Ketamine Increases Proliferation of Human iPSC-Derived Neuronal Progenitor Cells via Insulin-Like Growth Factor 2 and Independent of the NMDA Receptor

 and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Real-Time Cell Proliferation
2.4. Immunofluorescence Staining
2.5. Fluorescence-Based Calcium Mobilization Assay
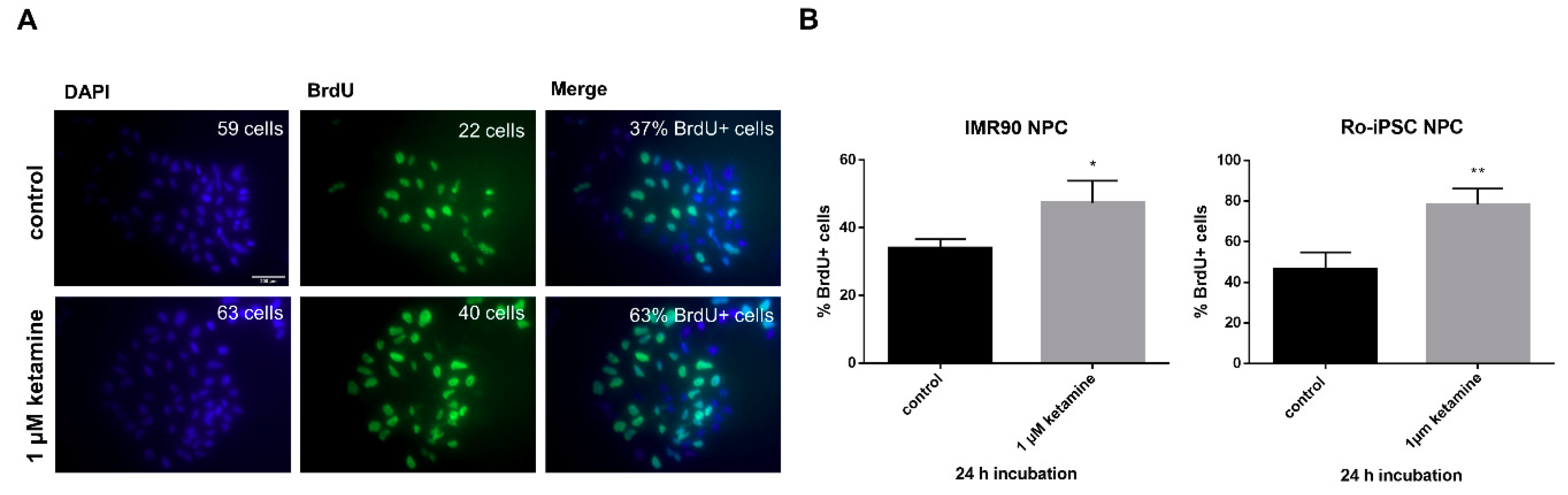
2.6. BrdU Analysis of Cell Proliferation In Vitro
2.7. RNA Extraction and Library Preparation for Transcriptome Sequencing
2.8. RNA-Seq Data Analysis
2.9. Quantitative Real-Time PCR
2.10. siRNA Transfection
2.11. Cyclic Adenosine Monophosphate (cAMP)-Glo Assay
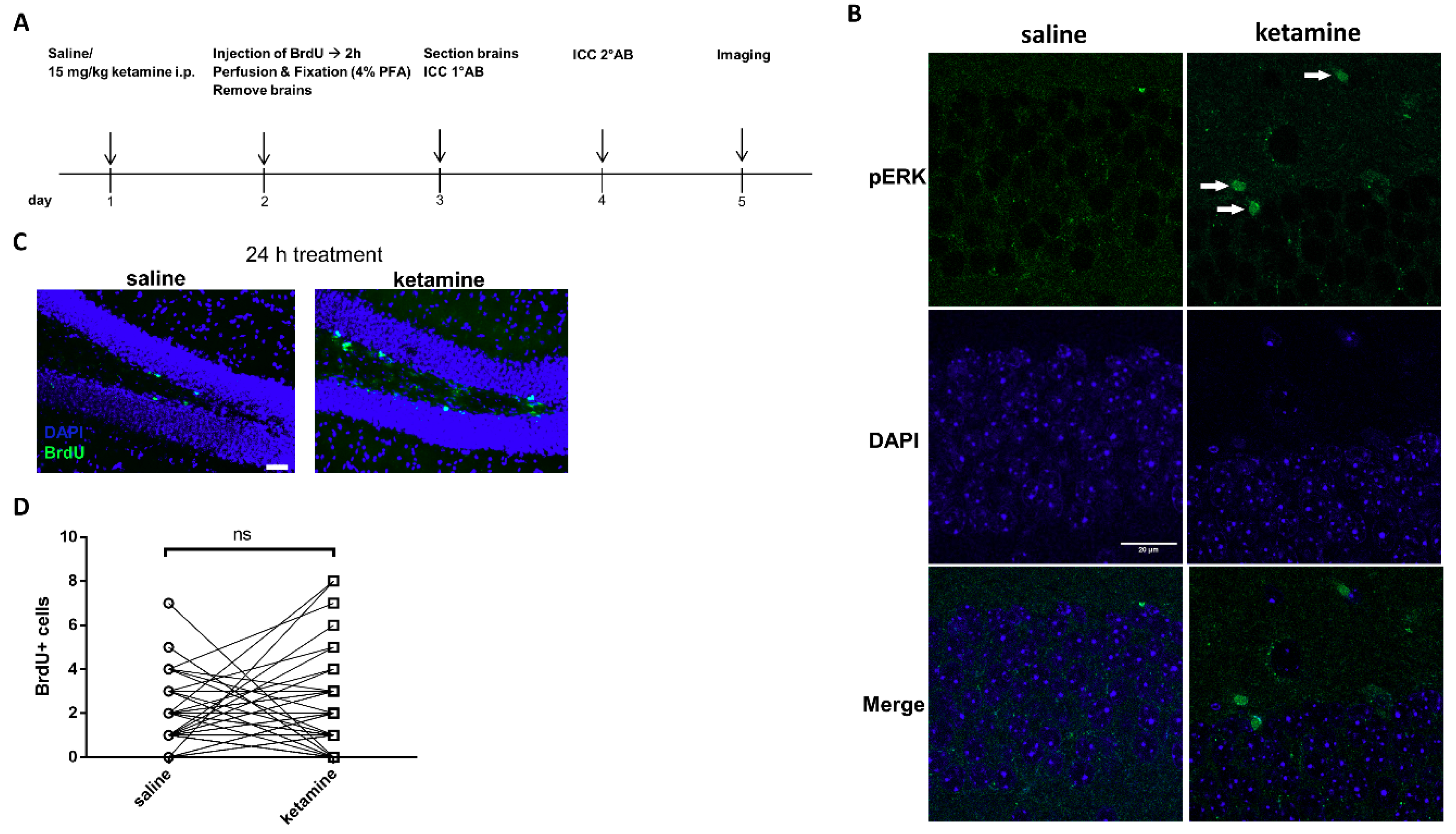
2.12. In Vivo Administration of Ketamine and Immunohistochemistry of BrdU and pERK1/2
2.13. Statistical Analysis
3. Results
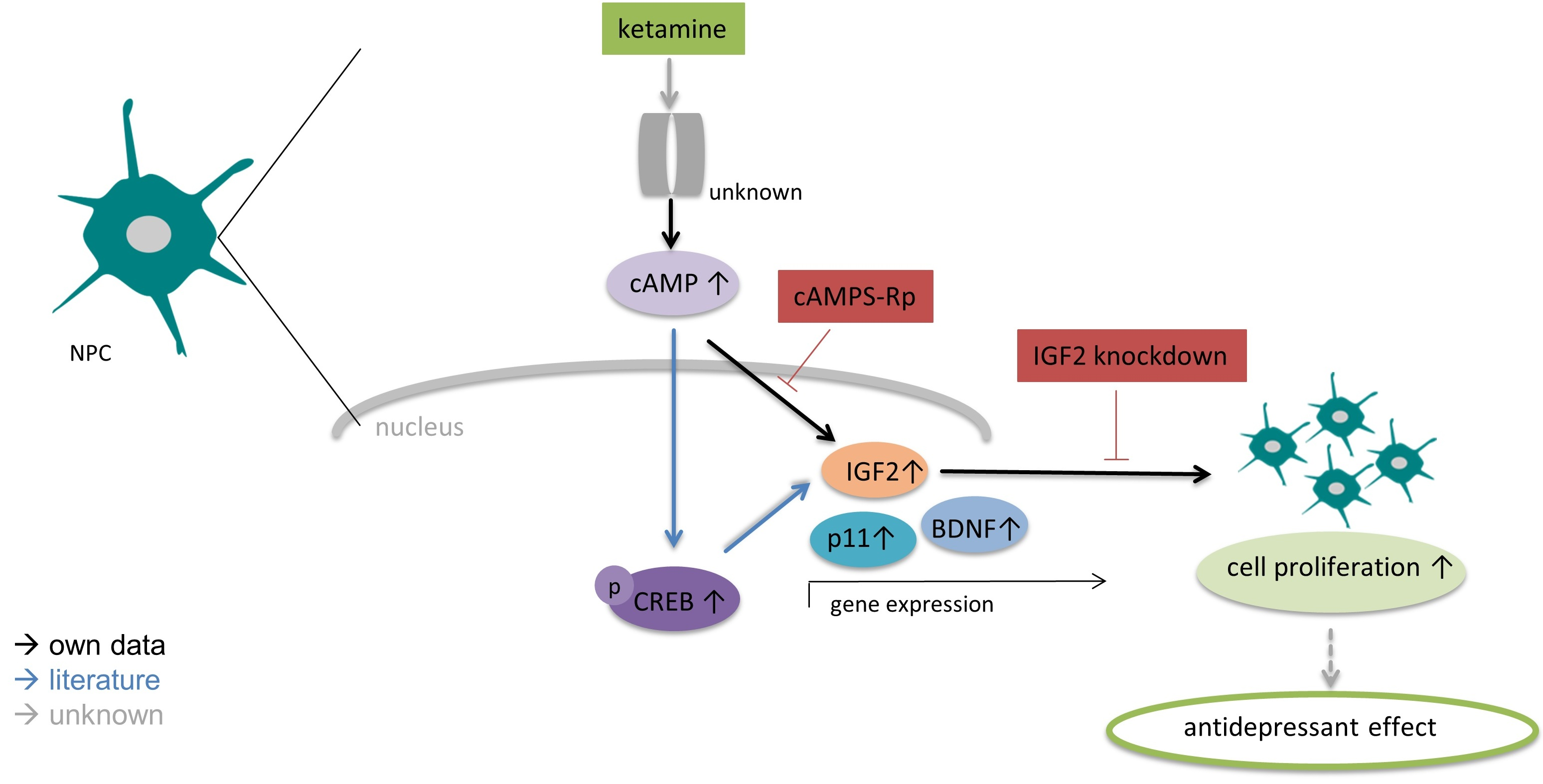
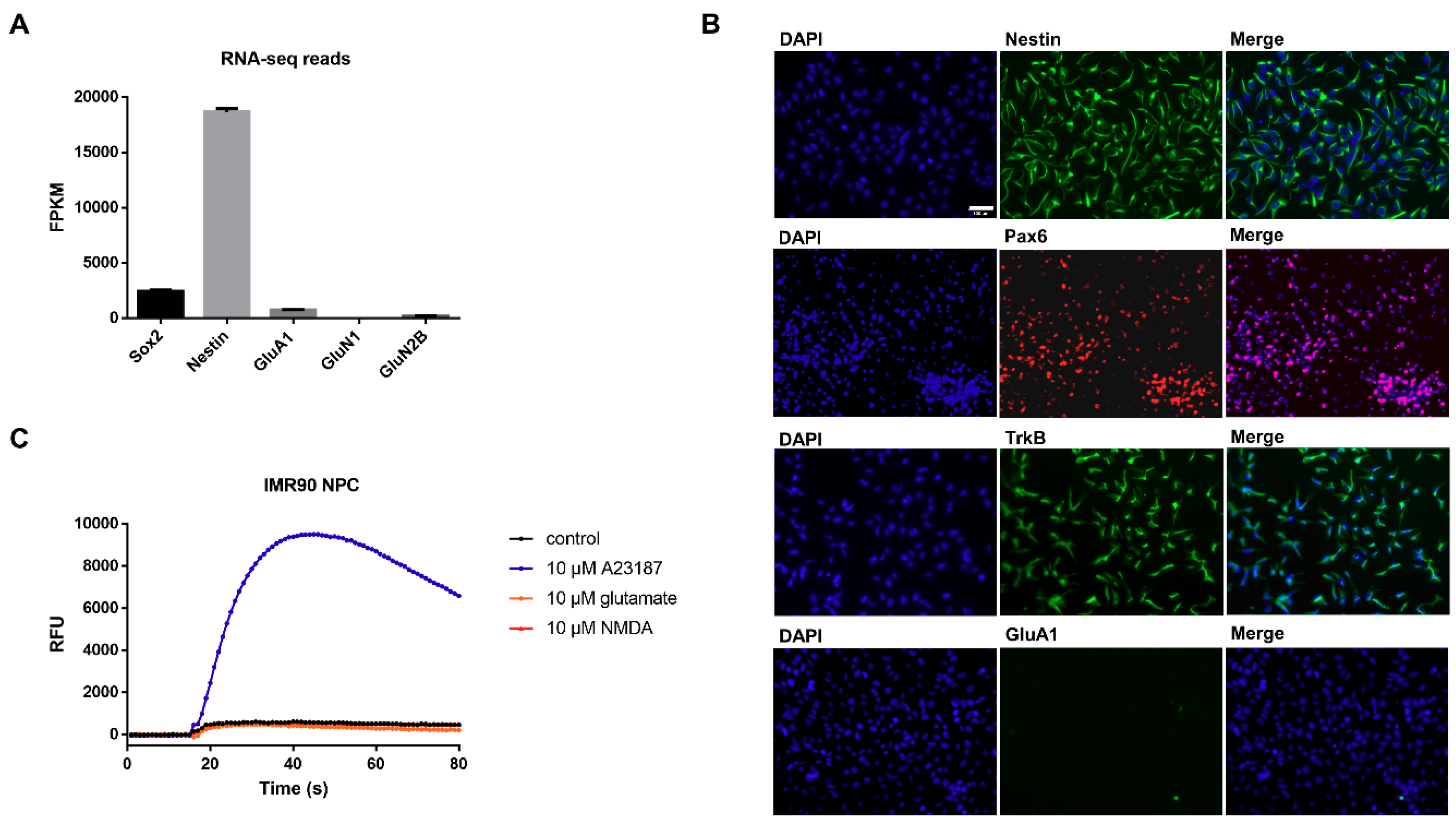
3.1. Characterization of Undifferentiated iPSC-Derived NPCs Proves Absence of Ionotropic Glutamate Receptors
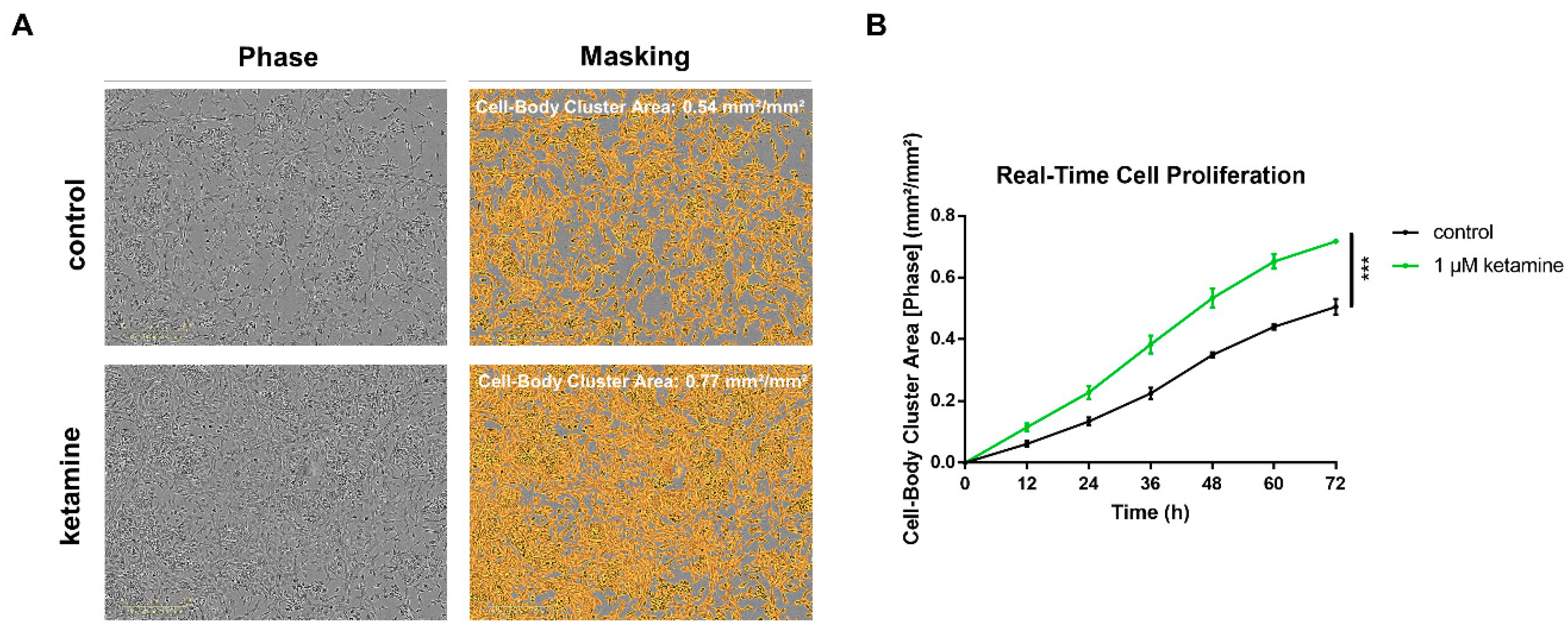
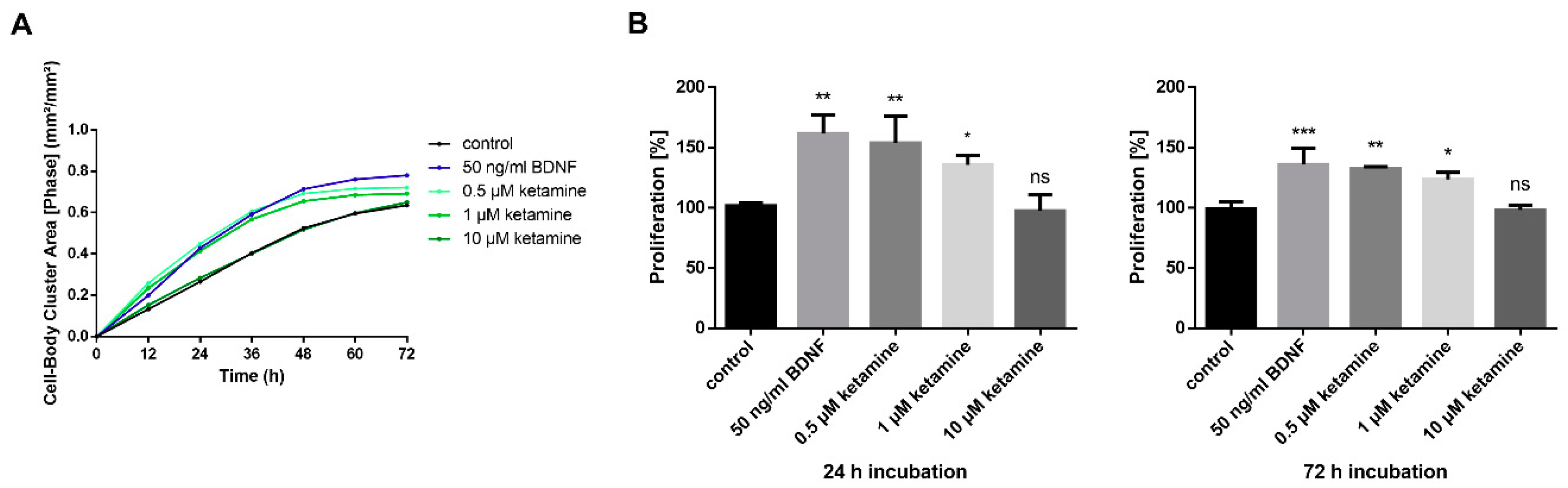
3.2. Ketamine Increases Cell Proliferation of Human iPSC-Derived NPCs
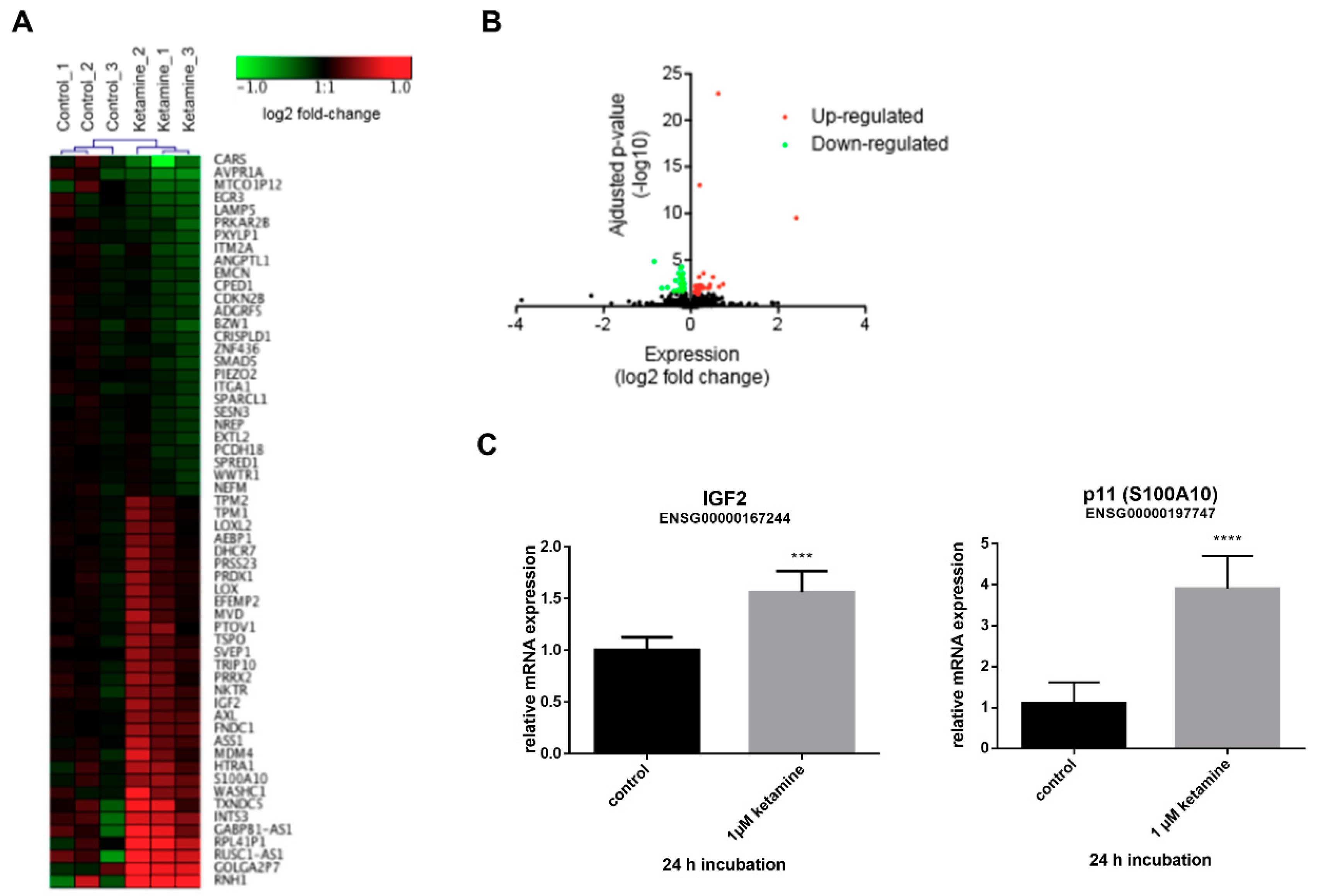
3.3. Ketamine Alters Gene Expression in Human iPSC-Derived NPCs
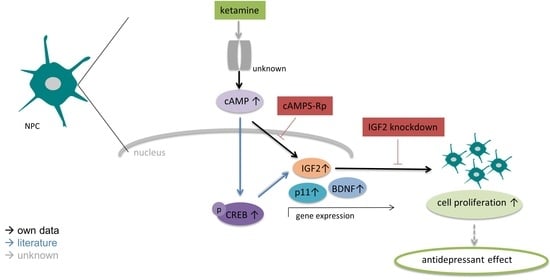
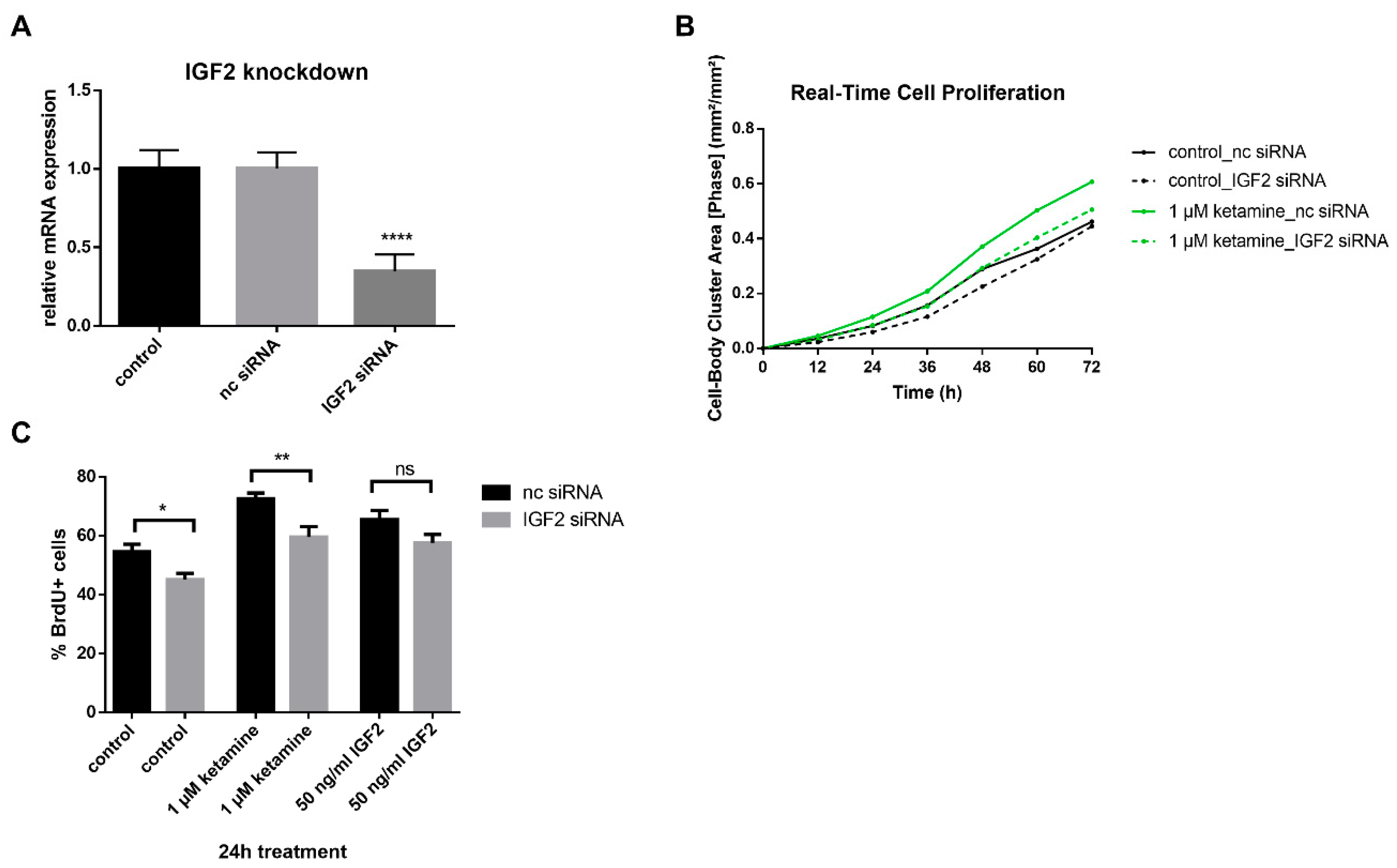
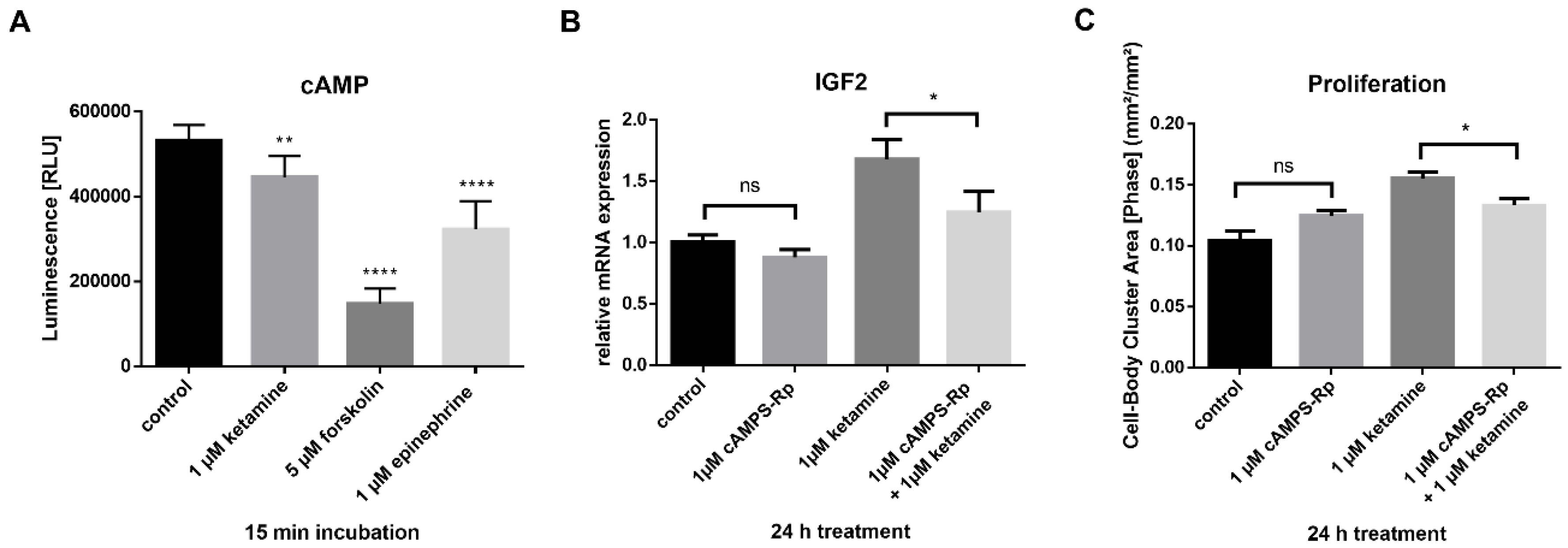
3.4. IGF2 Signaling is Involved in the Proliferative Effects of Ketamine
3.5. Ketamine Increases cAMP Levels in NPCs within 15 min
3.6. Phosphorylation of ERK1/2 is Enhanced by Ketamine in the SGZ of Mice
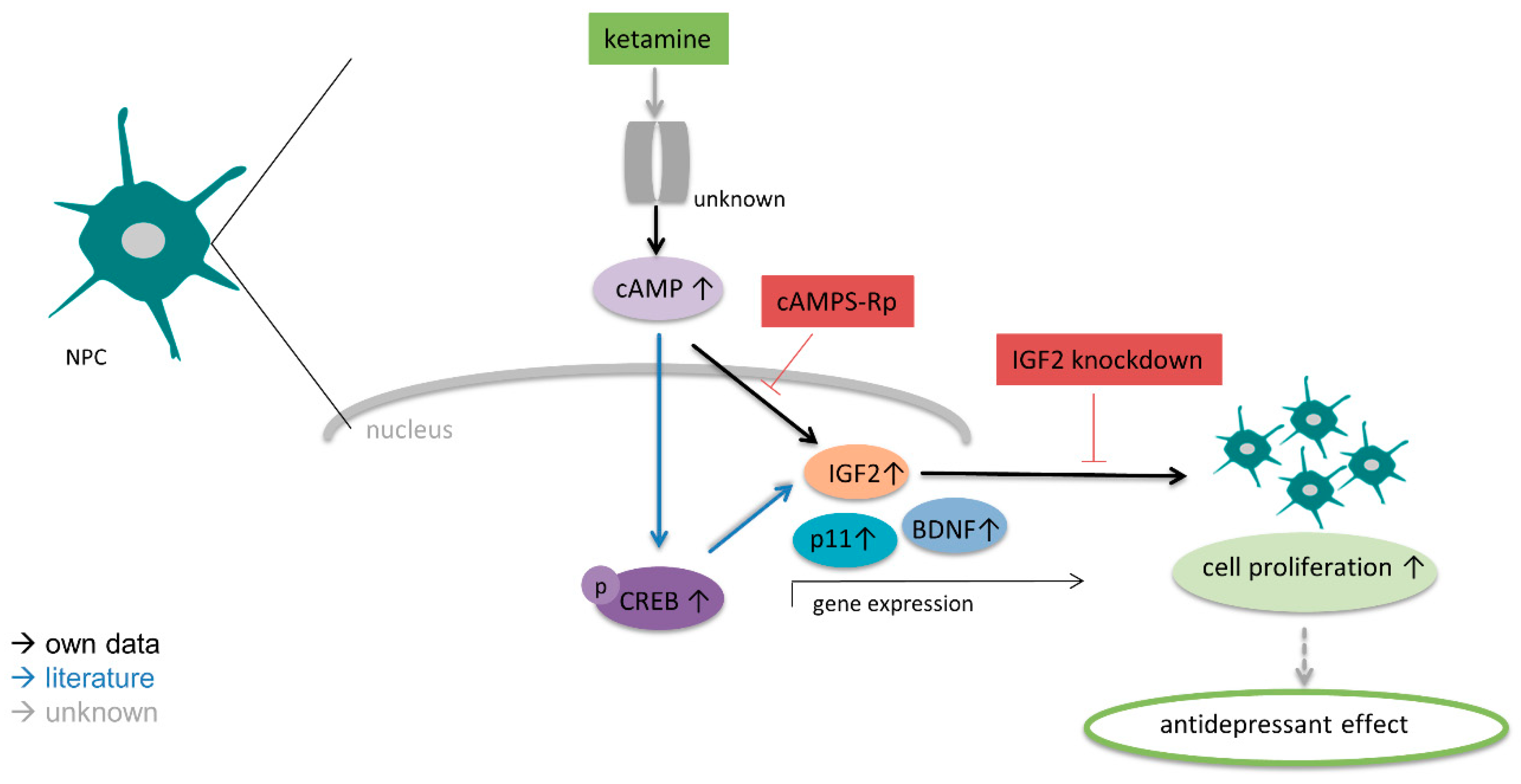
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Harmer, C.J.; Duman, R.S.; Cowen, P.J. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry 2017, 4, 409–418. [Google Scholar] [CrossRef]
- Nemeroff, C.B. Prevalence and management of treatment-resistant depression. J. Clin. Psychiatry 2007, 68, 17–25. [Google Scholar] [PubMed]
- Warner-Schmidt, J.L.; Duman, R.S. Hippocampal neurogenesis: Opposing effects of stress and antidepressant treatment. Hippocampus 2006, 16, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Sahay, A.; Hen, R. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 2007, 10, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, G.M.; Campbell, S.; McEwen, B.S.; Macdonald, K.; Amano, S.; Joffe, R.T.; Nahmias, C.; Young, L.T. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc. Natl. Acad. Sci. USA 2003, 100, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Pilar-Cuellar, F.; Vidal, R.; Díaz, A.; Castro, E.; Dos Anjos, S.; Pascual-Brazo, J.; Linge, R.; Vargas, V.; Blanco, H.; Martínez-Villayandre, B.; et al. Neural Plasticity and Proliferation in the Generation of Antidepressant Effects: Hippocampal Implication. Neural Plast. 2013, 2013, 537265. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic Antidepressant Treatment Increases Neurogenesis in Adult Rat Hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e5. [Google Scholar] [CrossRef]
- Ma, Z.; Zang, T.; Birnbaum, S.G.; Wang, Z.; Johnson, J.E.; Zhang, C.-L.; Parada, L.F. TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat. Commun. 2017, 8, 1668. [Google Scholar] [CrossRef]
- Andrus, B.M.; Blizinsky, K.; Vedell, P.T.; Dennis, K.; Shukla, P.K.; Schaffer, D.J.; Radulovic, J.; Churchill, G.A.; Redei, E.E. Gene expression patterns in the hippocampus and amygdala of endogenous depression and chronic stress models. Mol. Psychiatry 2012, 17, 49–61. [Google Scholar] [CrossRef]
- Bracko, O.; Singer, T.; Aigner, S.; Knobloch, M.; Winner, B.; Ray, J.; Clemenson, G.D.; Suh, H.; Couillard-Despres, S.; Aigner, L.; et al. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 2012, 32, 3376–3387. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Cheng, Y.; Sitbon, Y.; Lowell, J.; Grieco, S.; Worthen, R.; Desse, S.; Barreda-Diaz, A. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, P.; Juszczak, G.R.; Gościk, J.; Stankiewicz, A.M.; Wieczorek, M.; Zwierzchowski, L.; Swiergiel, A.H. Stress susceptibility-specific phenotype associated with different hippocampal transcriptomic responses to chronic tricyclic antidepressant treatment in mice. BMC Neurosci. 2013, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Kim, Y.; Warner-Schmidt, J.; Oh, Y.-S.; Greengard, P. p11 and its role in depression and therapeutic responses to antidepressants. Nat. Rev. Neurosci. 2013, 14, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Egeland, M.; Warner-Schmidt, J.; Greengard, P.; Svenningsson, P. Neurogenic Effects of Fluoxetine Are Attenuated in p11 (S100A10) Knockout Mice. Biol. Psychiatry 2010, 67, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J.L.; Chen, E.Y.; Zhang, X.; Marshall, J.J.; Morozov, A.; Svenningsson, P.; Greengard, P. A role for p11 in the antidepressant action of brain-derived neurotrophic factor. Biol. Psychiatry 2010, 68, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Richards, E.M.; Niciu, M.J.; Ionescu, D.F.; Zoghbi, S.S.; Hong, J.; Telu, S.; Hines, C.S.; Pike, V.W.; Zarate, C.A.; et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol. Psychiatry 2017, 22, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.G.; Deligiannidis, K.M.; Ulbricht, C.M.; Landolin, C.S.; Patel, J.K.; Rothschild, A.J. Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: A randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2013, 74, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Farchione, T.; Potter, A.; Chen, Q.; Temple, R. Esketamine for Treatment-Resistant Depression—First FDA-Approved Antidepressant in a New Class. N. Engl. J. Med. 2019, 381, 1–4. [Google Scholar] [CrossRef]
- Yao, N.; Skiteva, O.; Zhang, X.; Svenningsson, P.; Chergui, K. Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol. Psychiatry 2018, 23, 2066–2077. [Google Scholar] [CrossRef]
- Scheuing, L.; Chiu, C.-T.; Liao, H.-M.; Chuang, D.-M. Antidepressant mechanism of ketamine: Perspective from preclinical studies. Front. Mol. Neurosci. 2015, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Soumier, A.; Carter, R.M.; Schoenfeld, T.J.; Cameron, H.A. New Hippocampal Neurons Mature Rapidly in Response to Ketamine But Are Not Required for Its Acute Antidepressant Effects on Neophagia in Rats. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef] [PubMed]
- Simão, D.; Silva, M.M.; Terrasso, A.P.; Arez, F.; Sousa, M.F.; Mehrjardi, N.Z.; Šarić, T.; Gomes-Alves, P.; Raimundo, N.; Alves, P.M.; et al. Recapitulation of Human Neural Microenvironment Signatures in iPSC-Derived NPC 3D Differentiation. Stem Cell Rep. 2018, 11, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 2009, 106, 3225–3230. [Google Scholar] [CrossRef] [PubMed]
- Dage, J.L.; Colvin, E.M.; Fouillet, A.; Langron, E.; Roell, W.C.; Li, J.; Mathur, S.X.; Mogg, A.J.; Schmitt, M.G.; Felder, C.C.; et al. Pharmacological characterisation of ligand- and voltage-gated ion channels expressed in human iPSC-derived forebrain neurons. Psychopharmacology 2014, 231, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Tabata, Y.; Imaizumi, Y.; Kohmura, N.; Sugawara, M.; Sawada, K.; Yamazaki, K.; Ito, M. Characterization of Human Hippocampal Neural Stem/Progenitor Cells and Their Application to Physiologically Relevant Assays for Multiple Ionotropic Glutamate Receptors. J. Biomol. Screen. 2014, 19, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Perţea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiang, L.; Zhang, X.; Chen, H. In-vitro effects of brain-derived neurotrophic factor on neural progenitor/stem cells from rat hippocampus. NeuroReport 2009, 20, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.H.; Schappi, J.M.; Singh, H.; Senese, N.B.; Rasenick, M.M. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Gao, Y.; Xiao, Z.; Chen, B.; Han, J.; Zhang, J.; Wang, X.; Dai, J. Erk1/2 promotes proliferation and inhibits neuronal differentiation of neural stem cells. Neurosci. Lett. 2009, 461, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Salvadore, G.; Luckenbaugh, D.A.; Manji, H.K.; Zarate, C.A., Jr. Rapid Onset of Antidepressant Action: A New Paradigm in the Research and Treatment of Major Depression. J. Clin. Psychiatry 2008, 69, 946–958. [Google Scholar] [CrossRef]
- Zorumski, C.F.; Izumi, Y.; Mennerick, S. Ketamine: NMDA Receptors and Beyond. J. Neurosci. 2016, 36, 11158–11164. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Kanning, K.; Cavanaugh, J.E.; Xia, Z. Neuroprotection by Brain-derived Neurotrophic Factor Is Mediated by Extracellular Signal-regulated Kinase and Phosphatidylinositol 3-Kinase. J. Biol. Chem. 1999, 274, 22569–22580. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-L.; Zhou, Z.-Q.; Zhang, G.-F.; Yang, C.; Wang, X.-M.; Shen, J.-C.; Hashimoto, K.; Yang, J.-J. Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl. Psychiatry 2016, 6, e741. [Google Scholar] [CrossRef] [PubMed]
- Grieco, S.F.; Cheng, Y.; Eldar-Finkelman, H.; Jope, R.S.; Beurel, E. Up-regulation of insulin-like growth factor 2 by ketamine requires glycogen synthase kinase-3 inhibition. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 72, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Stern, S.A.; Garcia-Osta, A.; Saunier-Rebori, B.; Pollonini, G.; Bambah-Mukku, D.; Blitzer, R.D.; Alberini, C.M. A critical role for IGF-II in memory consolidation and enhancement. Nature 2011, 469, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Kim, J.-E.; Lee, R.; Malberg, J.E.; Chen, J.; Steffen, C.; Zhang, Y.-J.; Nestler, E.J.; Duman, R.S. Regulation of Neurogenesis in Adult Mouse Hippocampus by cAMP and the cAMP Response Element-Binding Protein. J. Neurosci. 2002, 22, 3673–3682. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossert, A.; Mehrjardi, N.Z.; Bailey, S.J.; Lindsay, M.A.; Hescheler, J.; Šarić, T.; Teusch, N. Ketamine Increases Proliferation of Human iPSC-Derived Neuronal Progenitor Cells via Insulin-Like Growth Factor 2 and Independent of the NMDA Receptor. Cells 2019, 8, 1139. https://doi.org/10.3390/cells8101139
Grossert A, Mehrjardi NZ, Bailey SJ, Lindsay MA, Hescheler J, Šarić T, Teusch N. Ketamine Increases Proliferation of Human iPSC-Derived Neuronal Progenitor Cells via Insulin-Like Growth Factor 2 and Independent of the NMDA Receptor. Cells. 2019; 8(10):1139. https://doi.org/10.3390/cells8101139
Chicago/Turabian StyleGrossert, Alessandra, Narges Zare Mehrjardi, Sarah J. Bailey, Mark A. Lindsay, Jürgen Hescheler, Tomo Šarić, and Nicole Teusch. 2019. "Ketamine Increases Proliferation of Human iPSC-Derived Neuronal Progenitor Cells via Insulin-Like Growth Factor 2 and Independent of the NMDA Receptor" Cells 8, no. 10: 1139. https://doi.org/10.3390/cells8101139
APA StyleGrossert, A., Mehrjardi, N. Z., Bailey, S. J., Lindsay, M. A., Hescheler, J., Šarić, T., & Teusch, N. (2019). Ketamine Increases Proliferation of Human iPSC-Derived Neuronal Progenitor Cells via Insulin-Like Growth Factor 2 and Independent of the NMDA Receptor. Cells, 8(10), 1139. https://doi.org/10.3390/cells8101139