WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
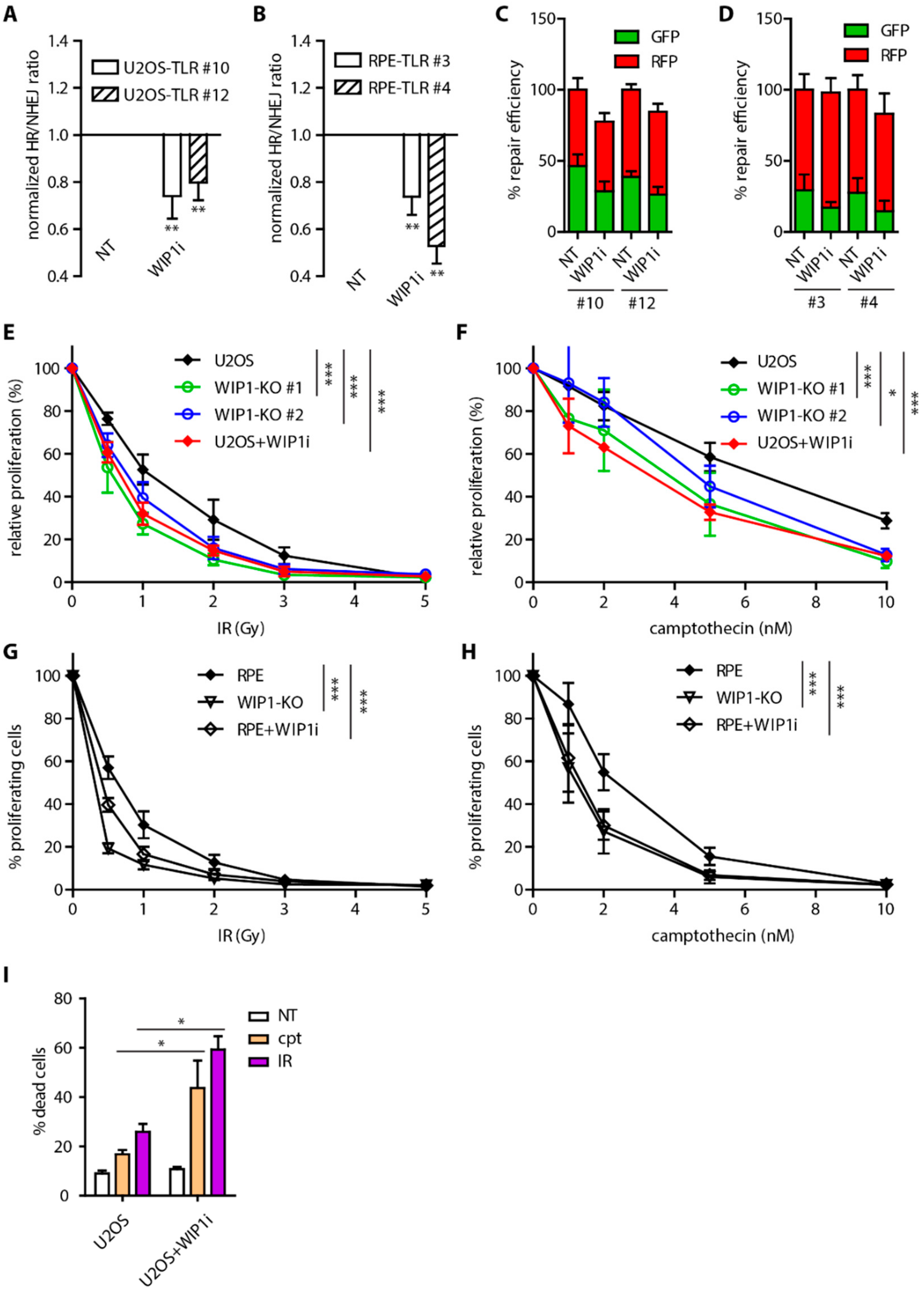
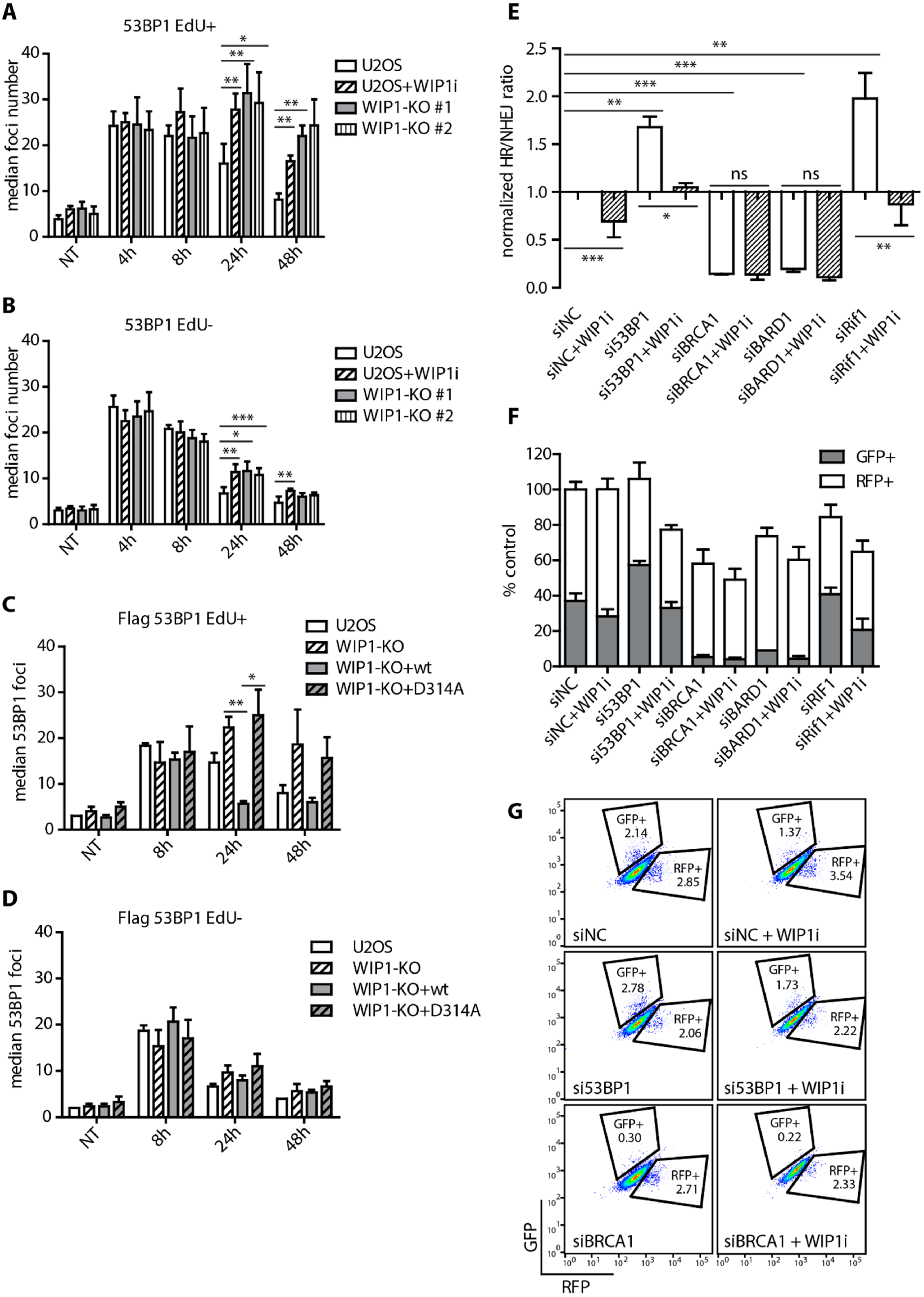
2.1. WIP1 Promotes DSB Repair by Homologous Recombination
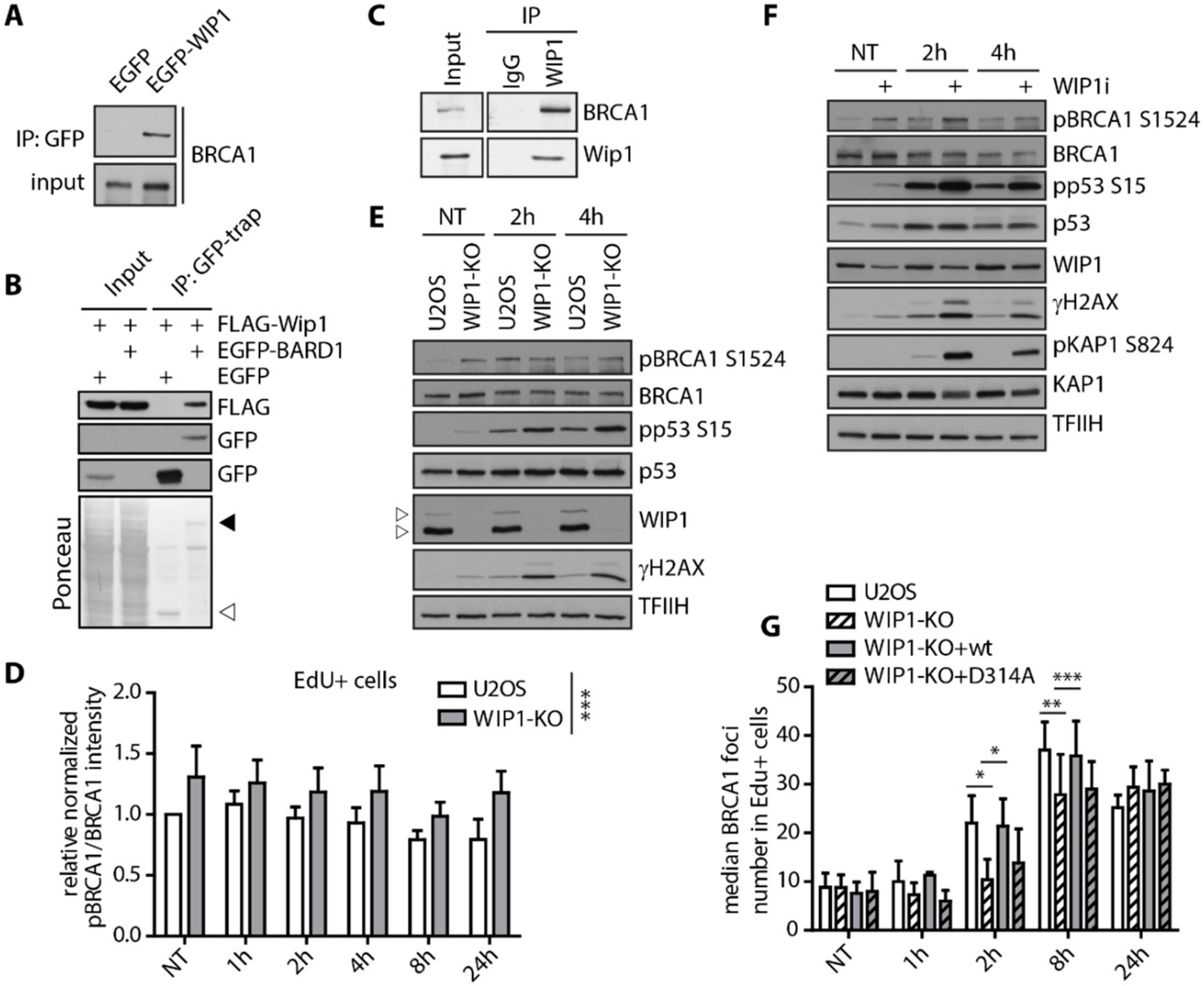
2.2. WIP1 Interacts with BRCA1 and Promotes its Recruitment to DSBs
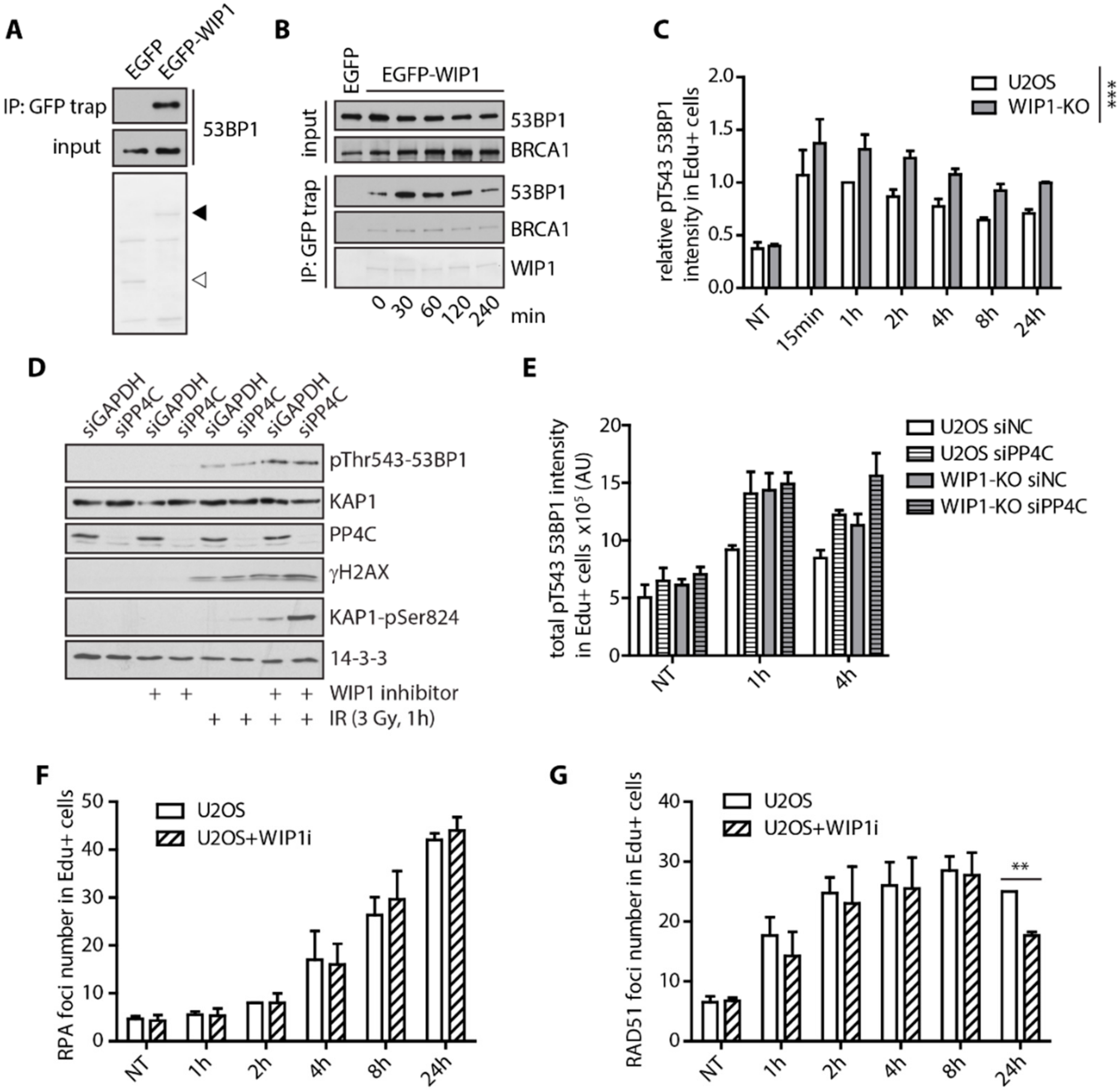
2.3. WIP1 Dephosphorylates 53BP1 at T543 Residue Needed for Interaction with RIF1
2.4. WIP1 Deficient Cells are Sensitive to PARP Inhibition in BRCA1 Dependent Manner
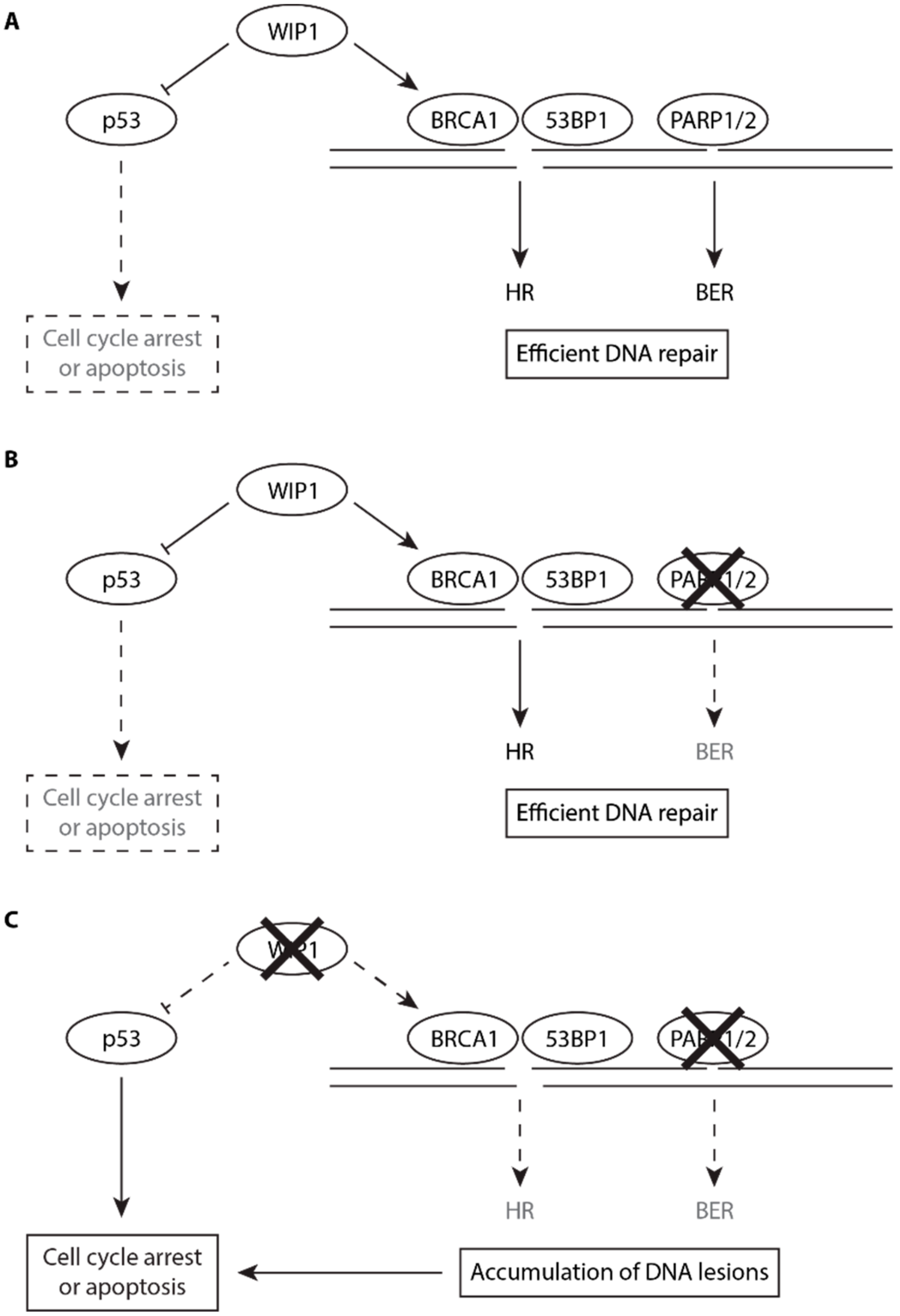
3. Discussion
4. Material and Methods
4.1. Cell Lines
4.2. Antibodies and Chemicals
4.3. Immunoprecipitation
4.4. Immunofluorescence
4.5. Flow Cytometry
4.6. Cell Survival Assays
4.7. Western Blot
4.8. In Vitro Phosphatase Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.; Lafranchi, L.; Sartori, A. Controlling DNA-end resection: A new task for CDKs. Front. Genet. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Jackson Stephen, P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Himmels, S.-F.; Sartori, A.A. Controlling DNA-End Resection: An. Emerging Task for Ubiquitin and SUMO. Front. Genet. 2016, 7, 152. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Chaudhuri, A.R. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 2019, 73, 1267–1281.e7. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Song, X. BRCA1–BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360. [Google Scholar] [CrossRef]
- Chapman, J.R.; Martin, R.G.; Taylor, J.; Simon, J.; Boulton, J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2013, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Greenberg, R.A. RAP80 Targets BRCA1 to Specific Ubiquitin Structures at DNA Damage Sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Chapman, J.R. H4K20me0 recognition by BRCA1–BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Durocher, D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef]
- Kleiner, R.E.; Verma, P.; Molloy, K.R.; Chait, B.T.; Kapoor, T.M. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 2015, 11, 807. [Google Scholar] [CrossRef]
- Callen, E.; di Virgilio, M.; Kruhlak, M.J.; Nieto-Soler, M.; Wong, N.; Chen, H.T.; Wesemann, D.R. 53BP1 Mediates Productive and Mutagenic DNA Repair through Distinct Phosphoprotein Interactions. Cell 2013, 153, 1266–1280. [Google Scholar] [CrossRef]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkáč, J.; Xu, D. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Petricci, E. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Pearl, L.H. Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Rodríguez, O. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Gogola, E.; Rottenberg, S.; Jonkers, J. Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annu. Rev. Cancer Biol. 2019, 3, 235–254. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Bitler, B.G. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef]
- Zhong, Q.; Hu, Z.; Li, Q.; Yi, T.; Li, J.; Yang, H. Cyclin D1 silencing impairs DNA double strand break repair, sensitizes BRCA1 wildtype ovarian cancer cells to olaparib. Gynecol. Oncol. 2019, 152, 157–165. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Coleman, R.L.; Westin, S.N. Using PARP Inhibitors in the Treatment of Patients with Ovarian Cancer. Curr. Treat. Opt. Oncol. 2018, 19, 1. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010, 15, 2281–2291. [Google Scholar]
- Macurek, L.; Benada, J.; Müllers, E.; Halim, V.A.; Krejčíková, K.; Burdová, K.; Bartek, J. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 2013, 12, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ma, O.; Nguyen, T.A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase Acts as a Gatekeeper in the p53-Mdm2 Autoregulatory Loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Minami, Y. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Minami, Y. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar]
- Shaltiel, I.A.; Aprelia, M.; Saurin, A.T.; Chowdhury, D.; Kops, G.J.; Voest, E.E.; Medema, R.H. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 7313–7318. [Google Scholar] [CrossRef]
- Cha, H.; Lowe, J.M.; Li, H.; Lee, J.S.; Belova, G.I.; Bulavin, D.V.; Fornace, A. JWip1 Directly Dephosphorylates γ-H2AX and Attenuates the DNA Damage Response. Cancer Res. 2010, 70, 4112–4122. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Demidov, O.N.; Saito, S.I.; Kauraniemi, P.; Phillips, C.; Amundson, S.A.; Kallioniemi, A. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 2002, 31, 210. [Google Scholar] [CrossRef]
- Tan, D.S.; Lambros, M.B.; Rayter, S.; Natrajan, R.; Vatcheva, R.; Gao, Q.; Fenwick, K. PPM1D Is a Potential Therapeutic Target in Ovarian Clear Cell Carcinomas. Clin. Cancer Res. 2009, 15, 2269–2280. [Google Scholar] [CrossRef]
- Castellino, R.C.; de Bortoli, M.; Lu, X.; Moon, S.H.; Nguyen, T.A.; Shepard, M.A.; Kim, J.Y. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J. Neurooncol. 2008, 86, 245–256. [Google Scholar] [CrossRef]
- Le Guezennec, X.; Bulavin, D.V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 2010, 35, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Minthorn, E. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Dayaram, T.; Gilmartin, A.G.; Ganji, G.; Pemmasani, S.K.; van der Key, H.; Kumar, R. WIP1 Phosphatase as a Potential Therapeutic Target in Neuroblastoma. PLoS ONE 2015, 10, e0115635. [Google Scholar]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [PubMed]
- Pecháčková, S.; Burdová, K.; Macurek, L. WIP1 phosphatase as pharmacological target in cancer therapy. J. Mol. Med. 2017, 95, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Iniguez, A.B.; Dharia, N.; Robichaud, A.; Conway, A.; Morgan, A.; Alexe, G. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef]
- Moon, S.H.; Lin, L.; Zhang, X.; Nguyen, T.A.; Darlington, Y.; Waldman, A.S.; Donehower, L.A. Wildtype p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J. Biol. Chem. 2010, 285, 12935–12947. [Google Scholar] [CrossRef]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Gunn, A.; Stark, J.M. I-SceI-Based Assays to Examine Distinct Repair Outcomes of Mammalian Chromosomal Double Strand Breaks. In DNA Repair Protocols; Bjergbæk, L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 379–391. [Google Scholar]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous Homologous Recombination Is Induced by Collapsed Replication Forks That Are Caused by Endogenous DNA Single-Strand Breaks. Mol. Cell. Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Annunziato, S. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Jackson, S.P. Systematic E2 screening reveals a UBE2D–RNF138–CtIP axis promoting DNA repair. Nat. Cell Biol. 2015, 17, 1458. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; O’Donnell, A.H.; Kim, S.T.; Kastan, M.B. Phosphorylation of Serine 1387 in Brca1 Is Specifically Required for the Atm-mediated S-Phase Checkpoint after Ionizing Irradiation. Cancer Res. 2002, 62, 4588–4591. [Google Scholar]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic Changes of BRCA1 Subnuclear Location and Phosphorylation State Are Initiated by DNA Damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Kim, H.S.; Li, H.; Cevher, M.; Parmelee, A.; Fonseca, D.; Kleiman, F.E.; Lee, S.B. DNA Damage–Induced BARD1 Phosphorylation Is Critical for the Inhibition of Messenger RNA Processing by BRCA1/BARD1 Complex. Cancer Res. 2006, 66, 4561–4565. [Google Scholar] [CrossRef]
- Filipponi, D.; Muller, J.; Emelyanov, A.; Bulavin, D.V. Wip1 Controls Global Heterochromatin Silencing via ATM/BRCA1-Dependent DNA Methylation. Cancer Cell 2013, 24, 528–541. [Google Scholar] [CrossRef]
- Chowdhury, D.; Xu, X.; Zhong, X.; Ahmed, F.; Zhong, J.; Liao, J.; Lieberman, J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol. Cell 2008, 31, 33–46. [Google Scholar] [CrossRef]
- Lee, D.H.; Goodarzi, A.A.; Adelmant, G.O.; Pan, Y.; Jeggo, P.A.; Marto, J.A.; Chowdhury, D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012, 31, 2403–2415. [Google Scholar] [CrossRef]
- Penning, T.D.; Zhu, G.D.; Gong, J.; Thomas, S.; Gandhi, V.B.; Liu, X.; Fry, E.H. Optimization of Phenyl-Substituted Benzimidazole Carboxamide Poly(ADP-Ribose) Polymerase Inhibitors: Identification of (S)-2-(2-Fluoro-4-(pyrrolidin-2-yl)phenyl)-1H-benzimidazole-4-carboxamide (A-966492), a Highly Potent and Efficacious Inhibitor. J. Med. Chem. 2010, 53, 3142–3153. [Google Scholar] [CrossRef]
- Lindqvist, A.; de Bruijn, M.; Macurek, L.; Brás, A.; Mensinga, A.; Bruinsma, W.; Medema, R. HWip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009, 28, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Bocangel, D.; Nannenga, B.; Yamaguchi, H.; Appella, E.; Donehower, L.A. The p53-Induced Oncogenic Phosphatase PPM1D Interacts with Uracil DNA Glycosylase and Suppresses Base Excision Repair. Mol. Cell 2004, 15, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Slattery, S.D.; Moon, S.H.; Darlington, Y.F.; Lu, X.; Donehower, L.A. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair 2010, 9, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Linke, S.P.; Sengupta, S.; Khabie, N.; Jeffries, B.A.; Buchhop, S.; Miska, S.; Yang, Q. p53 Interacts with hRAD51 and hRAD54, and Directly Modulates Homologous Recombination. Cancer Res. 2003, 63, 2596–2605. [Google Scholar] [PubMed]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Gatz, S.A.; Wiesmüller, L. p53 in recombination and repair. Cell Death Differ. 2006, 13, 1003–1016. [Google Scholar] [CrossRef]
- Sriraman, A.; Radovanovic, M.; Wienken, M.; Najafova, Z.; Li, Y.; Dobbelstein, M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 2016, 7, 31623–31638. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.; Yao, D.; Yang, T.; Cao, W.M.; Dou, J.; Zhao, Y. Wip1 inhibitor GSK2830371 inhibits neuroblastoma growth by inducing Chk2/p53-mediated apoptosis. Sci. Rep. 2016, 6, 38011. [Google Scholar] [CrossRef]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2017, 118, 495. [Google Scholar]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Staining of Suspension Cells with Hoechst 33258 to Detect. Apoptosis. Cold Spring Harb. Protoc. 2006, 2006, pdb.prot4492. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burdova, K.; Storchova, R.; Palek, M.; Macurek, L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019, 8, 1258. https://doi.org/10.3390/cells8101258
Burdova K, Storchova R, Palek M, Macurek L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells. 2019; 8(10):1258. https://doi.org/10.3390/cells8101258
Chicago/Turabian StyleBurdova, Kamila, Radka Storchova, Matous Palek, and Libor Macurek. 2019. "WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors" Cells 8, no. 10: 1258. https://doi.org/10.3390/cells8101258
APA StyleBurdova, K., Storchova, R., Palek, M., & Macurek, L. (2019). WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells, 8(10), 1258. https://doi.org/10.3390/cells8101258