MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy?



Abstract
1. Rearrangements of the MLL Gene in Leukemia
2. Leukemia with t(4;11)(q21;q23): Clinical Picture and Risk Stratification
3. Current Therapy for MLL-r Acute Leukemia Patients
3.1. Cytotoxic and Cytoreductive Chemotherapy
3.2. Other Treatment Strategies
3.3. Hematopoietic Stem Cell Transplant
3.4. Immunotherapy
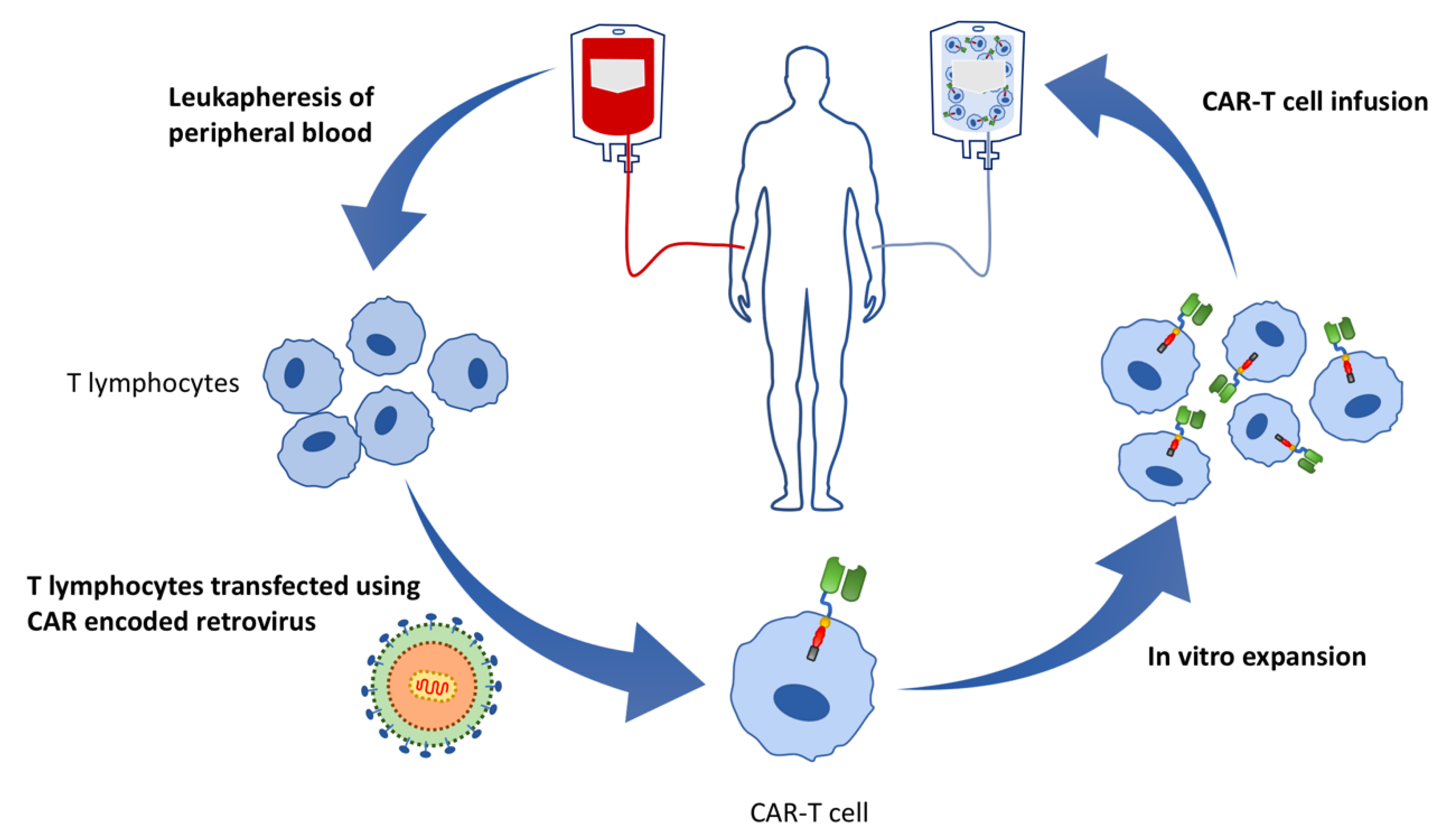
4. What Is CAR-T?
5. Treatment of MLL-r with CAR-T Cell Therapy: Advantages and Challenges
5.1. Immune Escape
5.2. Target Specificity and Alternative Antigens
5.3. Toxicity
5.4. Persistence and Long-Term Effects
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Moorman, A.V.; Ensor, H.M.; Richards, S.M.; Chilton, L.; Schwab, C.; Kinsey, S.E.; Vora, A.; Mitchell, C.D.; Harrison, C.J. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: Results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010, 11, 429–438. [Google Scholar] [CrossRef]
- Behm, F.G.; Raimondi, S.C.; Frestedt, J.L.; Liu, Q.; Crist, W.M.; Downing, J.R.; Rivera, G.K.; Kersey, J.H.; Pui, C.H. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood 1996, 87, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.C.; Frestedt, J.L.; Pui, C.H.; Downing, J.R.; Head, D.R.; Kersey, J.H.; Behm, F.G. Acute lymphoblastic leukemias with deletion of 11q23 or a novel inversion (11)(p13q23) lack MLL gene rearrangements and have favorable clinical features. Blood 1995, 86, 1881–1886. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Look, A.T. Molecular Genetics of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2005, 23, 6306–6315. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Link, M.P.; Shuster, J.J.; Carroll, A.J.; Hakami, N.; Frankel, L.S.; Pullen, D.J.; Cleary, M.L. Frequency and prognostic significance of HRX rearrangements in infant acute lymphoblastic leukemia: A Pediatric Oncology Group study. Blood 1994, 84, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Sam, T.N.; Kersey, J.H.; Linabery, A.M.; Johnson, K.J.; Heerema, N.A.; Hilden, J.M.; Davies, S.M.; Reaman, G.H.; Ross, J.A. MLL gene rearrangements in infant leukemia vary with age at diagnosis and selected demographic factors: A Children’s Oncology Group (COG) study. Pediatr. Blood Cancer 2011, 58, 836–839. [Google Scholar] [CrossRef]
- Shih, L.Y.; Liang, D.C.; Fu, J.F.; Wu, J.H.; Wang, P.N.; Lin, T.L.; Dunn, P.; Kuo, M.C.; Tang, T.C.; Lin, T.H.; et al. Characterization of fusion partner genes in 114 patients with de novo acute myeloid leukemia and MLL rearrangement. Leukemia 2006, 20, 218–223. [Google Scholar] [CrossRef]
- Palle, J.; Frost, B.M.; Forestier, E.; Gustafsson, G.; Nygren, P.; Hellebostad, M.; Jonsson, O.G.; Kanerva, J.; Schmiegelow, K.; Larsson, R.; et al. Cellular drug sensitivity in MLL-rearranged childhood acute leukaemia is correlated to partner genes and cell lineage. Br. J. Haematol. 2005, 129, 189–198. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1,612 Patients Entered Into the MRC AML 10 Trial. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef]
- Raimondi, S.C.; Chang, M.N.; Ravindranath, Y.; Behm, F.G.; Gresik, M.V.; Steuber, C.P.; Weinstein, H.J.; Carroll, A.J. Chromosomal abnormalities in 478 children with acute myeloid leukemia: Clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 1999, 94, 3707–3716. [Google Scholar] [PubMed]
- Pais, A.; Amare Kadam, P.; Raje, G.; Sawant, M.; Kabre, S.; Jain, H.; Advani, S.; Banavali, S. Identification of various MLL gene aberrations that lead to MLL gene mutation in patients with acute lymphoblastic leukemia (ALL) and infants with acute leukemia. Leuk. Res. 2005, 29, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Pession, A.; Locatelli, F. Acute Myeloid Leukemia in Infants: Biology and Treatment. Front. Pediatr. 2015, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Frankel, L.S.; Carroll, A.J.; Raimondi, S.C.; Shuster, J.J.; Head, D.R.; Crist, W.M.; Land, V.J.; Pullen, D.J.; Steuber, C.P. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia with the t(4;11)(q21;q23): A collaborative study of 40 cases. Blood 1991, 77, 440–447. [Google Scholar] [CrossRef]
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the WHO classification: Incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003, 102, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Tamai, H.; Inokuchi, K. 11q23/MLL acute leukemia: Update of clinical aspects. J. Clin. Exp. Hematop. 2010, 50, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer. 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Bueno, C.; Montes, R.; Catalina, P.; Rodríguez, R.; Menendez, P. Insights into the cellular origin and etiology of the infant pro-B acute lymphoblastic leukemia with MLL-AF4 rearrangement. Leukemia 2011, 25, 400–410. [Google Scholar] [CrossRef]
- Rossi, J.G.; Bernasconi, A.R.; Alonso, C.N.; Rubio, P.L.; Gallego, M.S.; Carrara, C.A.; Guitter, M.R.; Eberle, S.E.; Cocce, M.; Zubizarreta, P.A.; et al. Lineage switch in childhood acute leukemia: An unusual event with poor outcome. Am. J. Hematol. 2012, 87, 890–897. [Google Scholar] [CrossRef]
- Rayes, A.; McMasters, R.L.; O’Brien, M.M. Lineage Switch in MLL-Rearranged Infant Leukemia Following CD19-Directed Therapy. Pediatr. Blood Cancer 2016, 63, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, R.; Nilson, I.; Löchner, K.; Greim, R.; Siegler, G.; Greil, J.; Beck, J.D.; Fey, G.H. The structure of the human ALL-1/MLL/HRX gene. Leuk. Lymphoma 1997, 27, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, R. Systematic Classification of Mixed-Lineage Leukemia Fusion Partners Predicts Additional Cancer Pathways. Ann. Lab. Med. 2016, 36, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A. Molecular mechanisms of MLL-associated leukemia. Int. J. Hematol. 2015, 101, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef]
- Blütters-Sawatzki, R.; Borkhardt, A.; Grathwohl, J.; Repp, R.; Rheinisch-Becker, I.; Bohle, R.M.; Lampert, F. Secondary acute myeloid leukemia with translocation (4;11) and MLL/AF4 rearrangement in a 15-year-old boy treated for common acute lymphoblastic leukemia 11 years earlier. Ann. Hematol. 1995, 70, 31–35. [Google Scholar] [CrossRef]
- Aoki, Y.; Watanabe, T.; Saito, Y.; Kuroki, Y.; Hijikata, A.; Takagi, M.; Tomizawa, D.; Eguchi, M.; Eguchi-Ishimae, M.; Kaneko, A.; et al. Identification of CD34+ and CD34− leukemia-initiating cells in MLL-rearranged human acute lymphoblastic leukemia. Blood 2015, 125, 967–980. [Google Scholar] [CrossRef]
- Pui, C.H. Acute leukemias with the t(4;11)(q21;q23). Leuk. Lymphoma 1992, 7, 173–179. [Google Scholar] [CrossRef]
- Domer, P.H.; Fakharzadeh, S.S.; Chen, C.S.; Jockel, J.; Johansen, L.; Silverman, G.A.; Kersey, J.H.; Korsmeyer, S.J. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc. Natl. Acad. Sci. 1993, 90, 7884–7888. [Google Scholar] [CrossRef]
- Downing, J.R.; Head, D.R.; Raimondi, S.C.; Carroll, A.J.; Curcio-Brint, A.M.; Motroni, T.A.; Hulshof, M.G.; Pullen, D.J.; Domer, P.H. The der(11)-encoded MLL/AF-4 fusion transcript is consistently detected in t(4;11)(q21;q23)-containing acute lymphoblastic leukemia. Blood 1994, 83, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Montes, R.; Ayllón, V.; Gutierrez-Aranda, I.; Prat, I.; Hernández-Lamas, M.C.; Ponce, L.; Bresolin, S.; Te Kronnie, G.; Greaves, M.; Bueno, C.; et al. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood 2011, 117, 4746–4758. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tamai, H.; Miyake, K.; Takatori, M.; Miyake, N.; Yamaguchi, H.; Dan, K.; Shimada, T.; Inokuchi, K. Activated K-Ras protein accelerates human MLL AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia 2011, 25, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, S.E.; Sherborne, A.L.; Ma, Y.P.; Bardini, M.; Biondi, A.; Cazzaniga, G.; Lloyd, A.; Chubb, D.; Greaves, M.F.; Houlston, R.S. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosom. Cancer 2013, 52, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W. MLL-AF4 driven leukemogenesis: What are we missing? Cell Res. 2012, 22, 948–949. [Google Scholar] [CrossRef]
- Bursen, A.; Schwabe, K.; Rüster, B.; Henschler, R.; Ruthardt, M.; Dingermann, T.; Marschalek, R. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood 2010, 115, 3570–3579. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ballabio, E.; Geng, H.; North, P.; Tapia, M.; Kerry, J.; Biswas, D.; Roeder, R.G.; Allis, C.D.; Melnick, A.; et al. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013, 3, 116–127. [Google Scholar] [CrossRef]
- Kumar, A.R.; Yao, Q.; Li, Q.; Sam, T.A.; Kersey, J.H. t(4;11) leukemias display addiction to MLL-AF4 but not to AF4-MLL. Leuk. Res. 2011, 35, 305–309. [Google Scholar] [CrossRef][Green Version]
- Prieto, C.; Marschalek, R.; Kühn, A.; Bursen, A.; Bueno, C.; Menéndez, P. The AF4-MLL fusion transiently augments multilineage hematopoietic engraftment but is not sufficient to initiate leukemia in cord blood CD34+ cells. Oncotarget 2017, 8, 81936–81941. [Google Scholar] [CrossRef][Green Version]
- Pui, C.H.; Chessells, J.M.; Camitta, B.; Baruchel, A.; Biondi, A.; Boyett, J.M.; Carroll, A.; Eden, O.B.; Evans, W.E.; Gadner, H.; et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 2003, 17, 700–706. [Google Scholar] [CrossRef]
- Marks, D.I.; Moorman, A.V.; Chilton, L.; Paietta, E.; Enshaie, A.; DeWald, G.; Harrison, C.J.; Fielding, A.K.; Foroni, L.; Goldstone, A.H.; et al. The clinical characteristics, therapy and outcome of 85 adults with acute lymphoblastic leukemia and t(4;11)(q21;q23)/MLL-AFF1 prospectively treated in the UKALLXII/ECOG2993 trial. Haematologica 2013, 98, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.; Gaynon, P.S.; Boyett, J.M.; Chessells, J.M.; Baruchel, A.; Kamps, W.; Silverman, L.B.; Biondi, A.; Harms, D.; Vilmer, E.; et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 2002, 359, 1909–1915. [Google Scholar] [CrossRef]
- Mann, G.; Attarbaschi, A.; Schrappe, M.; De Lorenzo, P.; Peters, C.; Hann, I.; De Rossi, G.; Felice, M.; Lausen, B.; LeBlanc, T.; et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)–rearranged acute lymphoblastic leukemia: Results from the Interfant-99 Study. Blood 2010, 116, 2644–2650. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, P.; Moorman, A.V.; Pieters, R.; Dreyer, Z.E.; Heerema, N.A.; Carroll, A.J.; Hunger, S.P.; Harvey, R.; Willman, C.L.; Devidas, M.; et al. Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 2014, 28, 428–430. [Google Scholar] [CrossRef]
- Kang, H.; Wilson, C.S.; Harvey, R.C.; Chen, I.M.; Murphy, M.H.; Atlas, S.R.; Bedrick, E.J.; Devidas, M.; Carroll, A.J.; Robinson, B.W.; et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2012, 119, 1872–1881. [Google Scholar] [CrossRef]
- Kistler, M.; Schiller, G. Acute Leukemia and Myelodysplastic Syndromes. In Manual of Clinical Oncology, 8th ed.; Chmielowski, B., Territo, M., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2017; pp. 592–593. [Google Scholar]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef]
- Reaman, G.H.; Sposto, R.; Sensel, M.G.; Lange, B.J.; Feusner, J.H.; Heerema, N.A.; Leonard, M.; Holmes, E.J.; Sather, H.N.; Pendergrass, T.W.; et al. Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children’s Cancer Group. J. Clin. Oncol. 1999, 17, 445–455. [Google Scholar] [CrossRef]
- Heerema, N.A.; Arthur, D.C.; Sather, H.; Albo, V.; Feusner, J.; Lange, B.J.; Steinherz, P.G.; Zeltzer, P.; Hammond, D.; Reaman, G.H. Cytogenetic features of infants less than 12 months of age at diagnosis of acute lymphoblastic leukemia: Impact of the 11q23 breakpoint on outcome: A report of the Childrens Cancer Group. Blood 1994, 83, 2274–2284. [Google Scholar] [CrossRef]
- Chessells, J.M.; Hall, E.; Prentice, H.G.; Durrant, J.; Bailey, C.C.; Richards, S.M. The impact of age on outcome in lymphoblastic leukaemia; MRC UKALL X and XA compared: A report from the MRC Paediatric and Adult Working Parties. Leukemia 1998, 12, 463–473. [Google Scholar] [CrossRef]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction therapy for adults with acute lymphoblastic leukemia: Results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Burnett, A.K.; on behalf of the National Cancer Research Institute Adult Leukaemia Working Group; Hills, R.K.; Moorman, A.V.; Harrison, C.J.; et al. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Tomizawa, D.; Koh, K.; Hirayama, M.; Miyamura, T.; Hatanaka, M.; Saikawa, Y.; Ishii, E. Outcome of recurrent or refractory acute lymphoblastic leukemia in infants with MLL gene rearrangements: A report from the Japan infant leukemia study group. Pediatric. Blood Cancer 2009, 52, 808–813. [Google Scholar] [CrossRef]
- Badar, T.; Kantarjian, H.M.; O’Brien, S.; Garcia-Manero, G.; Jabbour, E.; Garris, R.; Pemmaraju, N.; Daver, N.; Ravandi, F.; Cortes, J.; et al. Clinical Outcome of De Novo Adult Acute Lymphoblastic Leukemia (ALL) with 11q23/Mixed Lineage Leukemia (MLL) Gene Rearrangements. Blood 2014, 124, 5342. [Google Scholar] [CrossRef]
- Kersey, J.H.; Wang, D.; Oberto, M. Resistance of t(4;11) (MLL-AF4 fusion gene) leukemias to stress-induced cell death: Possible mechanism for extensive extramedullary accumulation of cells and poor prognosis. Leukemia 1998, 12, 1561–1564. [Google Scholar] [CrossRef]
- Gaussmann, A.; Wenger, T.; Eberle, I.; Bursen, A.; Bracharz, S.; Herr, I.; Dingermann, T.; Marschalek, R. Combined effects of the two reciprocal t(4;11) fusion proteins MLL.AF4 and AF4.MLL confer resistance to apoptosis, cell cycling capacity and growth transformation. Oncogene 2007, 26, 3352–3363. [Google Scholar] [CrossRef]
- Stam, R.W.; den Boer, M.L.; Meijerink, J.P.P.; Ebus, M.E.G.; Peters, G.J.; Noordhuis, P.; Janka-Schaub, G.E.; Armstrong, S.A.; Korsmeyer, S.J.; Pieters, R. Differential mRNA expression of Ara-C–metabolizing enzymes explains Ara-C sensitivity in MLL gene–rearranged infant acute lymphoblastic leukemia. Blood 2003, 101, 1270–1276. [Google Scholar] [CrossRef]
- Brown, P.; Pieters, R.; Biondi, A. How I treat infant leukemia. Blood 2019, 133, 205–214. [Google Scholar] [CrossRef]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.H.; Chopra, R.; Buck, G.; Richards, M.; Franklin, J.M.; Lazarus, H.M.; Wiemik, P.H.; Tallman, M.S.; Prentice, H.G.; Durrant, J.; et al. The outcome of 267 Philadelphia positive adults in the international UKALL12/ECOG E 2993 study. Final analysis and the role of allogeneic transplant in those under 50 years. 2003, 102, 80A. [Google Scholar]
- Barry, E.; DeAngelo, D.J.; Neuberg, D.; Stevenson, K.; Loh, M.L.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Hurwitz, C.A.; Moghrabi, A.; et al. Favorable Outcome for Adolescents with Acute Lymphoblastic Leukemia Treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J. Clin. Oncol. 2007, 25, 813–819. [Google Scholar] [CrossRef]
- Kliman, D.S.; Barnett, M.J.; Broady, R.; Forrest, D.L.; Gerrie, A.S.; Hogge, D.E.; Nantel, S.H.; Narayanan, S.; Nevill, T.J.; Power, M.M.; et al. Pediatric-Based Versus Adult Treatment Protocols in Young Adults (18-40 years) with Standard Risk Acute Lymphoblastic Leukemia: The BC Cancer Agency Experience. Blood 2015, 126, 3770. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.; Hills, R.K.; Kell, J.; Cavenagh, J.; Kjeldsen, L.; McMullin, M.F.; Cahalin, P.; Dennis, M.; Milligan, D.; et al. A Randomised Comparison of Daunorubicin 90mg/m2 Vs 60mg/m2 in AML Induction: Results from the UK NCRI AML17 Trial in 1206 Patients. Blood 2014, 124, 7. [Google Scholar] [CrossRef]
- Dreyer, Z.E.; Hilden, J.M.; Jones, T.L.; Devidas, M.; Winick, N.J.; Willman, C.L.; Harvey, R.C.; Chen, I.; Behm, F.G.; Pullen, J.; et al. Intensified chemotherapy without SCT in infant ALL: Results from COG P9407 (Cohort 3). Pediatric. Blood Cancer 2015, 62, 419–426. [Google Scholar] [CrossRef]
- Salzer, W.L.; Jones, T.L.; Devidas, M.; Dreyer, Z.E.; Gore, L.; Winick, N.J.; Sung, L.; Raetz, E.; Loh, M.L.; Wang, C.Y.; et al. Decreased Induction Morbidity and Mortality Following Modification to Induction Therapy in Infants with Acute Lymphoblastic Leukemia Enrolled on AALL0631: A Report from the Children’s Oncology Group. Pediatric. Blood Cancer 2014, 62, 414–418. [Google Scholar] [CrossRef]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur. J. Pharmacol. 2014, 723, 276–287. [Google Scholar] [CrossRef]
- Stumpel, Dominique, J.P.M.; Schneider, P.; Pieters, R.; Stam, R.W. The potential of clofarabine in MLL -rearranged infant acute lymphoblastic leukaemia. Eur. J. Cancer 2015, 51, 2008–2021. [Google Scholar] [CrossRef]
- Roolf, C.; Richter, A.; Konkolefski, C.; Knuebel, G.; Sekora, A.; Krohn, S.; Stenzel, J.; Krause, B.J.; Vollmar, B.; Murua Escobar, H.; et al. Decitabine demonstrates antileukemic activity in B cell precursor acute lymphoblastic leukemia with MLL rearrangements. J. Hematol. Oncol. 2018, 11, 62. [Google Scholar] [CrossRef]
- Xu, X.; Schneider, B. Therapeutic targeting potential of chromatin-associated proteins in MLL-rearranged acute leukemia. Cell. Oncol. 2019, 42, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Wetmore, C.; O’Brien, M.M.; Silverman, L.B.; Brown, P.; Cooper, T.M.; Thomson, B.; Blakemore, S.J.; Daigle, S.; Suttle, B.; et al. Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood 2016, 128, 2780. [Google Scholar] [CrossRef]
- Bhatla, T.; Wang, J.; Morrison, D.J.; Raetz, E.A.; Burke, M.J.; Brown, P.; Carroll, W.L. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 2012, 119, 5201–5210. [Google Scholar] [CrossRef]
- Stumpel, D.J.P.M.; Schneider, P.; Seslija, L.; Osaki, H.; Williams, O.; Pieters, R.; Stam, R.W. Connectivity mapping identifies HDAC inhibitors for the treatment of t(4;11)-positive infant acute lymphoblastic leukemia. Leukemia 2012, 26, 682–692. [Google Scholar] [CrossRef]
- Garrido Castro, P.; van Roon, E.H.J.; Pinhanços, S.S.; Trentin, L.; Schneider, P.; Kerstjens, M.; te Kronnie, G.; Heidenreich, O.; Pieters, R.; Stam, R.W. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2018, 32, 323–331. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Prelle, C.; Bursen, A.; Dingermann, T.; Marschalek, R. Secondary mutations in t(4;11) leukemia patients. Leukemia 2013, 27, 1425–1427. [Google Scholar] [CrossRef]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Taketani, T.; Taki, T.; Sugita, K.; Furuichi, Y.; Ishii, E.; Hanada, R.; Tsuchida, M.; Sugita, K.; Ida, K.; Hayashi, Y. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood 2004, 103, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; den Boer, M.L.; Schneider, P.; Meier, M.; Beverloo, H.B.; Pieters, R. D-HPLC analysis of the entire FLT3 gene in MLL rearranged and hyperdiploid acute lymphoblastic leukemia. Haematologica 2007, 92, 1565–1568. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bardini, M.; Galbiati, M.; Lettieri, A.; Bungaro, S.; Gorletta, T.A.; Biondi, A.; Cazzaniga, G. Implementation of array based whole-genome high-resolution technologies confirms the absence of secondary copy-number alterations in MLL-AF4-positive infant ALL patients. Leukemia 2011, 25, 175–178. [Google Scholar] [CrossRef]
- Chillón, M.C.; Gómez-Casares, M.T.; López-Jorge, C.E.; Rodriguez-Medina, C.; Molines, A.; Sarasquete, M.E.; Alcoceba, M.; Miguel, J.D.G.-S.; Bueno, C.; Montes, R.; et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia 2012, 26, 2360–2366. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lawton, L.N.; Rozovskaia, T.; Frampton, G.M.; Levine, S.S.; Volkert, T.L.; Croce, C.M.; Nakamura, T.; Canaani, E.; Young, R.A. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008, 22, 3403–3408. [Google Scholar] [CrossRef]
- Bueno, C.; Ayllón, V.; Montes, R.; Navarro-Montero, O.; Ramos-Mejia, V.; Real, P.J.; Romero-Moya, D.; Araúzo-Bravo, M.J.; Menendez, P. FLT3 activation cooperates with MLL-AF4 fusion protein to abrogate the hematopoietic specification of human ESCs. Blood 2013, 121, 3867–3878. [Google Scholar] [CrossRef]
- Brown, P.; Kairalla, J.; Wang, C.; Dreyer, Z.; Salzer, W.; Sorenson, M.; Borowitz, M.; Carroll, A.; Heerema, N.; Rao, K.; et al. Addition of FLT3 inhibitor lestaurtinib to post-induction chemotherapy does not improve outcomes in MLL-rearranged infant acute lymphoblastic leukemia (ALL): AALL0631, a Children’s Oncology Group study. Pediatr. Blood Cancer 2016, 63, S7–S10. [Google Scholar]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A Phase I Study of Quizartinib Combined with Chemotherapy in Relapsed Childhood Leukemia: A Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Clin. Cancer Res. 2016, 22, 4014–4022. [Google Scholar]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Hunger, S.P.; Loh, K.M.; Baker, K.S.; Schultz, K.R. Controversies of and unique issues in hematopoietic cell transplantation for infant leukemia. Boil. Blood Marrow Transplant. 2009, 15, 79–83. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sison, E.A.R.; Brown, P. Does hematopoietic stem cell transplantation benefit infants with acute leukemia? Hematology ASH Educ. Program 2013, 2013, 601–604. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chessells, J.M.; Harrison, C.J.; Watson, S.L.; Vora, A.J.; Richards, S.M. Treatment of infants with lymphoblastic leukaemia: Results of the UK Infant Protocols 19871999. Br. J. Haematol. 2002, 117, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, Z.E.; Dinndorf, P.A.; Camitta, B.; Sather, H.; La, M.K.; Devidas, M.; Hilden, J.M.; Heerema, N.A.; Sanders, J.E.; McGlennen, R.; et al. Analysis of the Role of Hematopoietic Stem-Cell Transplantation in Infants With Acute Lymphoblastic Leukemia in First Remission and MLL Gene Rearrangements: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2010, 29, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.E.; Im, H.J.; Hoffmeister, P.A.; Gooley, T.A.; Woolfrey, A.E.; Carpenter, P.A.; Andrews, R.G.; Bryant, E.M.; Appelbaum, F.R. Allogeneic hematopoietic cell transplantation for infants with acute lymphoblastic leukemia. Blood 2005, 105, 3749–3756. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kosaka, Y.; Koh, K.; Kinukawa, N.; Wakazono, Y.; Isoyama, K.; Oda, T.; Hayashi, Y.; Ohta, S.; Moritake, H.; Oda, M.; et al. Infant acute lymphoblastic leukemia with MLL gene rearrangements: Outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 2004, 104, 3527–3534. [Google Scholar] [CrossRef] [PubMed]
- Marco, F.; Bureo, E.; Ortega, J.J.; Badell, I.; Verdaguer, A.; Martínez, A.; Muñoz, A.; Madero, L.; Olivé, T.; Cubells, J.; et al. High Survival Rate in Infant Acute Leukemia Treated With Early High-Dose Chemotherapy and Stem-Cell Support. J. Clin. Oncol. 2000, 18, 3256–3261. [Google Scholar] [CrossRef]
- Yu, W.; Qi-Fa, L.; Ya-Zhen, Q.; Dai-Hong, L.; Lan-Ping, X.; Bin, J.; Qian, J.; Min, D.; Si-Jian, Y.; Xin-Miao, J.; et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of patients with mixed-lineage-leukemia-rearranged acute leukemia: Results from a prospective, multi-center study. Am. J. Hematol. 2014, 89, 130–136. [Google Scholar] [CrossRef]
- Wetzler, M.; Dodge, R.K.; Mrózek, K.; Carroll, A.J.; Tantravahi, R.; Block, A.W.; Pettenati, M.J.; Le Beau, M.M.; Frankel, S.R.; Stewart, C.C.; et al. Prospective karyotype analysis in adult acute lymphoblastic leukemia: The cancer and leukemia Group B experience. Blood 1999, 93, 3983–3993. [Google Scholar]
- Thomas, X.; Boiron, J.; Huguet, F.; Dombret, H.; Bradstock, K.; Vey, N.; Kovacsovics, T.; Delannoy, A.; Fegueux, N.; Fenaux, P.; et al. Outcome of Treatment in Adults With Acute Lymphoblastic Leukemia: Analysis of the LALA-94 Trial. J. Clin. Oncol. 2004, 22, 4075–4086. [Google Scholar] [CrossRef]
- Konuma, T.; Mizuno, S.; Kondo, T.; Yamaguchi, H.; Fukuda, T.; Uchida, N.; Najima, Y.; Kanamori, H.; Ota, S.; Nakamae, H.; et al. Adult Acute Myeloid Leukemia Working Group of the Japan Society for Hematopoietic, Cell Transplantation Allogeneic hematopoietic cell transplantation in adult acute myeloid leukemia with 11q23 abnormality: A retrospective study of the Adult Acute Myeloid Leukemia Working Group of the Japan Society for Hematopoietic Cell Transplantation (JSHCT). Ann. Hematol. 2018, 97, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Spinelli, O.; Oldani, E.; Intermesoli, T.; Tosi, M.; Peruta, B.; Rossi, G.; Borlenghi, E.; Pogliani, E.M.; Terruzzi, E.; et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood 2009, 113, 4153–4162. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.; Hudson, M.; Zhu, Y.; Rivera, G.K.; Ribeiro, R.C.; Sandlund, J.T.; Bowman, L.C.; Evans, W.E.; Kun, L.; Pui, C.H. Late effects in survivors of infant leukemia. Leukemia 2000, 14, 1185–1190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kersey, J.; Kolb, H.J.; Rimm, A.A.; Ringdén, O.; Rozman, C.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar] [CrossRef]
- Dickinson, A.M.; Norden, J.; Li, S.; Hromadnikova, I.; Schmid, C.; Schmetzer, H.; Jochem-Kolb, H. Graft-versus-Leukemia Effect Following Hematopoietic Stem Cell Transplantation for Leukemia. Front. Immunol. 2017, 8, 496. [Google Scholar] [CrossRef]
- Tamai, H.; Miyake, K.; Yamaguchi, H.; Okabe, M.; Dan, K.; Inokuchi, K.; Shimada, T. MLL/AF4 Positive Acute Lymphoblastic Leukemia Has Resistance to Tumor Necrosis Factor-Alpha Caused by up-Regration of S100A6. Blood 2010, 116, 2477. [Google Scholar] [CrossRef]
- von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Boil. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- An, N.; Tao, Z.; Li, S.; Xing, H.; Tang, K.; Tian, Z.; Rao, Q.; Wang, M.; Wang, J. Construction of a new anti-CD19 chimeric antigen receptor and the anti-leukemia function study of the transduced T cells. Oncotarget 2016, 7, 10638–10649. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Shrestha, G.; Robison, R.A.; O’Neill, K.L. The expansion of targetable biomarkers for CAR T cell therapy. J. Exp. Clin. Cancer Res. 2018, 37, 163. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, R.H.; Racila, E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef]
- Park, J.; Riviere, I.; Wang, X.; Bernal, Y.; Purdon, T.; Halton, E.; Curran, K.; Sauter, C.; Sadelain, M.; Brentjens, R. Efficacy and safety of CD19-targeted 19-28z CAR modified T cells in adult patients with relapsed or refractory B-ALL. Haematologica 2015, 33, 7010. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Jain, M.D.; Bachmeier, C.A.; Phuoc, V.H.; Chavez, J.C. Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma. Ther. Clin. Risk Manag. 2018, 14, 1007–1017. [Google Scholar] [CrossRef]
- Muhammad, N.; Mao, Q.; Xia, H. CAR T-cells for cancer therapy. Biotechnol. Genet. Eng. Rev. 2017, 33, 190–226. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Haddox, C.L.; Mangaonkar, A.A.; Chen, D.; Shi, M.; He, R.; Oliveira, J.L.; Litzow, M.R.; Al-Kali, A.; Hogan, W.J.; Elliott, M.A. Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: Pre- and post-switch phenotypic, cytogenetic and molecular analysis. Blood Cancer J. 2017, 7, e607. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Luo, R.T.; Ptasinska, A.; Kerry, J.; Assi, S.A.; Wunderlich, M.; Imamura, T.; Kaberlein, J.J.; Rayes, A.; Althoff, M.J.; et al. Instructive Role of MLL-Fusion Proteins Revealed by a Model of t(4;11) Pro-B Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Lucero, O.M.; Parker, K.; Funk, T.; Dunlap, J.; Press, R.; Gardner, R.A.; Chang, B.H. Phenotype switch in acute lymphoblastic leukaemia associated with 3 years of persistent CAR T cell directed-CD19 selective pressure. Br. J. Haematol. 2019, 186, 333–336. [Google Scholar] [CrossRef]
- Lin, S.; Luo, R.T.; Shrestha, M.; Thirman, M.J.; Mulloy, J.C. The full transforming capacity of MLL-Af4 is interlinked with lymphoid lineage commitment. Blood 2017, 130, 903–907. [Google Scholar] [CrossRef]
- Heidenreich, O.; Tirtakusuma, R.; Bomken, S.; Williamson, D.; Fordham, S.; McNeill, H.; Meyer, C.; Marschalek, R.; Vormoor, J.; Hall, A.; et al. The Genomic Landscape of Lineage Switch Acute Leukemia. Blood 2013, 122, 2552. [Google Scholar] [CrossRef]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef]
- Tirtakusuma, R.; Milne, P.; Ptasinska, A.; Meyer, C.; Nakjang, S.; Komkov, A.; Williamson, D.; Cauchy, P.; Assi, S.; Blair, H.; et al. Epigenetic Regulator Genes Direct the Fate of Multipotent Progenitor Cell of Origin in Lineage Switched MLL-AF4 Leukaemia. Available online: http:// dx.doi.org/10.2139/ssrn.3432467 (accessed on 23 October 2019).
- Cohen, A.; Petsche, D.; Grunberger, T.; Freedman, M.H. Interleukin 6 induces myeloid differentiation of a human biphenotypic leukemic cell line. Leuk. Res. 1992, 16, 751–760. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther.-Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I.; et al. Long-Lived Plasma Cells Are Contained within the CD19(−)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity 2015, 43, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Arhontoulis, D.; Wertheim, G.; Capobianchi, J.; Callahan, C.A.; Ellebrecht, C.T.; Obstfeld, A.E.; Lacey, S.F.; Melenhorst, J.J.; Nazimuddin, F.; et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 2016, 128, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Maus, M.V.; Hwang, W.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Danhof, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Cox, C.V.; Evely, R.S.; Oakhill, A.; Pamphilon, D.H.; Goulden, N.J.; Blair, A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood 2004, 104, 2919–2925. [Google Scholar] [CrossRef]
- Hofmann, S.; Schubert, M.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J Clin. Med 2019, 8, 200. [Google Scholar] [CrossRef]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 1–13. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Stevenson, M.S.; Yuan, C.M.; Richards, K.; Delbrook, C.; Kreitman, R.J.; Pastan, I.; Wayne, A.S. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2015, 62, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Wang, Z.; Wang, Y.; Liu, Y.; Dai, H.; Tong, C.; Guo, Y.; Guo, B.; Ti, D.; Han, X.; et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hu, Y.; Jin, Z.; Zhai, Y.; Tan, Y.; Sun, Y.; Zhu, S.; Zhao, C.; Chen, B.; Zhu, J.; et al. TanCAR T cells targeting CD19 and CD133 efficiently eliminate MLL leukemic cells. Leukemia 2018, 32, 2012–2016. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.B.; Nixon, A.M.L.; Moffat, J. The mixed lineage leukemia (MLL) fusion-associated gene AF4 promotes CD133 transcription. Cancer Res. 2012, 72, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Velasco-Hernandez, T.; Gutiérrez-Agüera, F.; Zanetti, S.R.; Baroni, M.L.; Sánchez-Martínez, D.; Molina, O.; Closa, A.; Agraz-Doblás, A.; Marín, P.; et al. CD133-directed CAR T-cells for MLL leukemia: On-target, off-tumor myeloablative toxicity. Leukemia 2019, 33, 2090–2125. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Montes, R.; Martín, L.; Prat, I.; Hernandez, M.C.; Orfao, A.; Menendez, P. NG2 antigen is expressed in CD34+ HPCs and plasmacytoid dendritic cell precursors: Is NG2 expression in leukemia dependent on the target cell where leukemogenesis is triggered? Leukemia 2008, 22, 1475–1478. [Google Scholar] [CrossRef][Green Version]
- Lopez-Millan, B.; Sanchéz-Martínez, D.; Roca-Ho, H.; Gutiérrez-Agüera, F.; Molina, O.; de la Guardia, R.D.; Torres-Ruiz, R.; Fuster, J.L.; Ballerini, P.; Suessbier, U.; et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 1557–1569. [Google Scholar] [CrossRef]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar] [CrossRef]
- Harrer, D.C.; Schuler, G.; Dörrie, J.; Schaft, N. CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int. J. Mol. Sci. 2019, 20, 2764. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Rosado, F.G.N.; Kim, A.S. Hemophagocytic LymphohistiocytosisAn Update on Diagnosis and Pathogenesis. Am J Clin. Pathol. 2013, 139, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.D.; Roskams, T.; Van Damme-Lombaerts, R.; Matthys, P.; Wouters, C. Macrophage activation syndrome: Characteristic findings on liver biopsy illustrating the key role of activated, IFN-γ-producing lymphocytes and IL-6- and TNF-α-producing macrophages. Blood 2005, 105, 1648–1651. [Google Scholar] [CrossRef] [PubMed]
- Prudent, V.; Breitbart, W.S. Chimeric antigen receptor T-cell neuropsychiatric toxicity in acute lymphoblastic leukemia. Palliat. Support. Care 2017, 15, 499–503. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | Disadvantages/Challenges |
|---|---|
| Independent of HLA and major histocompatibility complex | Toxicity/Neurotoxicity:
|
| Avoid majority of unnecessary killing of healthy tissue | Lysis of some healthy cells expressing the target antigen |
| Numerous accounts of efficacy | Antigen escape/Lineage switch |
| Personalized treatment (many potential target antigens) | CAR-T cell persistence |
| Treatment of advanced ALL when there are no other effective available options | Cost of treatment is very high in addition to the need for hospitalization of the patients and close observation during and after treatment |
| Current regulatory approved age restriction of <25 and >1 year old limits accessibility to more patients |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Britten, O.; Ragusa, D.; Tosi, S.; Mostafa Kamel, Y. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. https://doi.org/10.3390/cells8111341
Britten O, Ragusa D, Tosi S, Mostafa Kamel Y. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells. 2019; 8(11):1341. https://doi.org/10.3390/cells8111341
Chicago/Turabian StyleBritten, Oliver, Denise Ragusa, Sabrina Tosi, and Yasser Mostafa Kamel. 2019. "MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy?" Cells 8, no. 11: 1341. https://doi.org/10.3390/cells8111341
APA StyleBritten, O., Ragusa, D., Tosi, S., & Mostafa Kamel, Y. (2019). MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells, 8(11), 1341. https://doi.org/10.3390/cells8111341