BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Generation of Stable Cell Lines
2.2. Immunoblotting Analysis
2.3. Xenograft Model
2.4. Immunoprecipitation
2.5. Immunostaining
2.6. siRNA Targeting of Genes
2.7. Cell Viability Assay
2.8. Annexin V Binding by FACS
2.9. Cell Invasion Assay
2.10. Statistical Analysis
3. Results
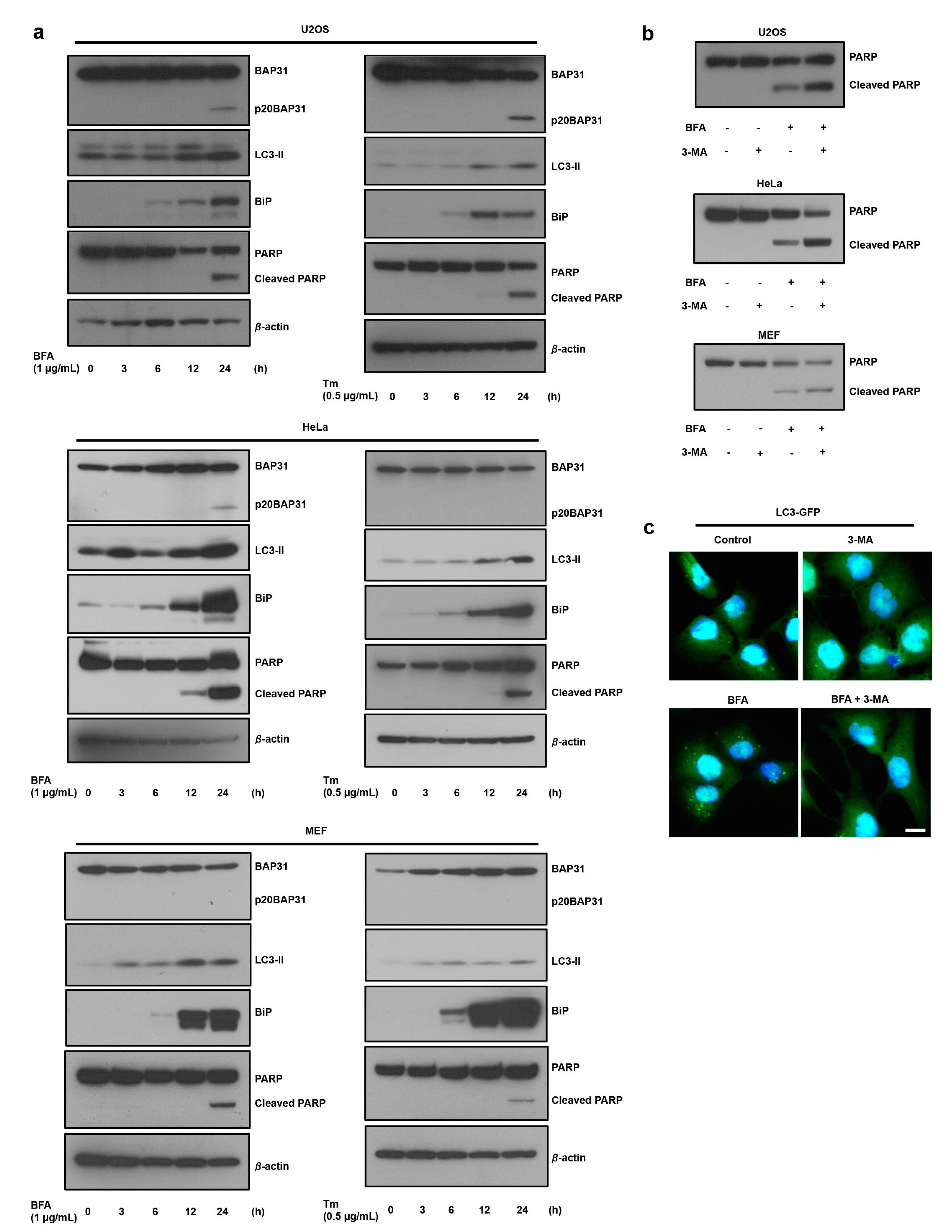
3.1. ER Stress Induces Autophagy, which Antagonizes Cell Death
3.2. The Loss of BAP31-Suppressed ER Stress-Induced Cell Death by Inducing Autophagy
3.3. Identification of STX17 as a BAP31 Binding Partner and How This Complex Regulates Induction of Autophagy via Suppression of STX17 and ATG14 Interaction
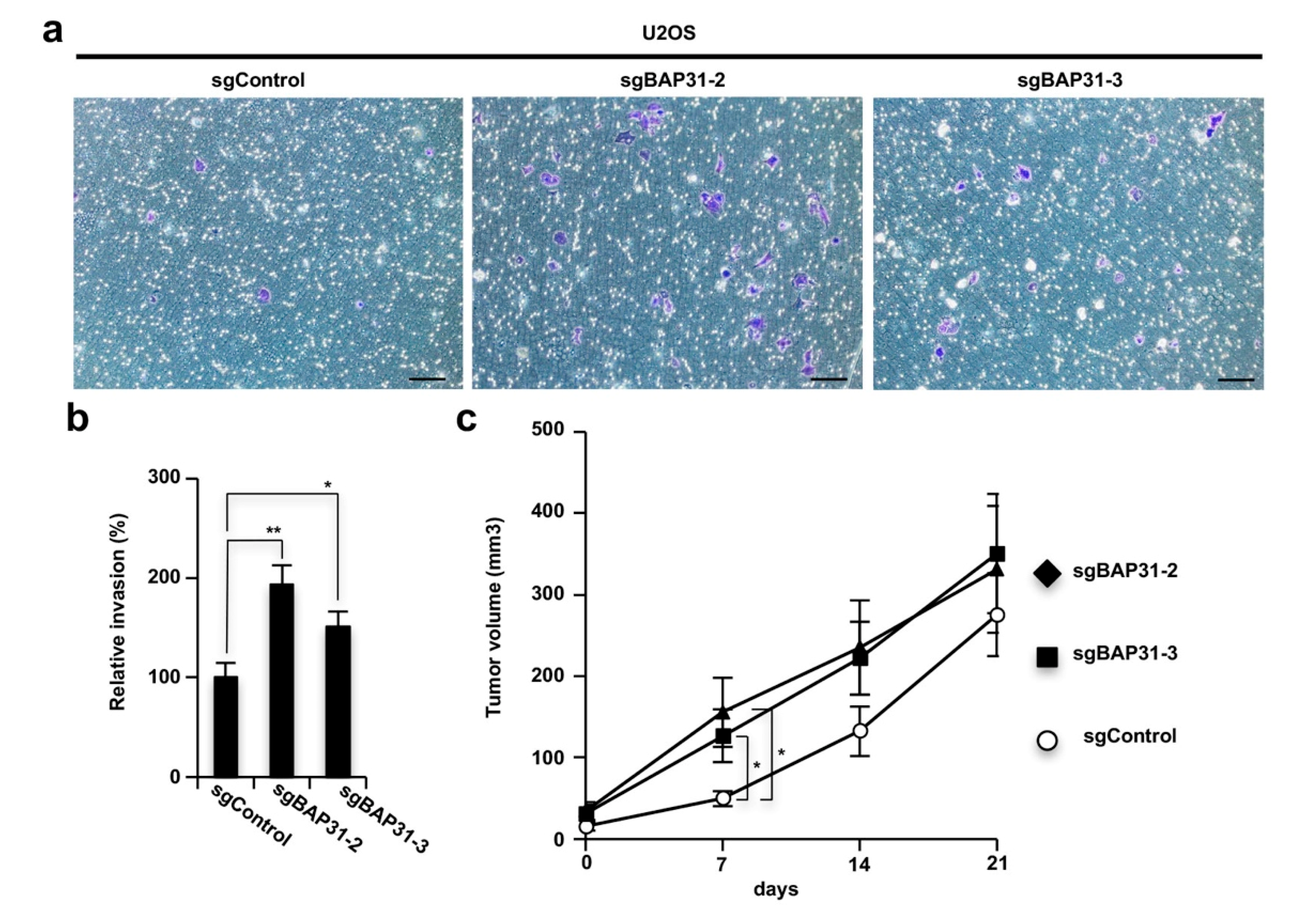
3.4. Loss of BAP31 Stimulates Tumor Invasion and Growth In Vitro and In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: Multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Nguyen, M.; Kuppig, S.; Reth, M.; Shore, G.C. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2002, 99, 4331–4336. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Ng, F.W.; Shore, G.C. Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J. Biol. Chem. 1998, 273, 3140–3143. [Google Scholar] [CrossRef]
- Ng, F.W.; Nguyen, M.; Kwan, T.; Branton, P.E.; Nicholson, D.W.; Cromlish, J.A.; Shore, G.C. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 1997, 139, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Heath-Engel, H.; Zhang, D.; Nguyen, N.; Thomas, D.Y.; Hanrahan, J.W.; Shore, G.C. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Kodama, R.; Moritomo, S.; Hoshino, T.; Mizushima, T. Zidovudine, an anti-viral drug, resensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine by inhibition of the Akt-GSK3beta-Snail pathway. Cell Death Dis. 2015, 6, e1795. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Rampino, G.; De Falco, F.; Di Ianni, M.; Falzetti, F.; Fettucciari, K.; Bartoli, A.; Screpanti, I.; Marconi, P. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 2010, 116, 2713–2723. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Arasaki, K.; Mikami, Y.; Shames, S.R.; Inoue, H.; Wakana, Y.; Tagaya, M. Legionella effector Lpg1137 shuts down ER-mitochondria communication through cleavage of syntaxin 17. Nat. Commun. 2017, 8, 15406. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, Y.; Cao, P. Inhibition of BAP31 expression inhibits cervical cancer progression by suppressing metastasis and inducing intrinsic and extrinsic apoptosis. Biochem. Biophys. Res. Commun. 2019, 508, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.; Yang, S.; Song, C.; Jiang, D.; Li, Z.; Fan, W.; Sun, Y.; Tao, L.; Wang, J.; Liu, T.; et al. BAP31, a newly defined cancer/testis antigen, regulates proliferation, migration, and invasion to promote cervical cancer progression. Cell Death Dis. 2018, 9, 791. [Google Scholar] [CrossRef]
- Regan, J.A.; Laimins, L.A. Bap31 is a novel target of the human papillomavirus E5 protein. J. Virol. 2008, 82, 10042–10051. [Google Scholar] [CrossRef]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef]
- Xu, K.; Han, B.; Bai, Y.; Ma, X.Y.; Ji, Z.N.; Xiong, Y.; Miao, S.K.; Zhang, Y.Y.; Zhou, L.M. MiR-451a suppressing BAP31 can inhibit proliferation and increase apoptosis through inducing ER stress in colorectal cancer. Cell Death Dis. 2019, 10, 152. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machihara, K.; Namba, T. BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17. Cells 2019, 8, 1350. https://doi.org/10.3390/cells8111350
Machihara K, Namba T. BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17. Cells. 2019; 8(11):1350. https://doi.org/10.3390/cells8111350
Chicago/Turabian StyleMachihara, Kayo, and Takushi Namba. 2019. "BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17" Cells 8, no. 11: 1350. https://doi.org/10.3390/cells8111350
APA StyleMachihara, K., & Namba, T. (2019). BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17. Cells, 8(11), 1350. https://doi.org/10.3390/cells8111350