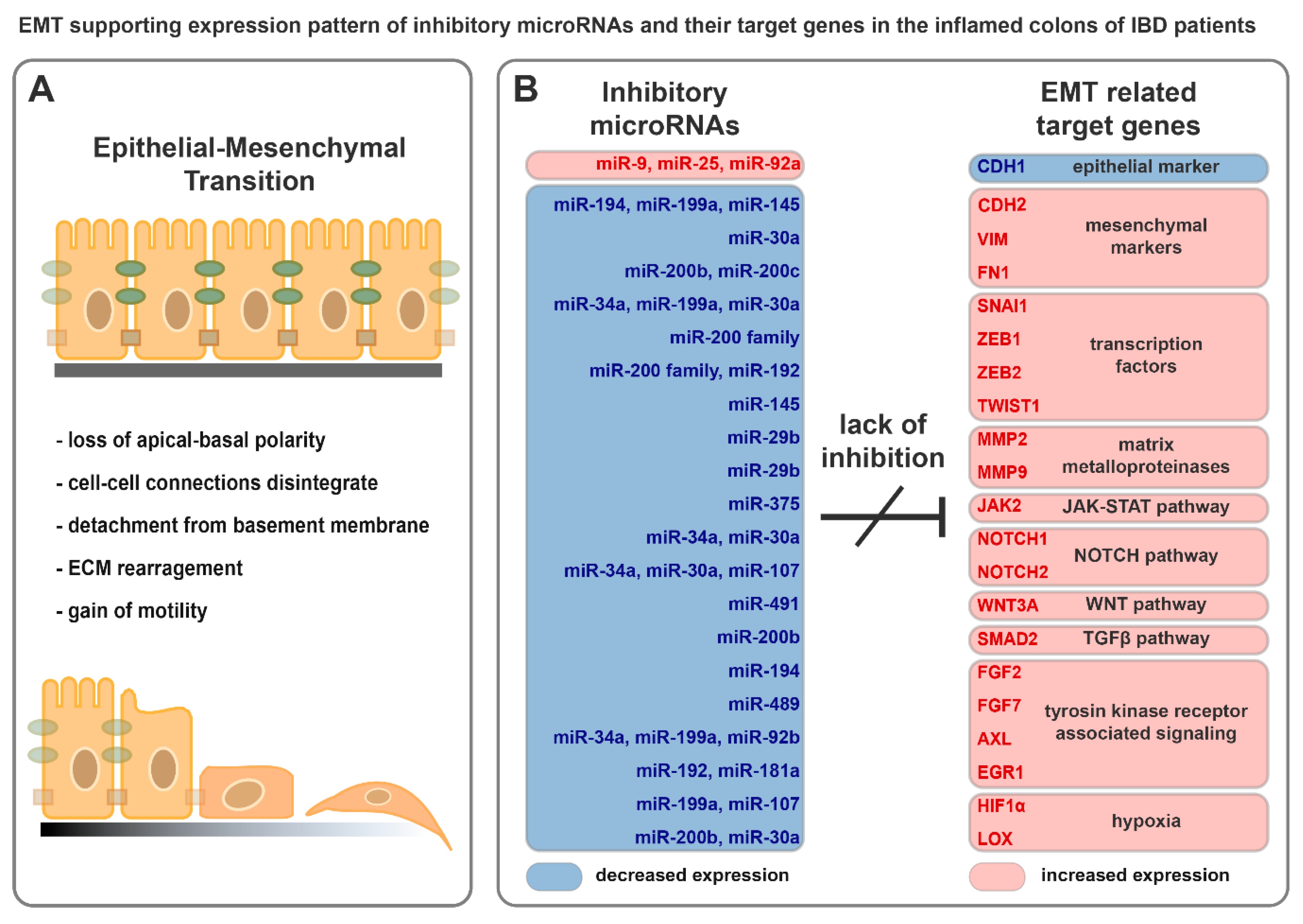
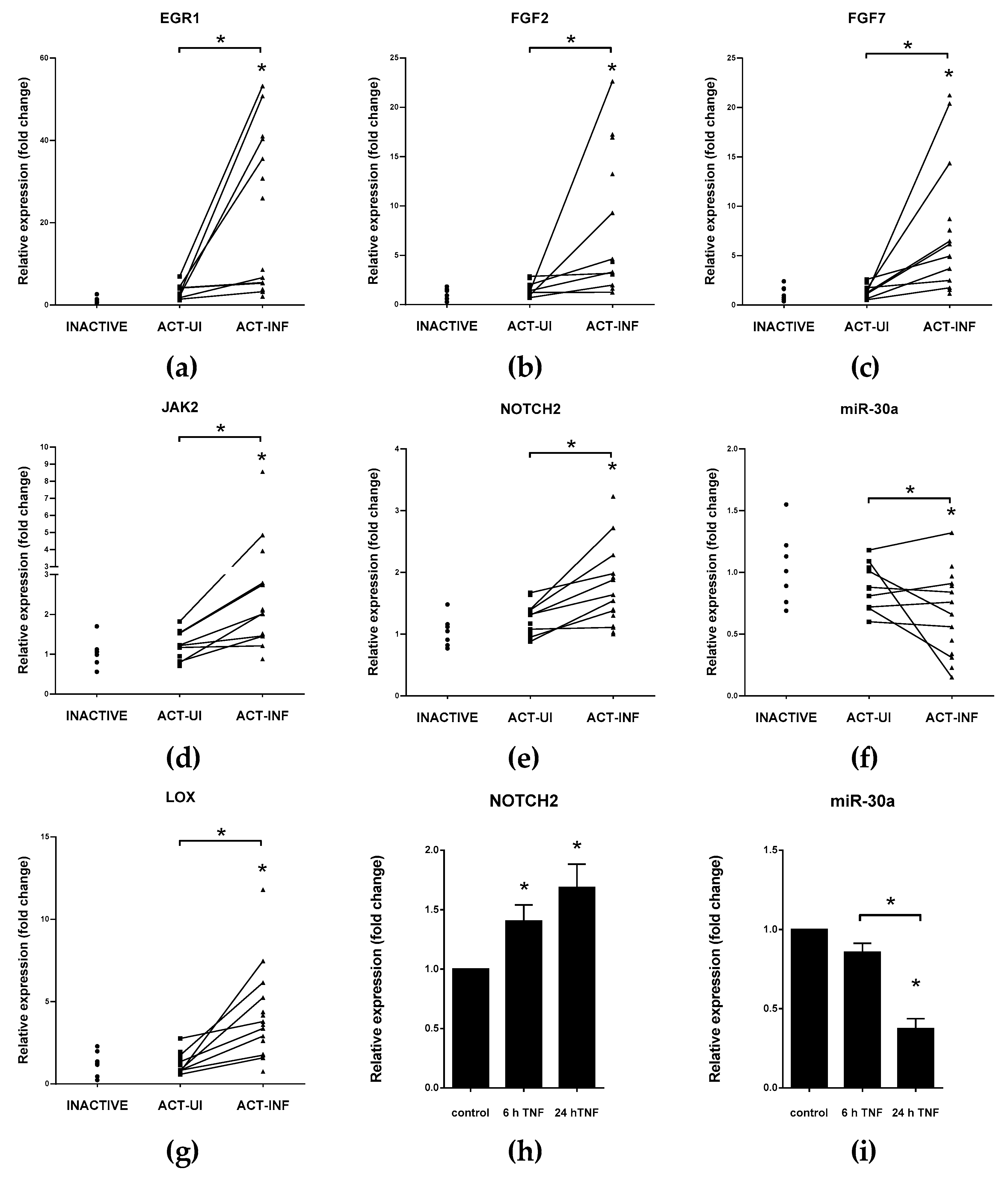
5.3. miRNAs Having Target mRNAs Related to EMT with Potential Role in IBD Pathogenesis
5.4. miRNAs Involved in CSC Function
6. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, D.K. Inflammatory bowel disease (1). N. Engl. J. Med. 1991, 325, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Werkstetter, K.J.; Ullrich, J.; Schatz, S.B.; Prell, C.; Koletzko, B.; Koletzko, S. Lean body mass, physical activity and quality of life in paediatric patients with inflammatory bowel disease and in healthy controls. J. Crohns Colitis 2012, 6, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hubenthal, M.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Ek, W.E.; D’Amato, M.; Halfvarson, J. The history of genetics in inflammatory bowel disease. Ann. Gastroenterol. 2014, 27, 294–303. [Google Scholar]
- Park, S.C.; Jeen, Y.T. Genetic Studies of Inflammatory Bowel Disease-Focusing on Asian Patients. Cells 2019, 8, 404. [Google Scholar] [CrossRef]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Shanahan, F. Disorders of a modern lifestyle: Reconciling the epidemiology of inflammatory bowel diseases. Gut 2008, 57, 1185–1191. [Google Scholar] [CrossRef]
- Borren, N.Z.; van der Woude, C.J.; Ananthakrishnan, A.N. Fatigue in IBD: Epidemiology, pathophysiology and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 247–259. [Google Scholar] [CrossRef]
- Sexton, K.A.; Walker, J.R.; Targownik, L.E.; Graff, L.A.; Haviva, C.; Beatie, B.E.; Petty, S.K.; Bernstein, M.T.; Singh, H.; Miller, N.; et al. The Inflammatory Bowel Disease Symptom Inventory: A Patient-report Scale for Research and Clinical Application. Inflamm. Bowel Dis. 2019, 25, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Stidham, R.W.; Higgins, P.D.R. Colorectal Cancer in Inflammatory Bowel Disease. Clin. Colon. Rectal. Surg. 2018, 31, 168–178. [Google Scholar] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518. [Google Scholar] [CrossRef]
- Herranz, H.; Cohen, S.M. MicroRNAs and gene regulatory networks: Managing the impact of noise in biological systems. Genes Dev. 2010, 24, 1339–1344. [Google Scholar] [CrossRef]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal Cancer and Dysplasia in Inflammatory Bowel Disease: A Review of Disease Epidemiology, Pathophysiology, and Management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Bio. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Romano, M.; De Francesco, F.; Zarantonello, L.; Ruffolo, C.; Ferraro, G.A.; Zanus, G.; Giordano, A.; Bassi, N.; Cillo, U. From Inflammation to Cancer in Inflammatory Bowel Disease: Molecular Perspectives. Anticancer Res. 2016, 36, 1447–1460. [Google Scholar]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Unal, N.G.; Ozutemiz, O.; Tekin, F.; Turan, I.; Osmanoglu, N. Colorectal cancer and dysplasia risk of ulcerative colitis patients in a tertiary referral center in Turkey. Turk. J. Gastroenterol. 2019, 30, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Hernandez, V.; Myrelid, P.; Kariv, R.; Tsianos, E.; Toruner, M.; Marti-Gallostra, M.; Spinelli, A.; van der Meulen-de Jong, A.E.; Yuksel, E.S.; et al. Colorectal cancer in inflammatory bowel disease: Results of the 3rd ECCO pathogenesis scientific workshop (I). J. Crohns Colitis 2014, 8, 5–18. [Google Scholar] [CrossRef]
- Wang, Z.H.; Fang, J.Y. Colorectal Cancer in Inflammatory Bowel Disease: Epidemiology, Pathogenesis and Surveillance. Gastrointest Tumors 2014, 1, 146–154. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Samadder, N.J.; Valentine, J.F.; Guthery, S.; Singh, H.; Bernstein, C.N.; Leighton, J.A.; Wan, Y.; Wong, J.; Boucher, K.; Pappas, L.; et al. Family History Associates with Increased Risk of Colorectal Cancer in Patients With Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2019, 17, 1807–1813. [Google Scholar] [CrossRef]
- Ryan, B.M.; Wolff, R.K.; Valeri, N.; Khan, M.; Robinson, D.; Paone, A.; Bowman, E.D.; Lundgreen, A.; Caan, B.; Potter, J.; et al. An analysis of genetic factors related to risk of inflammatory bowel disease and colon cancer. Cancer Epidemiol. 2014, 38, 583–590. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Jamontt, J.; Petit, S.; Clark, N.; Parkinson, S.J.; Smith, P. Nucleotide-binding oligomerization domain 2 signaling promotes hyperresponsive macrophages and colitis in IL-10-deficient mice. J. Immunol. 2013, 190, 2948–2958. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Boros, E.; Kellermayer, Z.; Balogh, P.; Strifler, G.; Voros, A.; Sarlos, P.; Vincze, A.; Varga, C.; Nagy, I. Elevated Expression of AXL May Contribute to the Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease Patients. Mediat. Inflamm. 2018, 2018, 3241406. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Van Staa, T.P.; Card, T.; Logan, R.F.; Leufkens, H.G. 5-Aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: A large epidemiological study. Gut 2005, 54, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Mei, Z.; Guo, Y.; Wang, G.; Wu, T.; Cui, X.; Huang, Z.; Zhu, Y.; Wen, D.; Song, J.; et al. Reduced Risk of Inflammatory Bowel Disease-associated Colorectal Neoplasia with Use of Thiopurines: A Systematic Review and Meta-analysis. J. Crohns Colitis 2018, 12, 546–558. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Boman, B.M.; Huang, E. Human colon cancer stem cells: A new paradigm in gastrointestinal oncology. J. Clin. Oncol. 2008, 26, 2828–2838. [Google Scholar] [CrossRef]
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeong, J.; Seo, J.; Kim, H.S.; Kim, S.J.; Jin, W. Dysregulated JAK2 expression by TrkC promotes metastasis potential, and EMT program of metastatic breast cancer. Sci. Rep. 2016, 6, 33899. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, W.; Zhang, L.; Chen, X.; Liu, F.; Zhang, J.; Guan, S.; Sun, Y.; Chen, P.; Wang, D.; et al. miR-146a-5p mediates epithelial-mesenchymal transition of oesophageal squamous cell carcinoma via targeting Notch2. Br. J. Cancer 2016, 115, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mateo, I.; Arruabarrena-Aristorena, A.; Artaza-Irigaray, C.; Lopez, J.A.; Calvo, E.; Belandia, B. HEY1 functions are regulated by its phosphorylation at Ser-68. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, T.; Guo, T.W.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J.A. The NOTCH4-HEY1 Pathway Induces Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 619–633. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Luo, J.; Xiao, W.; Ye, X.; Chen, M.; Li, Y.; Zhang, G.J. Notch3 inhibits epithelial-mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis 2016, 5, e269. [Google Scholar] [CrossRef]
- Vincan, E.; Barker, N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin. Exp. Metastasis 2008, 25, 657–663. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Lu, X.; Wu, J.; Yu, K.; Ji, A.; Lu, W.; Wang, Z.; Wu, J.; et al. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/beta-catenin signaling. Sci. Rep. 2019, 9, 7539. [Google Scholar] [CrossRef]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef]
- Richards, E.J.; Zhang, G.; Li, Z.P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) beta. LncRNA-HIT-MEDIATED TGF-INDUCED EPITHELIAL TO MESENCHYMAL TRANSITION IN MAMMARY EPITHELIA. J. Biol. Chem. 2016, 291, 22860. [Google Scholar] [CrossRef]
- Pang, M.F.; Georgoudaki, A.M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Maehara, O.; Suda, G.; Natsuizaka, M.; Ohnishi, S.; Komatsu, Y.; Sato, F.; Nakai, M.; Sho, T.; Morikawa, K.; Ogawa, K.; et al. Fibroblast growth factor-2-mediated FGFR/Erk signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis 2017, 38, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2016, 7, 5440–5460. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Mason, I.J.; Fuller-Pace, F.; Smith, R.; Dickson, C. FGF-7 (keratinocyte growth factor) expression during mouse development suggests roles in myogenesis, forebrain regionalisation and epithelial-mesenchymal interactions. Mech. Dev. 1994, 45, 15–30. [Google Scholar] [CrossRef]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef]
- Pagel, J.I.; Deindl, E. Early Growth Response 1-A Transcription Factor in the crossfire of Signal Transduction Cascades. Indian J. Biochem. Bio. 2011, 48, 226–235. [Google Scholar]
- Baron, V.; Adamson, E.D.; Calogero, A.; Ragona, G.; Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGF beta 1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006, 13, 115–124. [Google Scholar] [CrossRef]
- Wu, W.S.; You, R.I.; Cheng, C.C.; Lee, M.C.; Lin, T.Y.; Hu, C.T. Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep. 2017, 7, 17753. [Google Scholar] [CrossRef]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, H.; Yashiro, M.; Kinoshita, H.; Fukuoka, T.; Morisaki, T.; Masuda, G.; Sakurai, K.; Kubo, N.; Ohira, M.; Hirakawa, K. Lysyl oxidase is associated with the epithelial-mesenchymal transition of gastric cancer cells in hypoxia. Gastric. Cancer 2016, 19, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Genovese, G.; Danese, S. Role of Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease. J. Crohns Colitis 2019, 13, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Weber, A.; Furst, A.; Farkas, S.; Jehle, E.; Pesch, T.; Kellermeier, S.; Fried, M.; Rogler, G. Potential Role for SNAIL Family Transcription Factors in the Etiology of Crohn’s Disease-Associated Fistulae. Inflamm. Bowel Dis. 2011, 17, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Bataille, F.; Rohrmeier, C.; Bates, R.; Weber, A.; Rieder, F.; Brenmoehl, J.; Strauch, U.; Farkas, S.; Furst, A.; Hofstadter, F.; et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Huber, N.; Lang, S.; Furst, A.; Jehle, E.; Rogler, G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin. Transl. Med. 2015, 4, 1. [Google Scholar] [CrossRef]
- Boros, E.; Csatari, M.; Varga, C.; Balint, B.; Nagy, I. Specific Gene- and MicroRNA-Expression Pattern Contributes to the Epithelial to Mesenchymal Transition in a Rat Model of Experimental Colitis. Mediat. Inflamm. 2017, 2017, 5257378. [Google Scholar] [CrossRef]
- Mohammadi, A.; Mansoori, B.; Baradaran, B. The role of microRNAs in colorectal cancer. Biomed. Pharmacother. 2016, 84, 705–713. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Davis-Dusenbery, B.N.; Hata, A. Mechanisms of control of microRNA biogenesis. J. Biochem. 2010, 148, 381–392. [Google Scholar] [PubMed]
- Sontheimer, E.J. Assembly and function of RNA silencing complexes. Nat. Rev. Mol. Cell Biol. 2005, 6, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Therap. Adv. Gastroenterol. 2015, 8, 4–22. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Henao-Mejia, J.; Flavell, R.A. Regulation of the Antimicrobial Response by NLR Proteins. Immunity 2011, 34, 665–679. [Google Scholar] [CrossRef]
- Chuang, A.Y.; Chuang, J.C.; Zhai, Z.L.; Wu, F.; Kwon, J.H. NOD2 Expression is Regulated by microRNAs in Colonic Epithelial HCT116 Cells. Inflamm. Bowel Dis. 2014, 20, 126–135. [Google Scholar] [CrossRef]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, Y.; Ge, W.; Zhou, K.; Wen, J.; Yan, W.; Wang, Y.; Wang, B.; Qu, C.; Wu, J.; et al. miR-200b inhibits TGF-beta 1-induced epithelial-mesenchymal transition and promotes growth of intestinal epithelial cells. Cell Death Dis. 2013, 4, e541. [Google Scholar] [CrossRef]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–U252. [Google Scholar] [CrossRef]
- Li, M.L.; Guan, X.F.; Sun, Y.Q.; Mi, J.; Shu, X.H.; Liu, F.; Li, C.G. miR-92a family and their target genes in tumorigenesis and metastasis. Exp. Cell Res. 2014, 323, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, Z.; Zhao, X.; Wang, J.; Ding, D.; Wang, Z.; Tan, F.; Tan, X.; Zhou, F.; Sun, J.; et al. MicroRNA-25 promotes cell migration and invasion in esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2012, 421, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Zhao, X.H.; Wang, J.W.; Li, B.Z.; Wang, Z.; Sun, J.; Tan, F.W.; Ding, D.P.; Xu, X.H.; Zhou, F.; et al. microRNA-92a promotes lymph node metastasis of human esophageal squamous cell carcinoma via E-cadherin. J. Biol. Chem. 2011, 286, 10725–10734. [Google Scholar] [CrossRef] [PubMed]
- Fasseu, M.; Treton, X.; Guichard, C.; Pedruzzi, E.; Cazals-Hatem, D.; Richard, C.; Aparicio, T.; Daniel, F.; Soule, J.C.; Moreau, R.; et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS ONE 2010, 5, e13160. [Google Scholar] [CrossRef]
- Zhen, Y.; Luo, C.; Zhang, H. Early detection of ulcerative colitis-associated colorectal cancer. Gastroenterol. Rep. 2018, 6, 83–92. [Google Scholar] [CrossRef]
- Burke, J.P.; Cunningham, M.F.; Sweeney, C.; Docherty, N.G.; O’Connell, P.R. N-Cadherin Is Overexpressed in Crohn’s Stricture Fibroblasts and Promotes Intestinal Fibroblast Migration. Inflamm. Bowel Dis. 2011, 17, 1665–1673. [Google Scholar] [CrossRef]
- Meng, Z.P.; Fu, X.H.; Chen, X.S.; Zeng, S.; Tian, Y.; Jove, R.; Xu, R.Z.; Huang, W.D. miR-194 Is a Marker of Hepatic Epithelial Cells and Suppresses Metastasis of Liver Cancer Cells in Mice. Hepatology 2010, 52, 2148–2157. [Google Scholar] [CrossRef]
- Lin, J.M.; Welker, N.C.; Zhao, Z.J.; Li, Y.; Zhang, J.J.; Reuss, S.A.; Zhang, X.J.; Lee, H.J.; Liu, Y.L.; Bronner, M.P. Novel specific microRNA biomarkers in idiopathic inflammatory bowel disease unrelated to disease activity. Modern Pathol. 2014, 27, 602–608. [Google Scholar] [CrossRef]
- Suzuki, T.; Mizutani, K.; Minami, A.; Nobutani, K.; Kurita, S.; Nagino, M.; Shimono, Y.; Takai, Y. Suppression of the TGF-beta1-induced protein expression of SNAI1 and N-cadherin by miR-199a. Genes Cells 2014, 19, 667–675. [Google Scholar] [CrossRef]
- Pekow, J.R.; Dougherty, U.; Mustafi, R.; Zhu, H.; Kocherginsky, M.; Rubin, D.T.; Hanauer, S.B.; Hart, J.; Chang, E.B.; Fichera, A.; et al. miR-143 and miR-145 are downregulated in ulcerative colitis: Putative regulators of inflammation and protooncogenes. Inflamm. Bowel Dis. 2012, 18, 94–100. [Google Scholar] [CrossRef]
- Gao, P.; Xing, A.Y.; Zhou, G.Y.; Zhang, T.G.; Zhang, J.P.; Gao, C.; Li, H.; Shi, D.B. The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer. Oncogene 2013, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, L.; Zhang, X.; Xu, X.; Xing, H.; Zhang, Y.; Li, W.; Yu, H.; Zeng, J.; Jia, J. RUNX3 regulates vimentin expression via miR-30a during epithelial-mesenchymal transition in gastric cancer cells. J. Cell Mol. Med. 2014, 18, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Z.; Li, Y.; Fan, D.; Jiang, H. MicroRNA-200c binding to FN1 suppresses the proliferation, migration and invasion of gastric cancer cells. Biomed. Pharmacother. 2017, 88, 285–292. [Google Scholar] [CrossRef]
- Tang, O.; Chen, X.M.; Shen, S.; Hahn, M.; Pollock, C.A. MiRNA-200b represses transforming growth factor-beta 1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am. J. Physiol.-Renal. 2013, 304, F1266–F1273. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Bajaj, R.; Wang, L.; Yan, Y.; Ritzenthaler, J.D.; Gewirtz, A.T.; Roman, J.; Merlin, D.; Sitaraman, S.V. Epithelial-derived fibronectin expression, signaling, and function in intestinal inflammation. J. Biol. Chem. 2007, 282, 32965–32973. [Google Scholar] [CrossRef]
- Zidar, N.; Bostjancic, E.; Jerala, M.; Kojc, N.; Drobne, D.; Stabuc, B.; Glavac, D. Down-regulation of microRNAs of the miR-200 family and up-regulation of Snail and Slug in inflammatory bowel diseases—Hallmark of epithelial-mesenchymal transition. J. Cell. Mol. Med. 2016, 20, 1813–1820. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, M.; Lan, H.; Yu, X. miR-30a negatively regulates TGF-beta1-induced epithelial-mesenchymal transition and peritoneal fibrosis by targeting Snai1. Am. J. Pathol. 2013, 183, 808–819. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Mudduluru, G.; Ceppi, P.; Muppala, S.; Kozlowski, M.; Niklinski, J.; Papotti, M.; Allgayer, H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int. J. Cancer 2012, 130, 2044–2053. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H.; et al. A miR-192-EGR1-HOXB9 regulatory network controls the angiogenic switch in cancer. Nat. Commun. 2016, 7, 11169. [Google Scholar] [CrossRef] [PubMed]
- Nairismagi, M.L.; Fuchtbauer, A.; Labouriau, R.; Bramsen, J.B.; Fuchtbauer, E.M. The Proto-Oncogene TWIST1 Is Regulated by MicroRNAs. PLoS ONE 2013, 8, e66070. [Google Scholar] [CrossRef] [PubMed]
- Poudyal, D.; Cui, X.L.; Le, P.M.; Hofseth, A.B.; Windust, A.; Nagarkatti, M.; Nagarkatti, P.S.; Schetter, A.J.; Harris, C.C.; Hofseth, L.J. A Key Role of microRNA-29b for the Suppression of Colon Cancer Cell Migration by American Ginseng. PLoS ONE 2013, 8, e75034. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Lin, J.H.; Brenot, A.; Kim, J.W.; Provot, S.; Werb, Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat. Cell Biol. 2013, 15, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Xu, Y.; Zhang, W.; Deng, Y.; Si, M.; Du, Y.; Yao, H.; Liu, X.; Ke, Y.; Si, J.; et al. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res. 2010, 20, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jin, J.; Liu, Y.; Huang, Z.; Deng, Y.; You, T.; Zhou, T.; Si, J.; Zhuo, W. Snail-regulated MiR-375 inhibits migration and invasion of gastric cancer cells by targeting JAK2. PLoS ONE 2014, 9, e99516. [Google Scholar] [CrossRef]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gulla, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e194. [Google Scholar] [CrossRef]
- Ortega, M.; Bhatnagar, H.; Lin, A.P.; Wang, L.; Aster, J.C.; Sill, H.; Aguiar, R.C. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia 2015, 29, 968–976. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, R.; Li, P.; Liu, Y.; Qin, K.; Fa, Z.Q.; Liu, Y.J.; Ke, Y.Q.; Jiang, X.D. P53-induced microRNA-107 inhibits proliferation of glioma cells and down-regulates the expression of CDK6 and Notch-2. Neurosci. Lett. 2013, 534, 327–332. [Google Scholar] [CrossRef]
- Sun, R.F.; Liu, Z.G.; Tong, D.D.; Yang, Y.; Guo, B.; Wang, X.F.; Zhao, L.Y.; Huang, C. miR-491-5p, mediated by Foxi1, functions as a tumor suppressor by targeting Wnt3a/beta-catenin signaling in the development of gastric cancer. Cell Death Dis. 2017, 8, e2714. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lai, C.; Gu, H.; Zhao, L.; Xia, M.; Yang, P.; Wang, X. miR-194 Inhibits Innate Antiviral Immunity by Targeting FGF2 in Influenza H1N1 Virus Infection. Front. Microbiol. 2017, 8, 2187. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, L.Y.; Chong, Q.Y.; Yan, H.; Zhang, W.J.; Qian, W.C.; Tan, S.; Wu, Z.S.; Lobie, P.E.; Zhu, T. Long noncoding RNA Linc00460 promotes breast cancer progression by regulating the miR-489-5p/FGF7/AKT axis. Cancer Manag Res. 2019, 11, 5983–6001. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.Y.; Huang, J.S.; Shiah, S.G.; Chung, S.Y.; Lay, J.D.; Yang, Y.Y.; Lai, G.M.; Cheng, A.L.; Chen, L.T.; Chuang, S.E. Negative feedback regulation of AXL by miR-34a modulates apoptosis in lung cancer cells. RNA 2016, 22, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Bai, W.D.; Li, J.; Tao, K.; Wang, H.T.; Yang, X.K.; Liu, J.Q.; Wang, Y.C.; He, T.; Xie, S.T.; et al. Peroxisome proliferator-activated receptor-gamma agonist troglitazone suppresses transforming growth factor-beta1 signalling through miR-92b upregulation-inhibited Axl expression in human keloid fibroblasts in vitro. Am. J. Transl. Res. 2016, 8, 3460–3470. [Google Scholar]
- Verduci, L.; Azzalin, G.; Gioiosa, S.; Carissimi, C.; Laudadio, I.; Fulci, V.; Macino, G. microRNA-181a enhances cell proliferation in acute lymphoblastic leukemia by targeting EGR1. Leuk Res. 2015, 39, 479–485. [Google Scholar] [CrossRef]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef]
- Ding, G.; Huang, G.; Liu, H.D.; Liang, H.X.; Ni, Y.F.; Ding, Z.H.; Ni, G.Y.; Hua, H.W. MiR-199a suppresses the hypoxia-induced proliferation of non-small cell lung cancer cells through targeting HIF1alpha. Mol. Cell Biochem. 2013, 384, 173–180. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef]
- Sun, M.; Gomes, S.; Chen, P.; Frankenberger, C.A.; Sankarasharma, D.; Chung, C.H.; Chada, K.K.; Rosner, M.R. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene 2014, 33, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Boufraqech, M.; Nilubol, N.; Zhang, L.; Gara, S.K.; Sadowski, S.M.; Mehta, A.; He, M.; Davis, S.; Dreiling, J.; Copland, J.A.; et al. miR30a inhibits LOX expression and anaplastic thyroid cancer progression. Cancer Res. 2015, 75, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.Q.; Ma, C.H.; Wang, Q.; Song, Y.; Lv, T.F. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Niesner, U.; Albrecht, I.; Janke, M.; Doebis, C.; Loddenkemper, C.; Lexberg, M.H.; Eulenburg, K.; Kreher, S.; Koeck, J.; Baumgrass, R.; et al. Autoregulation of Th1-mediated inflammation by twist1. J. Exp. Med. 2008, 205, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, A.; Biancheri, P.; Lewis, A.; Bishop, C.L.; Giuffrida, P.; Chan, C.; Feakins, R.; Poulsom, R.; Di Sabatino, A.; Roberto-Corazza, G.; et al. In Crohn’s disease fibrosis-reduced expression of the miR-29 family enhances collagen expression in intestinal fibroblasts. Clin. Sci. 2014, 127, 341–350. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, R.; Zhang, X.; Dong, W.; Zhang, J.; Yan, Z.; Li, W.; Cui, J.; Lu, Y. miR-375 targets the p53 gene to regulate cellular response to ionizing radiation and etoposide in gastric cancer cells. DNA Repair 2013, 12, 741–750. [Google Scholar] [CrossRef]
- Zhang, N.; Lin, J.K.; Chen, J.; Liu, X.F.; Liu, J.L.; Luo, H.S.; Li, Y.Q.; Cui, S. MicroRNA 375 mediates the signaling pathway of corticotropin-releasing factor (CRF) regulating pro-opiomelanocortin (POMC) expression by targeting mitogen-activated protein kinase 8. J. Biol. Chem. 2013, 288, 10361–10373. [Google Scholar] [CrossRef]
- Selth, L.A.; Das, R.; Townley, S.L.; Coutinho, I.; Hanson, A.R.; Centenera, M.M.; Stylianou, N.; Sweeney, K.; Soekmadji, C.; Jovanovic, L.; et al. A ZEB1-miR-375-YAP1 pathway regulates epithelial plasticity in prostate cancer. Oncogene 2017, 36, 24–34. [Google Scholar] [CrossRef]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef]
- Chen, L.; Chen, X.R.; Chen, F.F.; Liu, Y.; Li, P.; Zhang, R.; Yan, K.; Yi, Y.J.; Xu, Z.M.; Jiang, X.D. MicroRNA-107 inhibits U87 glioma stem cells growth and invasion. Cell Mol. Neurobiol. 2013, 33, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Claessen, M.M.; Schipper, M.E.; Oldenburg, B.; Siersema, P.D.; Offerhaus, G.J.; Vleggaar, F.P. WNT-pathway activation in IBD-associated colorectal carcinogenesis: Potential biomarkers for colonic surveillance. Cell Oncol. 2010, 32, 303–310. [Google Scholar] [PubMed]
- Yu, T.; Wang, L.N.; Li, W.; Zuo, Q.F.; Li, M.M.; Zou, Q.M.; Xiao, B. Downregulation of miR-491-5p promotes gastric cancer metastasis by regulating SNAIL and FGFR4. Cancer Sci. 2018, 109, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.F.; Chen, X.J.; Cao, Y.W.; Huang, J.Y.; Xie, X.H.; Wei, Y.M. Expression of Wnt and Notch signaling pathways in inflammatory bowel disease treated with mesenchymal stem cell transplantation: Evaluation in a rat model. Stem Cell Res. Ther. 2015, 6, 101. [Google Scholar] [CrossRef]
- Ranjha, R.; Aggarwal, S.; Bopanna, S.; Ahuja, V.; Paul, J. Site-Specific MicroRNA Expression May Lead to Different Subtypes in Ulcerative Colitis. PLoS ONE 2015, 10, e0142869. [Google Scholar] [CrossRef]
- Shirakihara, T.; Horiguchi, K.; Miyazawa, K.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-beta regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.Q.; Kalluri, R.; Muller, G.A.; Neilson, E.G. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef]
- Iborra, M.; Bernuzzi, F.; Correale, C.; Vetrano, S.; Fiorino, G.; Beltran, B.; Marabita, F.; Locati, M.; Spinelli, A.; Nos, P.; et al. Identification of serum and tissue micro-RNA expression profiles in different stages of inflammatory bowel disease. Clin. Exp. Immunol. 2013, 173, 250–258. [Google Scholar] [CrossRef]
- Belfiore, A.; Genua, M.; Malaguarnera, R. PPAR-gamma agonists and their effects on IGF-I receptor signaling: Implications for cancer. PPAR Res. 2009, 2009, 830501. [Google Scholar] [CrossRef]
- Rokavec, M.; Wu, W.; Luo, J.L. IL6-mediated suppression of miR-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol. Cell 2012, 45, 777–789. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, F.L.; Wang, H.B.; Dong, N.; Zhu, X.M.; Wu, Y.; Wang, Y.T.; Yao, Y.M. TNF-alpha mRNA is negatively regulated by microRNA-181a-5p in maturation of dendritic cells induced by high mobility group box-1 protein. Sci. Rep. 2017, 7, 12239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shu, W.; Zhou, G.; Lin, J.; Chu, F.; Wu, H.; Liu, Z. Anti-TNF-alpha Therapy Suppresses Proinflammatory Activities of Mucosal Neutrophils in Inflammatory Bowel Disease. Mediat. Inflamm. 2018, 2018, 3021863. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, Z.; Rai, S.N.; Eichenberger, M.R.; Barnes, C.; Dworkin, A.M.; Weller, C.; Cohen, E.; Roberts, H.; Keskey, B.; Petras, R.E.; et al. Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease-associated colorectal cancer. Hum. Mutat. 2012, 33, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Shi, X.P.; Peng, Y.; Wu, M.Y.; Zhang, P.; Xie, R.Y.; Wu, Y.; Yan, Q.Q.; Liu, S.D.; Wang, J.D. HIF-1 alpha Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar]
- Zhu, Y.; Tan, J.; Xie, H.; Wang, J.; Meng, X.; Wang, R. HIF-1alpha regulates EMT via the Snail and beta-catenin pathways in paraquat poisoning-induced early pulmonary fibrosis. J. Cell Mol. Med. 2016, 20, 688–697. [Google Scholar] [CrossRef]
- Shah, Y.M. The role of hypoxia in intestinal inflammation. Mol. Cell Pediatr. 2016, 3, 1. [Google Scholar] [CrossRef]
- Shimshoni, E.; Yablecovitch, D.; Baram, L.; Dotan, I.; Sagi, I. ECM remodelling in IBD: Innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut 2015, 64, 367–372. [Google Scholar] [CrossRef]
- Polytarchou, C.; Iliopoulos, D.; Struhl, K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc. Natl. Acad. Sci. USA 2012, 109, 14470–14475. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Relation to EMT | Target Gene | Experimentally Validated Inhibitory miRNA(s) | |
|---|---|---|---|
| epithelial markers | CDH1 | E-cadherin | miR-9 [100], miR-25 [102], miR-92a [103] |
| mesenchymal markers | CDH2 | N-cadherin | miR-194 [107], miR-199a [109], miR-145 [111] |
| VIM | vimentin | miR-30a [112] | |
| FN1 | fibronectin | miR-200b [115], miR-200c [114] | |
| transcription factors | SNAI1 | snail family transcriptional repressor 1 | miR-34a [118], miR-199a [109], miR-30a [119,120] |
| ZEB1 | zinc finger E-box binding homeobox 1 | miR-200 family [18,121] | |
| ZEB2 | zinc finger E-box binding homeobox 2 | miR-200 family [18,121], miR-192 [122] | |
| TWIST1 | twist family bHLH transcription factor 1 | miR-145 [123] | |
| matrix metalloproteinases | MMP2 | matrix metallopeptidase 2 | miR-29b [124] |
| MMP9 | matrix metallopeptidase 9 | miR-29b [125] | |
| JAK-STAT pathway | JAK2 | Janus kinase 2 | miR-375 [126,127] |
| NOTCH pathway | NOTCH1 | notch receptor 1 | miR-34a [128], miR-30a [129] |
| NOTCH2 | notch receptor 2 | miR-34a [128], miR-30a [129], miR-107 [130] | |
| WNT pathway | WNT3A | Wnt family member 3A | miR-491 [131] |
| TGFβ pathway | SMAD2 | SMAD family member 2 | miR-200b [99] |
| Tyrosin kinase receptor associated signaling | FGF2 | fibroblast growth factor 2 | miR-194 [132] |
| FGF7 | fibroblast growth factor 7 | miR-489 [133] | |
| AXL | AXL receptor tyrosine kinase | miR-34a [134,135], miR-199a [135], miR-92b [136] | |
| EGR1 | early growth response 1 | miR-192 [122], miR-181a [137] | |
| hypoxia | HIF1α | hypoxia inducible factor 1 | miR-199a [138,139], miR-107 [140] |
| LOX | lysyl oxidase | miR-200b [141], miR-30a [142] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boros, É.; Nagy, I. The Role of MicroRNAs upon Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease. Cells 2019, 8, 1461. https://doi.org/10.3390/cells8111461
Boros É, Nagy I. The Role of MicroRNAs upon Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease. Cells. 2019; 8(11):1461. https://doi.org/10.3390/cells8111461
Chicago/Turabian StyleBoros, Éva, and István Nagy. 2019. "The Role of MicroRNAs upon Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease" Cells 8, no. 11: 1461. https://doi.org/10.3390/cells8111461
APA StyleBoros, É., & Nagy, I. (2019). The Role of MicroRNAs upon Epithelial-to-Mesenchymal Transition in Inflammatory Bowel Disease. Cells, 8(11), 1461. https://doi.org/10.3390/cells8111461