The Bright and Dark Side of DNA Methylation: A Matter of Balance


Abstract
1. DNA Methylation: More than One Purpose
2. DNA Methylation Analysis: Think Globally, Act Locally
3. DNA Methylation in Health: A Matter of Location and Timing
4. DNA Methylation and Disease: Too Little, too Much, or Both?
4.1. Germ-Line Associated Diseases
4.2. Somatic Diseases
4.2.1. Colorectal Cancer
4.2.2. Myelodysplastic Syndrome
5. Conclusions: The Best Is yet to Come
Funding
Acknowledgments
Conflicts of Interest
References
- Deichmann, U. Epigenetics: The origins and evolution of a fashionable topic. Dev. Biol. 2016, 416, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Nicoglou, A.; Merlin, F. Epigenetics: A way to bridge the gap between biological fi elds. Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 66, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.E.A.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; CSHL Press: Cold Spring Harbor, NY, USA, 1996. [Google Scholar]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The Role of DNA Methylation in Mammalian Epigenetics. Science 2001, 293, 1068–1071. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef]
- Fazzari, M.J.; Greally, J.M. Epigenomics: Beyond CpG islands. Nat. Rev. Genet. 2004, 5, 446–455. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
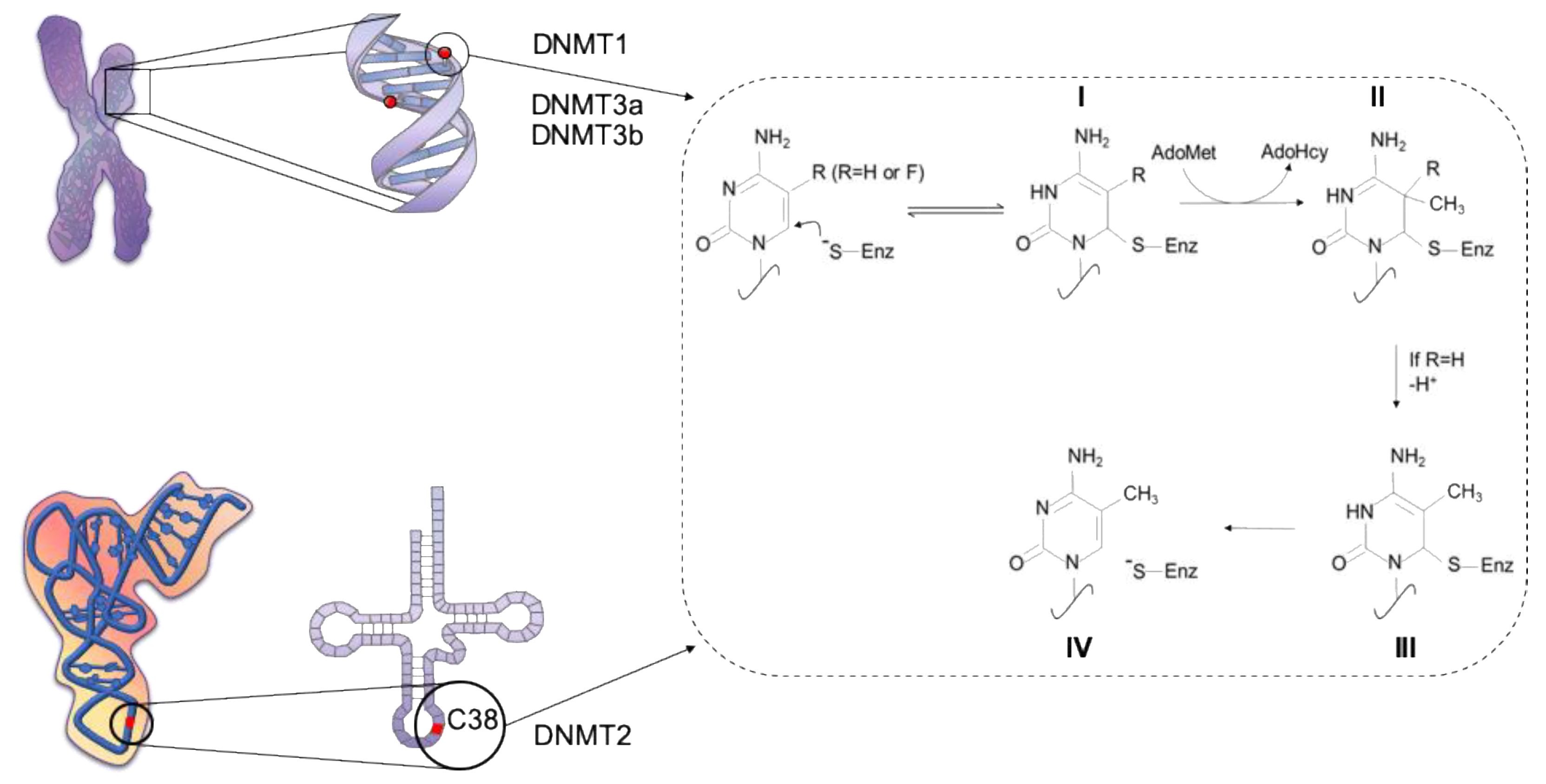
- Jeltsch, A.; Ehrenhofer-murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Santi, D.V. m5C RNA and m5C DNA methyl transferases use different cysteine residues as catalysts. Proc. Natl. Acad. Sci. USA 2000, 97, 8263–8265. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Krepelova, A.; Incarnato, D.; Maldotti, M.; Parlato, C.; Galvagni, F.; Matarese, F.; Stunnenberg, H.G.; Oliviero, S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013, 155, 121–134. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and chromatin—Unraveling the tangled web. Oncogene 2002, 21, 5361–5379. [Google Scholar] [CrossRef]
- Yarychkivska, O.; Shahabuddin, Z.; Comfort, N.; Boulard, M.; Bestor, T.H. BAH domains and a histone-like motif in DNA methyltransferase 1 (DNMT1) regulate de novo and maintenance methylation in vivo. J. Biol. Chem. 2018, 293, 19466–19475. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- Elliott, E.N.; Sheaffer, K.L.; Kaestner, K.H. The ‘de novo’ DNA methyltransferase Dnmt3b compensates the Dnmt1-deficient intestinal epithelium. Elife 2016, 5, e12975. [Google Scholar] [CrossRef]
- Song, Y.; van den Berg, P.R.; Markoulaki, S.; Soldner, F.; Dall’Agnese, A.; Henninger, J.E.; Drotar, J.; Rosenau, N.; Cohen, M.A.; Young, R.A.; et al. Dynamic Enhancer DNA Methylation as Basis for Transcriptional and Cellular Heterogeneity of ESCs. Mol. Cell 2019, 75, 905–920. [Google Scholar] [CrossRef]
- Liebl, K.; Zacharias, M. How methyl-sugar interactions determine DNA structure and flexibility. Nucleic Acids Res. 2019, 47, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Masle-Farquhar, E.; Luu, P.L.; Song, J.Z.; Qu, W.; Zotenko, E.; Gould, C.M.; Du, Q.; Peters, T.J.; Colino-Sanguino, Y.; et al. DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 2019, 35, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Ismail, K.; Gervin, K.; Kyllonen, A.; Hakkarainen, A.; Lundbom, J.; Jarvinen, E.A.; Harris, J.R.; Lundbom, N.; Rissanen, A.; et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenetics 2015, 7, 39. [Google Scholar] [PubMed]
- Visone, R.; Bacalini, M.G.; Di Franco, S.; Ferracin, M.; Colorito, M.L.; Pagotto, S.; Laprovitera, N.; Licastro, D.; Di Marco, M.; Scavo, E.; et al. DNA methylation of shelf, shore and open sea CpG positions distinguish high microsatellite instability from low or stable microsatellite status colon cancer stem cells. Epigenomics 2019, 11, 587–604. [Google Scholar]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Teissandier, A.; Bourc’his, D. Gene body DNA methylation conspires with H3K36me3 to preclude aberrant transcription. EMBO J. 2017, 36, 1471–1473. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Sartor, R.C.; Noshay, J.; Springer, N.M.; Briggs, S.P. Identification of the expressome by machine learning on omics data. Proc. Natl. Acad. Sci. USA 2019, 116, 18119–18125. [Google Scholar] [CrossRef]
- Sleutels, F.; Barlow, D.P. The Origins of Genomic. Adv. Genet. 2002, 46, 119–163. [Google Scholar]
- Gaszner, M.; Felsenfeld, G. Insulators: Exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006, 7, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Alasoo, K.; Rodrigues, J.; Mukhopadhyay, S.; Knights, A.J.; Mann, A.L.; Kundu, K.; Hale, C.; Dougan, G.; Gaffney, D.J. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 2018, 50, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hellman, A. DNA Methylation of Transcriptional Enhancers and Cancer Predisposition. Cell 2013, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Tatetsu, H.; Ueno, S.; Hata, H.; Yamada, Y.; Takeya, M.; Mitsuya, H.; Tenen, D.G.; Okuno, Y. Down-regulation of PU.1 by methylation of distal regulatory elements and the promoter is required for myeloma cell growth. Cancer Res. 2007, 67, 5328–5336. [Google Scholar] [CrossRef]
- West, A.G.; Gaszner, M.; Felsenfeld, G. Insulators: Many functions, many mechanisms. Genes Dev. 2002, 16, 271–288. [Google Scholar] [CrossRef]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 2–5. [Google Scholar] [CrossRef]
- Rao, X.; Evans, J.; Chae, H.; Pilrose, J.; Kim, S.; Yan, P.; Huang, R.l.; Lai, H.c.; Lin, H.; Liu, Y.; et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene 2013, 4519–4528. [Google Scholar] [CrossRef]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799. [Google Scholar] [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Cholewa-Waclaw, J.; Shah, R.; Webb, S.; Chhatbar, K.; Ramsahoye, B.; Pusch, O.; Yu, M.; Greulich, P.; Waclaw, B.; Bird, A.P. Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II traffic. Proc. Natl. Acad. Sci. USA 2019, 116, 14995–15000. [Google Scholar] [CrossRef] [PubMed]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Guldberg, P. DNA methylation analysis techniques. Biogerontology 2003, 4, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collist, C.M.; Wattt, F.; Griggt, G.W.; Molloyt, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohde, C.; Tierling, S.; Stamerjohanns, H.; Reinhardt, R.; Walter, J.; Jeltsch, A. DNA methylation analysis by bisulfite conversion, cloning, and sequencing of individual clones. Methods Mol. Biol. 2009, 507, 177–187. [Google Scholar]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Analysis Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Sant, K.E.; Nahar, M.S.; Dolinoy, D.C. DNA methylation screening and analysis. Methods Mol. Biol. 2012, 889, 385–406. [Google Scholar]
- Carmona, J.J., Jr.; Accomando, W.P.; Binder, A.M.; Hutchinson, J.N.; Pantano, L.; Izzi, B.; Just, A.C.; Lin, X.; Schwartz, J.; Vokonas, P.S.; et al. Empirical comparison of reduced representation bisul fi te sequencing and In fi nium BeadChip reproducibility and coverage of DNA methylation in humans. NPJ Genom. Med. 2017, 2, 13. [Google Scholar] [CrossRef]
- Li, N.; Ye, M.; Li, Y.; Yan, Z.; Butcher, L.M.; Sun, J.; Han, X.; Chen, Q.; Wang, J. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 2010, 52, 203–212. [Google Scholar] [CrossRef]
- Suzuki, M.; Liao, W.; Wos, F.; Johnston, A.D.; Degrazia, J.; Ishii, J.; Bloom, T.; Zody, M.C.; Germer, S.; Greally, J.M. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res. 2018, 28, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Bei, Y.; Church, H.E.; Dai, N.; Dimalanta, E.T.; Ettwiller, L.M.; Evans, T.C., Jr.; Langhorst, B.W.; Borgaro, J.G.; Guan, S.; et al. Enzymatic Methyl-Seq: The Next Generation of Methylome Analysis. 2019. Available online: https://www.neb.com/tools-and-resources/feature-articles/enzymatic-methyl-seq-the-next-generation-of-methylome-analysis (accessed on 15 September 2019).
- Clark, S.J.; Smallwood, S.A.; Lee, H.J.; Krueger, F.; Reik, W.; Kelsey, G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat. Protoc. 2017, 12, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Karemaker, I.D.; Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Guo, F.; Li, X.; Wu, X.; Fan, X.; Wen, L.; Tang, F. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc. 2015, 10, 645–659. [Google Scholar] [CrossRef]
- Gaiti, F.; Chaligne, R.; Gu, H.; Brand, R.M.; Kothen-hill, S.; Schulman, R.C.; Grigorev, K.; Risso, D.; Kim, K.-T.; Pastore, A.; et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 2019, 576–599. [Google Scholar] [CrossRef]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C.-X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar]
- Skvortsova, K.; Zotenko, E.; Luu, P.-L.; Gould, C.M.; Nair, S.S.; Clark, S.J.; Stirzaker, C. Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics Chromatin 2017, 10, 1–20. [Google Scholar] [CrossRef]
- Song, C.-X.; Diao, J.; Brunger, A.T.; Quake, S.R. Simultaneous single-molecule epigenetic imaging of DNA methylation and hydroxymethylation. PNAS 2016, 3–8. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, R.; Tabib, A.; Keshet, I.; Moss, J.; Sabag, O.; Goren, A.; Cedar, H. Role of transcription complexes in the formation of the basal methylation pattern in early development. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Dor, P.Y.; Cedar, P.H. Review Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, E.; et al. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 2019, 47, e46. [Google Scholar] [CrossRef]
- Bourc’his, D.; Xu, G.-L.; Lin, C.-S.; Bollman, B.; Bestor, T.H. Dnmt3L and the Establishment of Maternal Genomic Imprints. Science 2001, 294, 2536–2540. [Google Scholar] [CrossRef]
- Ferguson-smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12. [Google Scholar] [CrossRef]
- Barlow, D.P.; Bartolomei, M.S. Genomic Imprinting in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 31, 493–525. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Deal, C.; Paquette, J.; Goodyer, C.G.; Polychronakos, C. Parental genomic imprinting of the. Nat. Genet. 1993, 4, 98–101. [Google Scholar] [CrossRef]
- Park, K.-S.; Mitra, A.; Rahat, B.; Kim, K.; Pfeifer, K. Loss of imprinting mutations define both distinct and overlapping roles for misexpression of IGF2 and of H19 lncRNA. Nucleic Acids Res. 2017, 45, 12766–12779. [Google Scholar] [CrossRef][Green Version]
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Asp. Med. 2013, 34, 826–840. [Google Scholar] [CrossRef]
- Xu, Q.; Xiang, Y.; Wang, Q.; Wang, L.; Brind’Amour, J.; Bogutz, A.B.; Zhang, Y.; Zhang, B.; Yu, G.; Xia, W.; et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat. Genet. 2019, 51, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA Methylation in Health and Disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lou, H.; Xie, K.; Wang, H.; Chen, N.; Aparicio, O.M.; Zhang, M.Q.; Jiang, R.; Chen, T. Reconstructing cell cycle pseudo time-series via single-cell transcriptome data. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.E.; Fraga, M.F.; Weaver, I.C.G.; Berdasco, M.; Brown, S.E.; Fraga, M.F.; Weaver, I.C.G.; Berdasco, M.; Szyf, M. Variations in DNA Methylation Patterns During the Cell Cycle of HeLa Cells. Epigenetics 2007, 2, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Desjobert, C.; Maï, M.E.; Gérard-hirne, T.; Guianvarc, D.; Carrier, A.; Pottier, C.; Arimondo, P.B.; Riond, J.; Desjobert, C.; Maï, M.E.; et al. Combined analysis of DNA methylation and cell cycle in cancer cells Combined analysis of DNA methylation and cell cycle in cancer cells. Epigenetics 2015, 2294, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, A.R.; Idrizi, A.; Rizzardi, L.; Feinberg, A.P.; Hansen, K.D. DNA methylation is stable during replication and cell cycle arrest. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. Dna methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Li, C.; Zhao, S.; Zhang, N.; Zhang, S.; Hou, Y. Differences of DNA methylation profiles between monozygotic twins’ blood samples. Mol. Biol. Rep. 2013, 40, 5275–5280. [Google Scholar] [CrossRef]
- Souren, N.Y.; Gerdes, L.A.; Lutsik, P.; Gasparoni, G.; Beltran, E.; Salhab, A.; Kumpfel, T.; Weichenhan, D.; Plass, C.; Hohlfeld, R.; et al. DNA methylation signatures of monozygotic twins clinically discordant for multiple sclerosis. Nat. Commun. 2019, 10, 2094. [Google Scholar] [CrossRef] [PubMed]
- Castillo-fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Garrett-Bakelman, F.E.; Manjul, D.; Stefan, J.G. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science 2019, 144, eaau8650. [Google Scholar]
- Beayno, A.; Hayek, S.E.; Noufi, P.; Tarabay, Y.; Shamseddee, W. The Role of Epigenetics in Addiction: Clinical Overview and Recent Updates. In Psychiatric Disorders, 2nd ed.; Human Press: Hertfordshire, UK, 2019; pp. 609–631. [Google Scholar]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure-Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Horsthemke, B. In Brief: Genomic imprinting and imprinting diseases. J. Patol. 2014, 485–487. [Google Scholar] [CrossRef]
- Eggermann, T.; Nanclares, G.P.D.; Maher, E.R.; Temple, I.K.; Tümer, Z.; Monk, D.; Mackay, D.J.G.; Grønskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting disorders: A group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenet. 2015. [Google Scholar] [CrossRef]
- Muurinen, M.; Hannula-jouppi, K.; Reinius, L.E.; Söderhäll, C. Hypomethylation of HOXA4 promoter is common in Silver-Russell syndrome and growth restriction and associates with stature in healthy children. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Wakeling, E.L.; Brioude, F.; Lokulo-sodipe, O.; Connell, S.M.O.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Gerhardt, J. Epigenetic modi fi cations in human fragile X pluripotent stem cells; Implications in fragile X syndrome modeling. Brain Res. 2017, 1656, 55–62. [Google Scholar] [CrossRef]
- Korb, E.; Herre, M.; Zucker-scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B.; Korb, E.; Herre, M.; Zucker-scharff, I.; Gresack, J.; et al. Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition Article Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 2017, 170, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Tabib, A.; Kahan, T.; Orlanski, S.; Gropp, M.; Tabach, Y.; Yanuka, O.; Benvenisty, N.; Keshet, I.; Cedar, H. Epigenetic mechanism of FMR1 inactivation in Fragile X syndrome. Int. J. Dev. Biol. 2017, 61, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Baglivo, I.; Esposito, S.; De, C.L.; Sparago, A.; Anvar, Z.; Riso, V.; Cammisa, M.; Fattorusso, R.; Grimaldi, G.; Riccio, A.; et al. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1. FEBS Lett. 2013, 587, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Boonen, S.E.; Dahl, C.; Hahnemann, J.M.D.; Mackay, D.J.D.G.; Tümer, Z.; Grønskov, K.; Temple, I.K.; Guldberg, P.; Tommerup, N. Genome-wide DNA methylation analysis of transient neonatal diabetes type 1 patients with mutations in ZFP57. BMC Med. Genet. 2016, 17, 29. [Google Scholar] [CrossRef]
- Fontana, L.; Bedeschi, M.F.; Maitz, S.; Cereda, A.; Faré, C.; Motta, S.; D’Ursi, P.; Orro, A.; Pecile, V. Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15. 5 related imprinting disorders. Epigenetics 2018, 13, 897–909. [Google Scholar] [CrossRef]
- Krzyzewska, I.M.; Alders, M.; Maas, S.M.; Bliek, J.; Venema, A.; Henneman, P.; Rezwan, F.I.; Lip, K.V.D.; Mul, A.N.; Mackay, D.J.G.; et al. Genome-wide methylation profiling of Beckwith-Wiedemann syndrome patients without molecular confirmation after routine diagnostics. Clin. Epigenet. 2019, 11, 53. [Google Scholar] [CrossRef]
- Margolis, S.S.; Sell, G.L.; Zbinden, M.A.; Bird, L.M. Angelman Syndrome. Neurotherapeutics 2015, 12, 641–650. [Google Scholar] [CrossRef]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocronol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Wang, S.E.; Jiang, Y.-h. Potential of Epigenetic Therapy for Prader-Willi Syndrome. Trends Pharmacol. Sci. 2019, 40, 605–608. [Google Scholar] [CrossRef]
- Rochtus, A.; Martin-Trujillo, A.; Izzi, B.; Elli, F.; Garin, I.; Linglart, A.; Mantovani, G.; Perez de Nanclares, G.; Thiele, S.; Decallonne, B.; et al. Genome-wide DNA methylation analysis of pseudohypoparathyroidism patients with GNAS imprinting defects. Clin. Epigenet. 2016, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Tafaj, O.; Jüppner, H. Pseudohypoparathyroidism: One gene, several syndromes. J. Endocronol. Investig. 2017, 40, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Wainscoat, J.S.; Molecular, L.R.F.; Unit, H.; Hospital, J.R. Gene silencing by DNA methylation in haematological malignancies. Br. J. Haematol. 2007, 138, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Erola, P.; Torabi, K.; Miró, R.; Camps, J. The non-random landscape of somatically-acquired uniparental disomy in cancer. Oncotarget 2019, 10, 3982–3984. [Google Scholar] [CrossRef]
- Gelli, E.; Pinto, A.M.; Somma, S.; Imperatore, V.; Mari, F.; Cannone, M.G.; Galimberti, D.; Renieri, A.; Hadjistilianou, T.; Currò, A.; et al. Evidence of predisposing epimutation in retinoblastoma. Hum. Mutat. 2019, 40, 201–206. [Google Scholar] [CrossRef]
- Issa, J.P.; Baylin, S.B.; Herman, J.G. DNA methylation changes in hematologic malignancies: Biologic and clinical implications. Leukemia 1997, 11 (Suppl. 1), S7–S11. [Google Scholar]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322–e328. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 4–11. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Kurkjian, C.; Kummar, S.; Murgo, A.J. DNA methylation: Its role in cancer development and therapy. Curr. Probl. Cancer 2008, 32, 187–235. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Baretti, M.; Azad, N.S. The role of epigenetic therapies in colorectal cancer. Curr. Probl. Cancer 2018, 42, 530–547. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.W.T.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target ? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018, 1063, 1060–1063. [Google Scholar] [CrossRef] [PubMed]
- Römermann, D.; Hasemeier, B.; Metzig, K.; Göhring, G.; Schlegelberger, B.; Länger, F.; Lehmann, U. Global increase in DNA methylation in patients with myelodysplastic syndrome. Leukemia 2008, 1954–1956. [Google Scholar] [CrossRef]
- Aleshin, A.; Greenberg, P.L. Molecular pathophysiology of the myelodysplastic syndromes: Insights for targeted therapy. Blood Adv. 2018, 2, 2787–2797. [Google Scholar] [CrossRef] [PubMed]
- Nolte, F.; Hofmann, W.-K. Myelodysplastic syndromes: Molecular pathogenesis and genomic changes. Ann. Hmatol. 2008, 777–795. [Google Scholar] [CrossRef]
- Hu, X.-m.W.J.-b.; Yang, J.; Qian, W. CEBPA methylation and mutation in myelodysplastic syndrome. Med. Oncol. 2015, 32, 192. [Google Scholar]
- Figueroa, M.E.; Skrabanek, L.; Li, Y.; Jiemjit, A.; Fandy, T.E.; Paietta, E.; Fernandez, H.; Tallman, M.S.; Greally, J.M.; Carraway, H.; et al. MYELOID NEOPLASIA MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009, 114, 3448–3459. [Google Scholar] [CrossRef]
- Stosch, J.M.; Rothenberg-thurley, M.; Riba, J.; Renz, N.; Szarc, K.; Pfeifer, D.; Follo, M.; Pahl, H.L.; Zimmermann, S.; Duyster, J.; et al. Gene mutations and clonal architecture in myelodysplastic syndromes and changes upon progression to acute myeloid leukaemia and under treatment. Br. J. Haematol. 2018, 830–842. [Google Scholar] [CrossRef]
- Heuser, M.; Yun, H.; Thol, F. Epigenetics in myelodysplastic syndromes. Semin. Cancer Biol. 2018, 51, 170–179. [Google Scholar] [CrossRef]
- Hasegawa, N.; Oshima, M.; Sashida, G.; Matsui, H.; Koide, S.; Saraya, A.; Wang, C.; Muto, T.; Takane, K.; Kaneda, A.; et al. Impact of combinatorial dysfunctions of Tet2 and Ezh2 on the epigenome in the pathogenesis of myelodysplastic syndrome. Leukemia 2017, 31, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Garcia-manero, G.; Montalban-bravo, G. Annual Clinical Updates in Hematological Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 129–147. [Google Scholar] [CrossRef]
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Nguyen, A.; Shi, T.; Tang, L.; Ni, X.; Escoubet, L.; Macbeth, K.J. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. USA 2018, 116, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Yamada, H.; Ushijima, T. Chapter 5 Cancer Epigenetics Aberrant DNA Methylation in Cancer Diagnosis and Treatment. In Oncogenomics; Academic Press: Cambridge, MA, USA, 2019; pp. 65–76. [Google Scholar] [CrossRef]


 ] mC and/or hmC from the deamination by APOBEC. On the right hand-side, single-cell bisulfite-based methods as indicated: single-cell bisulfite sequencing (scBS) and single-cell reduced representation bisulfite sequencing (scRRBS). The red dot (●) indicates methylated cytosine.
] mC and/or hmC from the deamination by APOBEC. On the right hand-side, single-cell bisulfite-based methods as indicated: single-cell bisulfite sequencing (scBS) and single-cell reduced representation bisulfite sequencing (scRRBS). The red dot (●) indicates methylated cytosine.
] mC and/or hmC from the deamination by APOBEC. On the right hand-side, single-cell bisulfite-based methods as indicated: single-cell bisulfite sequencing (scBS) and single-cell reduced representation bisulfite sequencing (scRRBS). The red dot (●) indicates methylated cytosine.
] mC and/or hmC from the deamination by APOBEC. On the right hand-side, single-cell bisulfite-based methods as indicated: single-cell bisulfite sequencing (scBS) and single-cell reduced representation bisulfite sequencing (scRRBS). The red dot (●) indicates methylated cytosine.

{kind=link}
{kind=link}
{kind=link}
| Imprinting Diseases | Epigenetic Lesions | Reference |
|---|---|---|
| Transient Neonatal Diabetes Mellitus Type 1 (TNDM1) | Hypomethylation of the maternally imprinted genes PLAGL1 and HYMAI | [96,97] |
| Silver-Russell Syndrome | Hypomethylation of the paternally imprinted locus H19/IGF2 and promoter hypomethylation of HOXA4 | [91,92] |
| Beckwith-Wiedemann | Imprinting defects within two imprinted domains, IGF2/H19 and CDKN1C/KCNQ1OT1 | [98,99] |
| Fragile X Syndrome | De novo methylation of the FMR1 gene | [93,95] |
| Angelman Syndrome | Imprinting defects within chromosome 15q11-q13 that alter the expression of the maternally inherited UBE3A | [100,101] |
| Prader-Willi Syndrome | Loss of expression of the paternally inherited chromosome 15q11.2-q13 due to imprinting defects | [102,103] |
| Pseudohypoparathyroidism | Epigenetic defects in the imprinted GNAS cluster on chromosome 20q13.3 | [104,105] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borchiellini, M.; Ummarino, S.; Di Ruscio, A. The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells 2019, 8, 1243. https://doi.org/10.3390/cells8101243
Borchiellini M, Ummarino S, Di Ruscio A. The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells. 2019; 8(10):1243. https://doi.org/10.3390/cells8101243
Chicago/Turabian StyleBorchiellini, Marta, Simone Ummarino, and Annalisa Di Ruscio. 2019. "The Bright and Dark Side of DNA Methylation: A Matter of Balance" Cells 8, no. 10: 1243. https://doi.org/10.3390/cells8101243
APA StyleBorchiellini, M., Ummarino, S., & Di Ruscio, A. (2019). The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells, 8(10), 1243. https://doi.org/10.3390/cells8101243