Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response

 , ,
, ,  , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animal Studies
2.3. Cell Culture
2.3.1. Generation of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 Constructs for CypD Knockout and Establishment of Cell Lines
2.3.2. Modulation of NO Concentrations and sGC/PKG Pathway
2.3.3. Insulin and Wortmannin Stimulation
2.4. Assessment of NO Concentrations
2.5. Isolation of MAMs
2.6. Transmission Electronic Microscopy
2.7. Fluorescence Imaging
2.7.1. In Situ Proximity Ligation Assay (PLA)
2.7.2. MitoTracker Green®
2.8. Western Blot
2.9. Gene Expression
2.9.1. mRNA Extraction and Reverse Transcription
2.9.2. DNA Extraction (for Mitochondrial DNA Analysis)
2.9.3. Measurement of Gene Expression by PCR
2.10. Oxygraphy
2.11. Statistical Analysis
3. Results
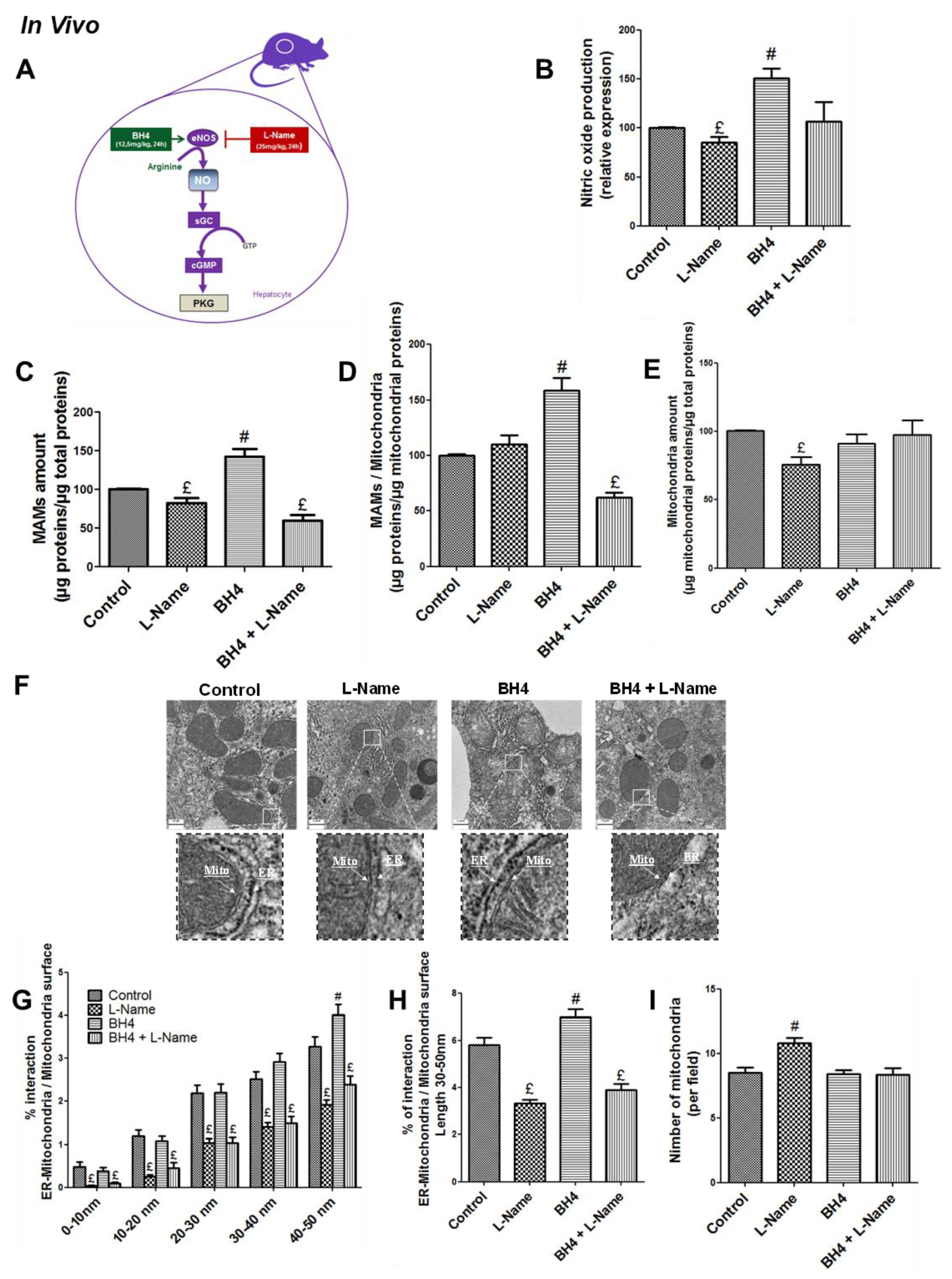
3.1. NO Regulates MAM Integrity and Insulin Response in the Liver in Vivo
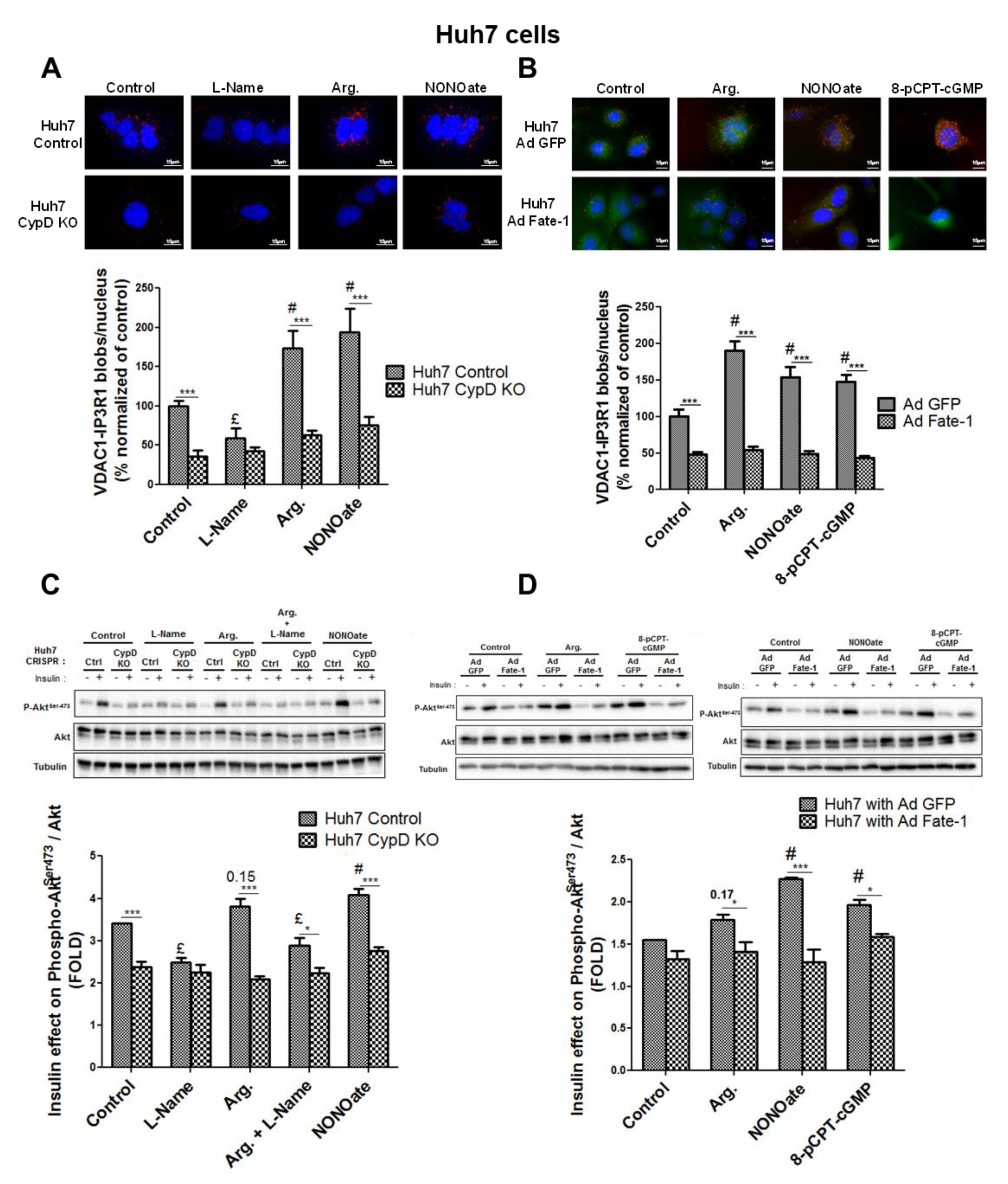
3.2. NO Regulates MAM Integrity and Insulin Response in Hepatocytes In Vitro
3.3. NO Regulates MAM Integrity and Insulin Response in Hepatocytes Through the sGC/PKG Pathway
3.4. The Insulin Signaling Pathway is Not Required in the Regulation of MAM Integrity by NO
3.5. MAM Integrity is Necessary for NO to Impact on Insulin Response In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT = PKB | protein kinase B |
| ANOVA | analysis of variance |
| AR | aspect ratio |
| Arg. | arginine |
| BAY41-2272 | 3-(4-amino-5-cyclopropylpyrimidin-2-yl)-1-(2-fluorobenzyl)-1H-pyrazolo [3–b]pyridine |
| BH4 | tetrahydrobiopterin |
| cGMP | cyclic guanosine monophosphate |
| COX-1 | cyclooxygenase-1 |
| CRISPR/cas9 | clustered regularly interspaced short palindromic repeats: CRISPR associated protein 9 |
| CypD | cyclophilin D |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | dimethyl Sulfoxide |
| DNA | deoxyribonucleic acid |
| DRP1 | dynamin-related GTPase |
| DTT | dithiothreitol |
| EDTA | ethylenediaminetetraacetic acid |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| eNOS = NOS3 | endothelial NO synthase |
| ER | endoplasmic reticulum |
| FACS | fluorescence-activated cell sorting |
| FATE-1 | fetal and adult testis-expressed 1 |
| FCS | fetal calf serum |
| FF | form factor |
| GFP | green fluorescent protein |
| Grp75 | glucose-regulated protein 75 |
| GSK3β | glycogen synthase kinase 3 beta |
| GTP | cyclic guanosine monophosphate |
| HCL | hydrochloric acid |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| iNOS = NOS2 | inducible NO synthase |
| IP3R | inositol 1,4,5-trisphosphate receptor |
| KO | knockout |
| l-Name | Nω-nitro-l-arginine methyl ester hydrochloride |
| MAMs | mitochondria-associated ER membranes |
| Mfn 2 | mitofusin 2 |
| mRNA | messenger ribonucleic acid |
| mtDNA | mitochondrial deoxyribonucleic acid |
| Na3VO4 | sodium orthovanadate |
| NaF | sodium fluoride |
| NO | nitric oxide |
| NONOate | diethylamine NONOate sodium salt hydrate |
| OPA1 | optic atrophy type 1 |
| PBS | phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PI3K | phosphoinositide 3-kinases |
| PKG | protein kinase G |
| PLA | proximity ligation assay |
| PPIA | peptidylprolyl isomerase A |
| 8-pCPT-cGMP | 8-(4-chlorophenylthio)-guanosine 3′,5′-cyclic monophosphate sodium salt |
| RNA | ribonucleic acid |
| RT | reverse transcription |
| SDS | sodium dodecyl sulfate |
| SEM | standard error of the mean |
| Ser | serine |
| sGC | soluble guanylate cyclase |
| siRNA | silencing ribonucleic acid |
| TBP | TATA-binding protein |
| TBS-Tween | tris-buffered saline (TBS) and polysorbate 20 (Tween) |
| TEM | transmission electron microscopy |
| VDAC | voltage dependent anion channel |
References
- Rieusset, J. Endoplasmic reticulum-mitochondria calcium signaling in hepatic metabolic diseases. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.-A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Shinjo, S.; Jiang, S.; Nameta, M.; Suzuki, T.; Kanai, M.; Nomura, Y.; Goda, N. Disruption of the mitochondria-associated ER membrane (MAM) plays a central role in palmitic acid–induced insulin resistance. Exp. Cell Res. 2017, 359, 86–93. [Google Scholar] [CrossRef]
- Meshkani, R.; Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem. 2009, 42, 1331–1346. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Culotta, E.; Koshland, D.E. NO news is good news. Science 1992, 258, 1862–1865. [Google Scholar] [CrossRef]
- Shankar, R.R.; Wu, Y.; Shen, H.Q.; Zhu, J.S.; Baron, A.D. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 2000, 49, 684–687. [Google Scholar] [CrossRef]
- Alexander, B. The role of nitric oxide in hepatic metabolism. Nutrition 1998, 14, 376–390. [Google Scholar] [CrossRef]
- Rafikov, R.; Fonseca, F.V.; Kumar, S.; Pardo, D.; Darragh, C.; Elms, S.; Fulton, D.; Black, S.M. eNOS activation and NO function: Structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011, 210, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.; Tapia, G.; Varela, P.; Videla, L.A. Redox regulation of thyroid hormone-induced Kupffer cell-dependent IkappaB-alpha phosphorylation in relation to inducible nitric oxide synthase expression. Free Radic. Res. 2005, 39, 411–418. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Green, A.K.; Stratton, R.C.; Squires, P.E.; Simpson, A.W.M. Atrial natriuretic peptide attenuates elevations in Ca2+ and protects hepatocytes by stimulating net plasma membrane Ca2+ efflux. J. Biol. Chem. 2007, 282, 34542–34554. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowicz, J.E.; Stafford, S.J.; Garg, N.J. Protein Cysteinyl-S-Nitrosylation: Analysis and Quantification. Meth. Enzymol. 2017, 586, 1–14. [Google Scholar]
- Thippeswamy, T.; McKay, J.S.; Quinn, J.P.; Morris, R. Nitric oxide, a biological double-faced janus--is this good or bad? Histol. Histopathol. 2006, 21, 445–458. [Google Scholar]
- Mejía-García, T.A.; Portugal, C.C.; Encarnação, T.G.; Prado, M.A.M.; Paes-de-Carvalho, R. Nitric oxide regulates AKT phosphorylation and nuclear translocation in cultured retinal cells. Cell. Signal. 2013, 25, 2424–2439. [Google Scholar] [CrossRef]
- Numajiri, N.; Takasawa, K.; Nishiya, T.; Tanaka, H.; Ohno, K.; Hayakawa, W.; Asada, M.; Matsuda, H.; Azumi, K.; Kamata, H.; et al. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc. Natl. Acad. Sci. USA 2011, 108, 10349–10354. [Google Scholar] [CrossRef]
- De Palma, C.; Falcone, S.; Pisoni, S.; Cipolat, S.; Panzeri, C.; Pambianco, S.; Pisconti, A.; Allevi, R.; Bassi, M.T.; Cossu, G.; et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010, 17, 1684–1696. [Google Scholar] [CrossRef]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar] [PubMed]
- Berry, M.N.; Friend, D.S. High-yield preparation of isolated rat liver parenchymal cells: A biochemical and fine structural study. J. Cell Biol. 1969, 43, 506–520. [Google Scholar] [CrossRef]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.-A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.-L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.-A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 2008, 118, 789–800. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Verkaart, S.; Visch, H.-J.; van der Westhuizen, F.H.; Murphy, M.P.; van den Heuvel, L.W.P.J.; Smeitink, J.A.M.; Willems, P.H.G.M. Inhibition of complex I of the electron transport chain causes O2−. -mediated mitochondrial outgrowth. Am. J. Physiol., Cell Physiol. 2005, 288, C1440–C1450. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Rieusset, J. Study of Endoplasmic Reticulum and Mitochondria Interactions by In Situ Proximity Ligation Assay in Fixed Cells. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tengan, C.H.; Moraes, C.T. NO control of mitochondrial function in normal and transformed cells. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Doghman-Bouguerra, M.; Granatiero, V.; Sbiera, S.; Sbiera, I.; Lacas-Gervais, S.; Brau, F.; Fassnacht, M.; Rizzuto, R.; Lalli, E. FATE-1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 2016, 17, 1264–1280. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.; Charles, I.G.; Moncada, S. Nitric oxide induces coupling of mitochondrial signaling with the endoplasmic reticulum stress response. Nat. Cell Biol. 2004, 6, 1129–1134. [Google Scholar] [CrossRef]
- Yang, L.; Calay, E.S.; Fan, J.; Arduini, A.; Kunz, R.C.; Gygi, S.P.; Yalcin, A.; Fu, S.; Hotamisligil, G.S. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015, 349, 500–506. [Google Scholar] [CrossRef]
- Miller, M.W.; Knaub, L.A.; Olivera-Fragoso, L.F.; Keller, A.C.; Balasubramaniam, V.; Watson, P.A.; Reusch, J.E.B. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1624–H1633. [Google Scholar] [CrossRef]
- Nisoli, E. Mitochondrial Biogenesis in Mammals: The Role of Endogenous Nitric Oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Cappellini, E.; Benfante, R.; Ragni, M.; Omodeo-Salè, F.; Nisoli, E.; Borgese, N.; Vicentini, L.M. Chronic Deficiency of Nitric Oxide Affects Hypoxia Inducible Factor-1α (HIF-1α) Stability and Migration in Human Endothelial Cells. PLoS ONE 2011, 6, e29680. [Google Scholar] [CrossRef]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.-H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. -Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; Ortiz-Sandoval, C.; Navarro-Marquez, M.; Rodríguez, A.E.; Diaz-Valdivia, N.; Sanhueza, C.; Lopez-Crisosto, C.; Tahbaz, N.; Rothermel, B.A.; et al. Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER–mitochondria communication during the early phase of ER stress. Cell Death Differ. 2019, 26, 1195–1212. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA. 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Gomez, L.; Thiebaut, P.-A.; Paillard, M.; Ducreux, S.; Abrial, M.; Crola Da Silva, C.; Durand, A.; Alam, M.R.; Van Coppenolle, F.; Sheu, S.-S.; et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016, 23, 313–322. [Google Scholar] [CrossRef]
- Feriod, C.N.; Gustavo Oliveira, A.; Guerra, M.T.; Nguyen, L.; Mitchell Richards, K.; Jurczak, M.J.; Ruan, H.-B.; Paulo Camporez, J.; Yang, X.; Shulman, G.I.; et al. Hepatic inositol 1,4,5 trisphosphate receptor type 1 mediates fatty liver. Hepatol. Commun. 2017, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Ha, C.-M.; Leem, J.; Chanda, D.; Park, J.-S.; Kim, H.-J.; Jeon, J.-H.; Choi, Y.-K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER–Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhao, L.; Gao, P.; Zhu, X.; Han, Y.; Chen, X.; Li, L.; Xiao, Y.; Wei, L.; Li, C.; et al. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAM integrity. EBioMedicine 2019, 43, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassot, A.; Chauvin, M.-A.; Bendridi, N.; Ji-Cao, J.; Vial, G.; Monnier, L.; Bartosch, B.; Alves, A.; Cottet-Rousselle, C.; Gouriou, Y.; et al. Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response. Cells 2019, 8, 1319. https://doi.org/10.3390/cells8111319
Bassot A, Chauvin M-A, Bendridi N, Ji-Cao J, Vial G, Monnier L, Bartosch B, Alves A, Cottet-Rousselle C, Gouriou Y, et al. Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response. Cells. 2019; 8(11):1319. https://doi.org/10.3390/cells8111319
Chicago/Turabian StyleBassot, Arthur, Marie-Agnès Chauvin, Nadia Bendridi, Jingwei Ji-Cao, Guillaume Vial, Léa Monnier, Birke Bartosch, Anaïs Alves, Cécile Cottet-Rousselle, Yves Gouriou, and et al. 2019. "Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response" Cells 8, no. 11: 1319. https://doi.org/10.3390/cells8111319
APA StyleBassot, A., Chauvin, M.-A., Bendridi, N., Ji-Cao, J., Vial, G., Monnier, L., Bartosch, B., Alves, A., Cottet-Rousselle, C., Gouriou, Y., Rieusset, J., & Morio, B. (2019). Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response. Cells, 8(11), 1319. https://doi.org/10.3390/cells8111319