Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
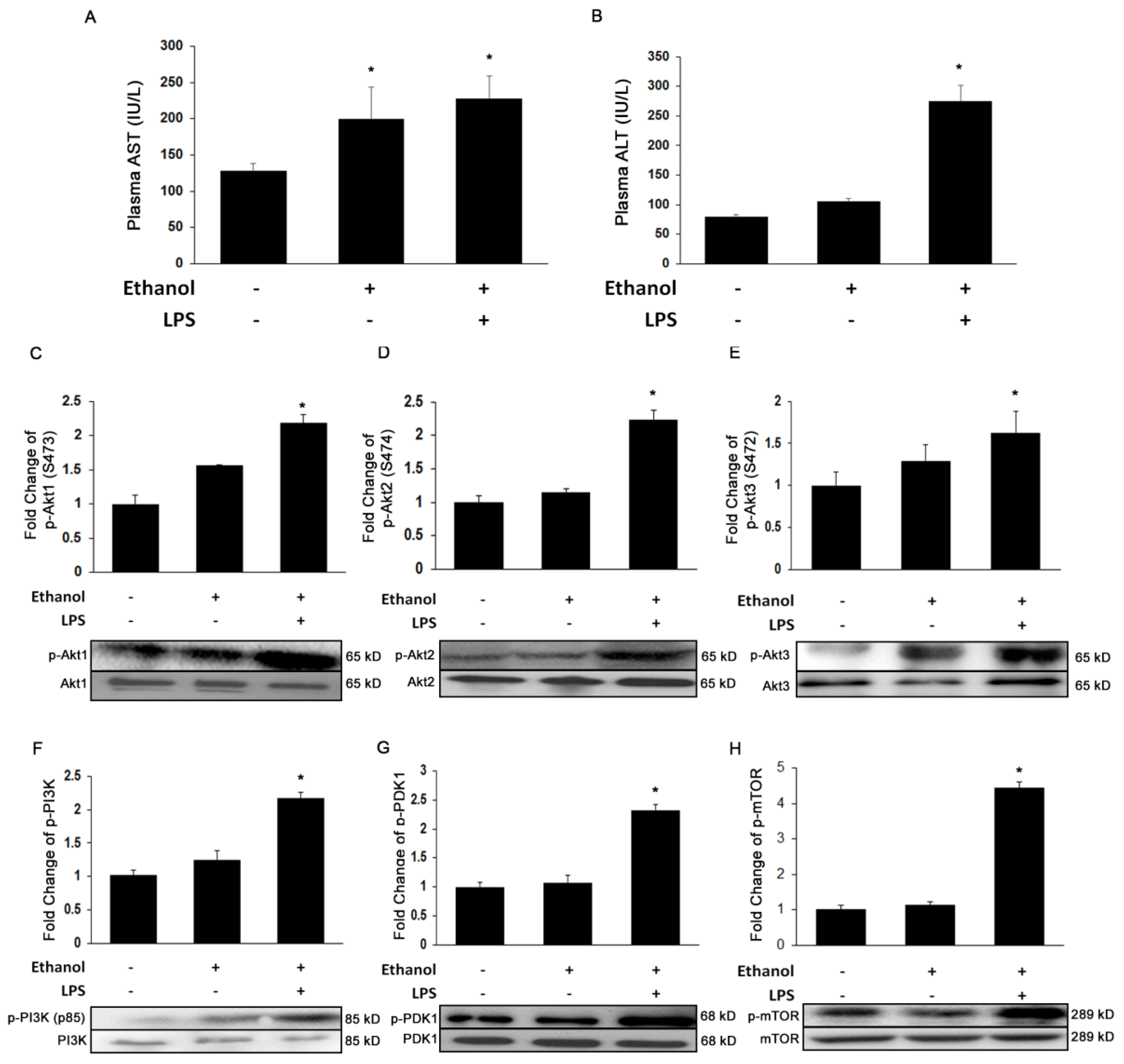
3.1. Ethanol and LPS Induce Liver Injury and Activate Akt Signaling Pathways
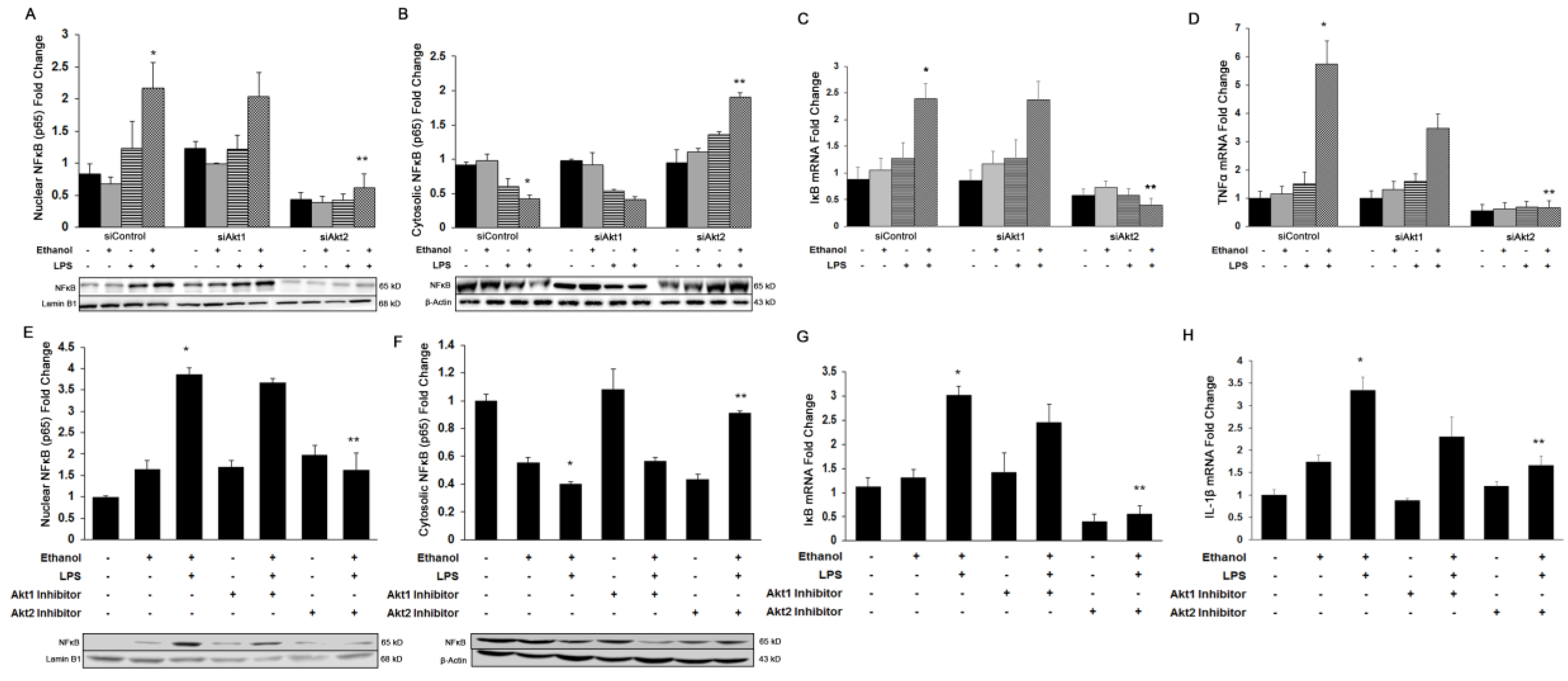
3.2. Akt2, But Not Akt1, Is Involved in Ethanol and LPS-Induced Nuclear Translocation of NFκB-p65 and Induction of Pro-Inflammatory Cytokines In Vitro and In Vivo
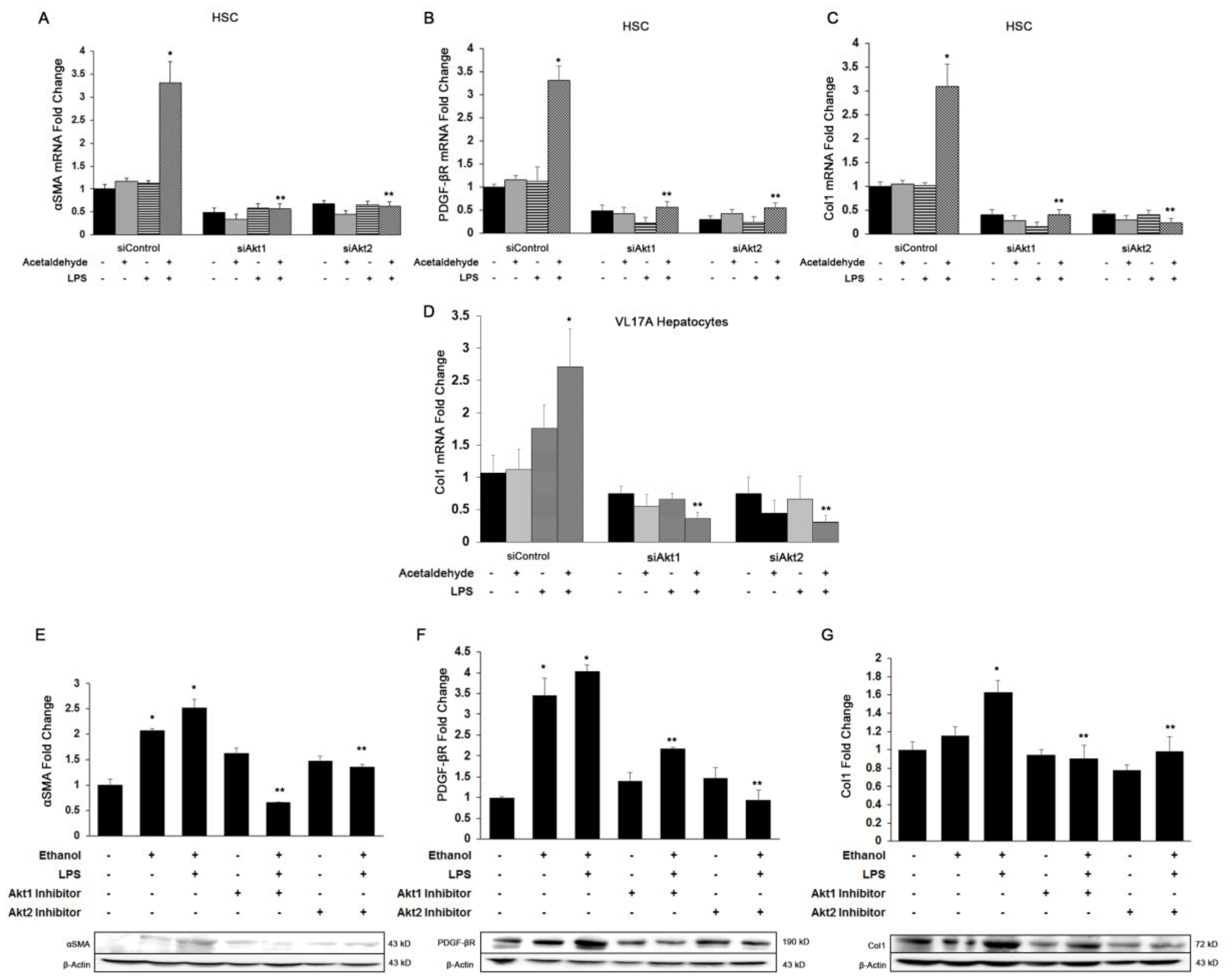
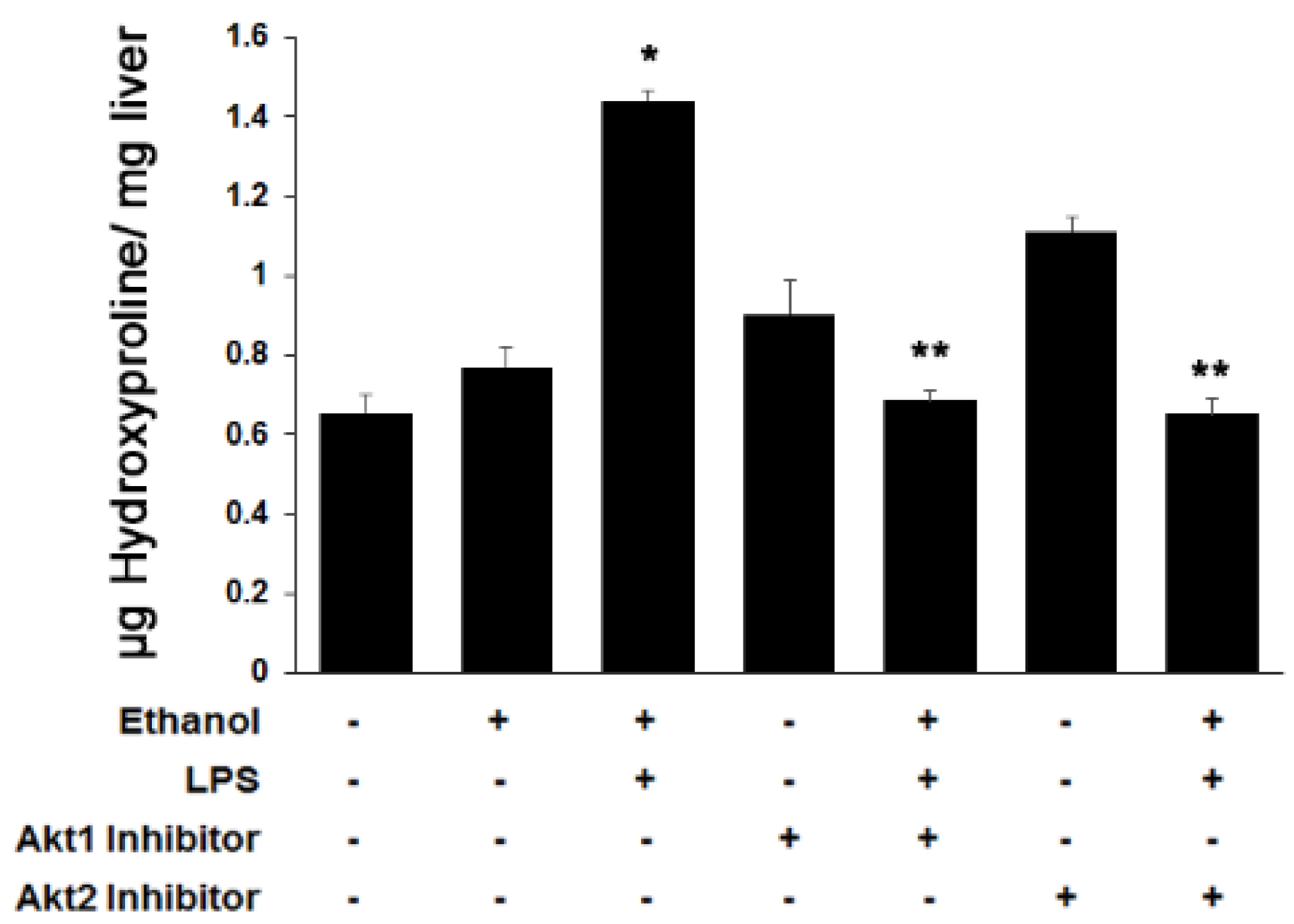
3.3. Both Akt1 and Akt2 Are Involved in Ethanol and LPS Induced Fibrosis
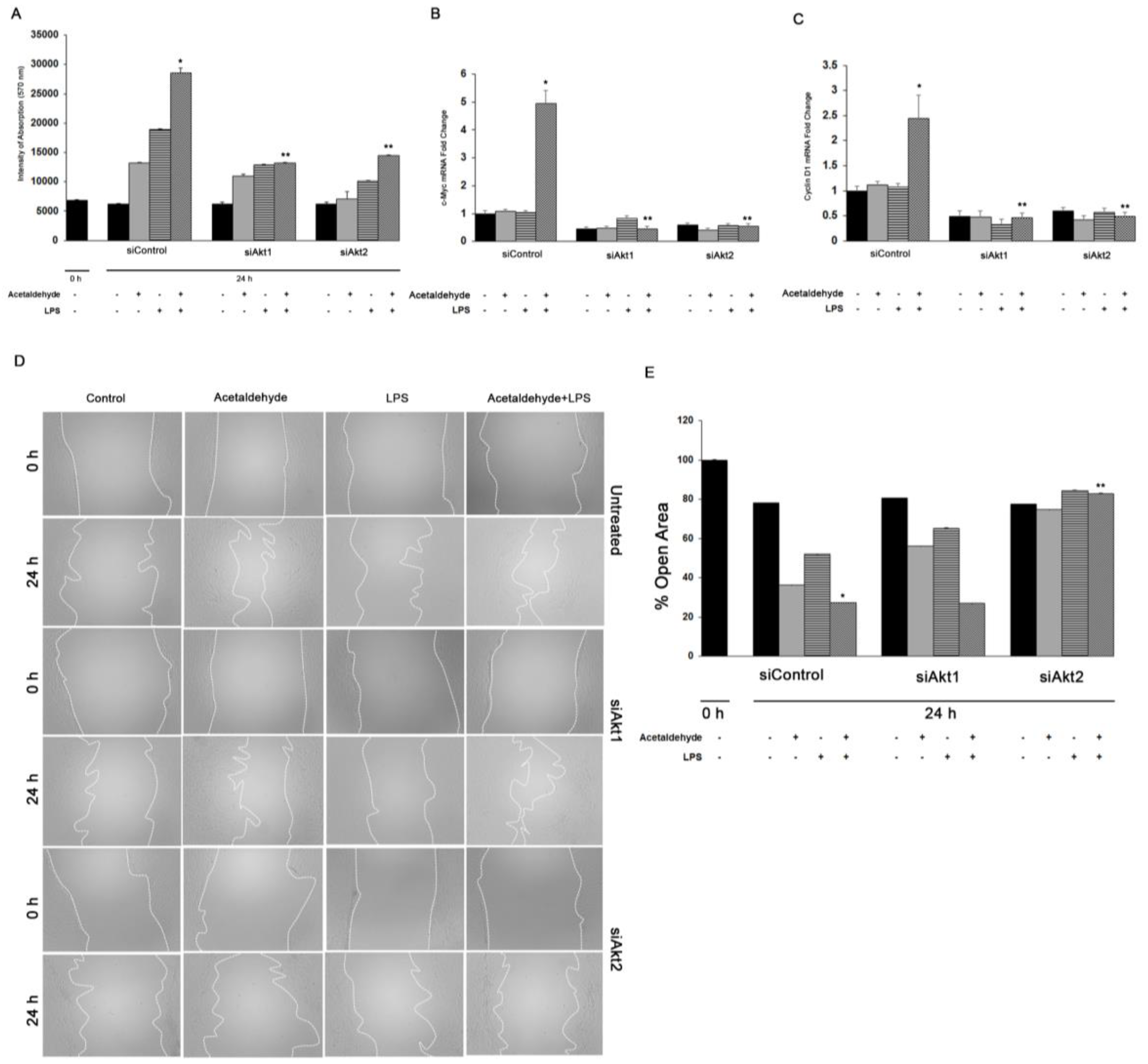
3.4. Akt1 and Akt2 Are Involved in HSC Proliferation, but Only Akt2 Regulates Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Orman, E.S.; Odena, G.; Bataller, R. Alcoholic liver disease: Pathogenesis, management, and novel targets for therapy. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 77–84. [Google Scholar] [CrossRef]
- Cohen-Naftaly, M.; Friedman, S.L. Current status of novel antifibrotic therapies in patients with chronic liver disease. Ther. Adv. Gastroenterol. 2011, 4, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Nagy, L.E. Pathogenesis of alcoholic liver disease: Interactions between parenchymal and non-parenchymal cells. J. Dig. Dis. 2011, 12, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, C.S.; Duryee, M.J.; Hunter, C.D.; Hamilton, B.C., 3rd; DeVeney, A.L.; Huerter, M.M.; Klassen, L.W.; Thiele, G.M. Alcohol metabolites and lipopolysaccharide: Roles in the development and/or progression of alcoholic liver disease. World J. Gastroenterol. 2009, 15, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Tamai, H.; Horie, Y.; Kato, S.; Yokoyama, H.; Ishii, H. Long-term ethanol feeding enhances susceptibility of the liver to orally administered lipopolysaccharides in rats. Alcohol. Clin. Exp. Res. 2002, 26, 75S–80S. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef]
- Hoek, J.B. Endotoxin and alcoholic liver disease: Tolerance and susceptibility. Hepatology 1999, 29, 1602–1604. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yokoyama, H.; Higuchi, S.; Hara, S.; Kato, S.; Ishii, H. Acetaldehyde accumulation suppresses Kupffer cell release of TNF-Alpha and modifies acute hepatic inflammation in rats. J. Gastroenterol. 2004, 39, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yokoyama, H.; Okamura, Y.; Ohgo, H.; Fukuda, M.; Horie, Y.; Kato, S.; Ishii, H. Evidence for ethanol oxidation by Kupffer cells. Alcohol. Clin. Exp. Res. 1999, 23, 92S–95S. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Nieto, N. Kupffer cells and alcoholic liver disease. Rev. Esp. De Enferm. Dig. Organo De La Soc. Esp. De Patol. Dig. 2006, 98, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.D. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2003, 27, 300–306. [Google Scholar]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4- hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Stefanovic, B.; Giordano, F.; Burchardt, E.R.; Brenner, D.A. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999, 30, 77–87. [Google Scholar] [CrossRef]
- Ma, K.L.; Ruan, X.Z.; Powis, S.H.; Chen, Y.; Moorhead, J.F.; Varghese, Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 2008, 48, 770–781. [Google Scholar] [CrossRef]
- Shafiei, M.S.; Shetty, S.; Scherer, P.E.; Rockey, D.C. Adiponectin regulation of stellate cell activation via PPARgamma-dependent and -independent mechanisms. Am. J. Pathol. 2011, 178, 2690–2699. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Zhu, N.L.; Asahina, K.; Mann, D.A.; Mann, J. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2011, 41, 675–682. [Google Scholar] [CrossRef]
- Dillon, R.L.; White, D.E.; Muller, W.J. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Das, S.; Yu, M.B.; Deyle, K.M.; Millward, S.W.; Heath, J.R. A chemical epitope-targeting strategy for protein capture agents: The serine 474 epitope of the kinase Akt2. Angew. Chem. 2013, 52, 13975–13979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riaz, A.; Zeller, K.S.; Johansson, S. Receptor-specific mechanisms regulate phosphorylation of AKT at Ser473: Role of RICTOR in beta1 integrin-mediated cell survival. PLoS ONE 2012, 7, e32081. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar]
- Mukherjee, A.; Larson, E.A.; Klein, R.F.; Rotwein, P. Distinct actions of akt1 on skeletal architecture and function. PLoS ONE 2014, 9, e93040. [Google Scholar] [CrossRef]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef]
- Hay, N. Akt isoforms and glucose homeostasis the leptin connection. Trends Endocrinol. Metab. TEM 2011, 22, 66–73. [Google Scholar] [CrossRef]
- Riggio, M.; Perrone, M.C.; Polo, M.L.; Rodriguez, M.J.; May, M.; Abba, M.; Lanari, C.; Novaro, V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017, 7, 44244. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Pellinen, T.; Rantala, J.K.; Perala, M.; Ivaska, J. Distinct roles of AKT isoforms in regulating beta1-integrin activity, migration, and invasion in prostate cancer. Mol. Biol. Cell 2012, 23, 3357–3369. [Google Scholar] [CrossRef]
- Cariaga-Martinez, A.E.; Lopez-Ruiz, P.; Nombela-Blanco, M.P.; Motino, O.; Gonzalez- Corpas, A.; Rodriguez-Ubreva, J.; Lobo, M.V.; Cortes, M.A.; Colas, B. Distinct and specific roles of AKT1 and AKT2 in androgen-sensitive and androgen-independent prostate cancer cells. Cell. Signal. 2013, 25, 1586–1597. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. The feeding of alcohol in liquid diets: Two decades of applications and 1982 update. Alcohol. Clin. Exp. Res. 1982, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Woods, A.J.; Dale, T.C.; Van Der Sluijs, P.; Norman, J.C. Protein kinase B/Akt acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol. Cell. Biol. 2004, 24, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Shoemaker, A.R.; Liu, X.; Woods, K.W.; Thomas, S.A.; de Jong, R.; Han, E.K.; Li, T.; Stoll, V.S.; Powlas, J.A.; et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol. Cancer Ther. 2005, 4, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Walton, M.I.; Hunter, L.J.; Valenti, M.; de Haven Brandon, A.; Eve, P.D.; Ruddle, R.; Heaton, S.P.; Henley, A.; Pickard, L.; et al. Preclinical pharmacology, antitumor activity, and development of pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930. Mol. Cancer Ther. 2011, 10, 360–371. [Google Scholar] [CrossRef]
- Zhu, Q.S.; Ren, W.; Korchin, B.; Lahat, G.; Dicker, A.; Lu, Y.; Mills, G.; Pollock, R.E.; Lev, D. Soft tissue sarcoma cells are highly sensitive to AKT blockade: A role for p53- independent up-regulation of GADD45 alpha. Cancer Res. 2008, 68, 2895–2903. [Google Scholar] [CrossRef]
- Donohue, T.M.; Osna, N.A.; Clemens, D.L. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int. J. Biochem. Cell Biol. 2006, 38, 92–101. [Google Scholar] [CrossRef]
- Swaminathan, K.; Clemens, D.L.; Dey, A. Inhibition of CYP2E1 leads to decreased malondialdehyde-acetaldehyde adduct formation in VL-17A cells under chronic alcohol exposure. Life Sci. 2013, 92, 325–336. [Google Scholar] [CrossRef]
- Olinga, P.; Merema, M.T.; de Jager, M.H.; Derks, F.; Melgert, B.N.; Moshage, H.; Slooff, M.J.; Meijer, D.K.; Poelstra, K.; Groothuis, G.M. Rat liver slices as a tool to study LPS-induced inflammatory response in the liver. J. Hepatol. 2001, 35, 187–194. [Google Scholar] [CrossRef]
- Jones, A.W. Measuring and reporting the concentration of acetaldehyde in human breath. Alcohol Alcohol. 1995, 30, 271–285. [Google Scholar]
- Gong, M.; Garige, M.; Hirsch, K. Liver Galbeta1,4GlcNAc alpha2,6-sialyltransferase is down-regulated in human alcoholics: Possible cause for the appearance of asialoconjugates. Metabolism 2007, 56, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Calvello, R.; Porro, C.; Trotta, T.; Salvatore, R.; Panaro, M.A. PI3k/Akt signalling pathway plays a crucial role in the anti-inflammatory effects of curcumin in LPS-activated microglia. Int. Immunopharmacol. 2016, 36, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Greenwel, P.; Dominguez-Rosales, J.A.; Mavi, G.; Rivas-Estilla, A.M.; Rojkind, M. Hydrogen peroxide: A link between acetaldehyde-elicited alpha1(I) collagen gene up- regulation and oxidative stress in mouse hepatic stellate cells. Hepatology 2000, 31, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Arias, I.M.; Alter, H.J.; Boyer, J.L.; Cohen, D.E.; Fausto, N.; Shafritz, D.A.; Wolkoff, A.W. (Eds.) Hepatic Stellate Cells. In The Liver: Biology and Pathobiology, 5th ed.; Wiley-Blackwell: Oxford, UK, 2009. [Google Scholar]
- Staveley, B.E.; Ruel, L.; Jin, J.; Stambolic, V.; Mastronardi, F.G.; Heitzler, P.; Woodgett, J.R.; Manoukian, A.S. Genetic analysis of protein kinase B (AKT) in Drosophila. Curr. Biol. CB 1998, 8, 599–602. [Google Scholar] [CrossRef]
- Grabinski, N.; Bartkowiak, K.; Grupp, K.; Brandt, B.; Pantel, K.; Jucker, M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell. Signal. 2011, 23, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Tschopp, O.; Hemmings-Mieszczak, M.; Feng, J.; Brodbeck, D.; Perentes, E.; Hemmings, B.A. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 2003, 278, 32124–32131. [Google Scholar] [CrossRef]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Fernandez-Hernando, C.; Cirino, G.; Sessa, W.C. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc. Natl. Acad. Sci. USA 2009, 106, 14552–14557. [Google Scholar] [CrossRef]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef]
- Nie, Y.; Sun, L.; Wu, Y.; Yang, Y.; Wang, J.; He, H.; Hu, Y.; Chang, Y.; Liang, Q.; Zhu, J.; et al. AKT2 Regulates Pulmonary Inflammation and Fibrosis via Modulating Macrophage Activation. J. Immunol. 2017, 198, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Bibaki, E.; Tsitoura, E.; Vasarmidi, E.; Margaritopoulos, G.; Trachalaki, A.; Koutoulaki, C.; Georgopoulou, T.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. miR-185 and miR-29a are similarly expressed in the bronchoalveolar lavage cells in IPF and lung cancer but common targets DNMT1 and COL1A1 show disease specific patterns. Mol. Med. Rep. 2018, 17, 7105–7112. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Zhang, J.; Xiao, Z.; Peng, X.; Qi, Y.; Du, J. Akt2 is involved in loss of epithelial cells and renal fibrosis following unilateral ureteral obstruction. PLoS ONE 2014, 9, e105451. [Google Scholar] [CrossRef] [PubMed]
- Ock, S.; Lee, W.S.; Kim, H.M.; Park, K.S.; Kim, Y.K.; Kook, H.; Park, W.J.; Lee, T.J.; Abel, E.D.; Kim, J. Connexin43 and zonula occludens-1 are targets of Akt in cardiomyocytes that correlate with cardiac contractile dysfunction in Akt deficient hearts. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Gordillo, K.; Shah, R.; Popratiloff, A.; Fu, S.; Hindle, A.; Brody, F.; Rojkind, M. Thymosin-beta4 (Tbeta4) blunts PDGF-dependent phosphorylation and binding of AKT to actin in hepatic stellate cells. Am. J. Pathol. 2011, 178, 2100–2108. [Google Scholar] [CrossRef] [PubMed]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Gordillo, K.; Shah, R.; Arellanes-Robledo, J.; Cheng, Y.; Ibrahim, J.; Tuma, P.L. Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease. Cells 2019, 8, 1337. https://doi.org/10.3390/cells8111337
Reyes-Gordillo K, Shah R, Arellanes-Robledo J, Cheng Y, Ibrahim J, Tuma PL. Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease. Cells. 2019; 8(11):1337. https://doi.org/10.3390/cells8111337
Chicago/Turabian StyleReyes-Gordillo, Karina, Ruchi Shah, Jaime Arellanes-Robledo, Ying Cheng, Joseph Ibrahim, and Pamela L. Tuma. 2019. "Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease" Cells 8, no. 11: 1337. https://doi.org/10.3390/cells8111337
APA StyleReyes-Gordillo, K., Shah, R., Arellanes-Robledo, J., Cheng, Y., Ibrahim, J., & Tuma, P. L. (2019). Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease. Cells, 8(11), 1337. https://doi.org/10.3390/cells8111337