HtrA4 Protease Promotes Chemotherapeutic-Dependent Cancer Cell Death


Abstract
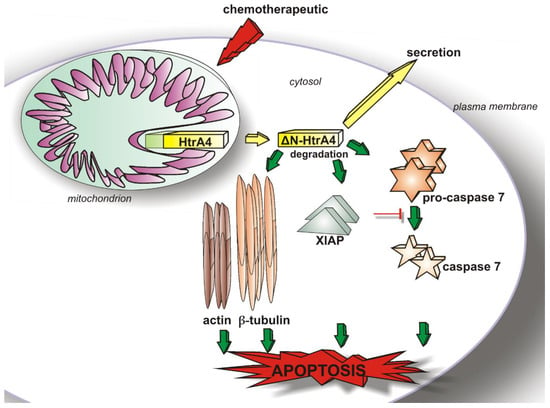
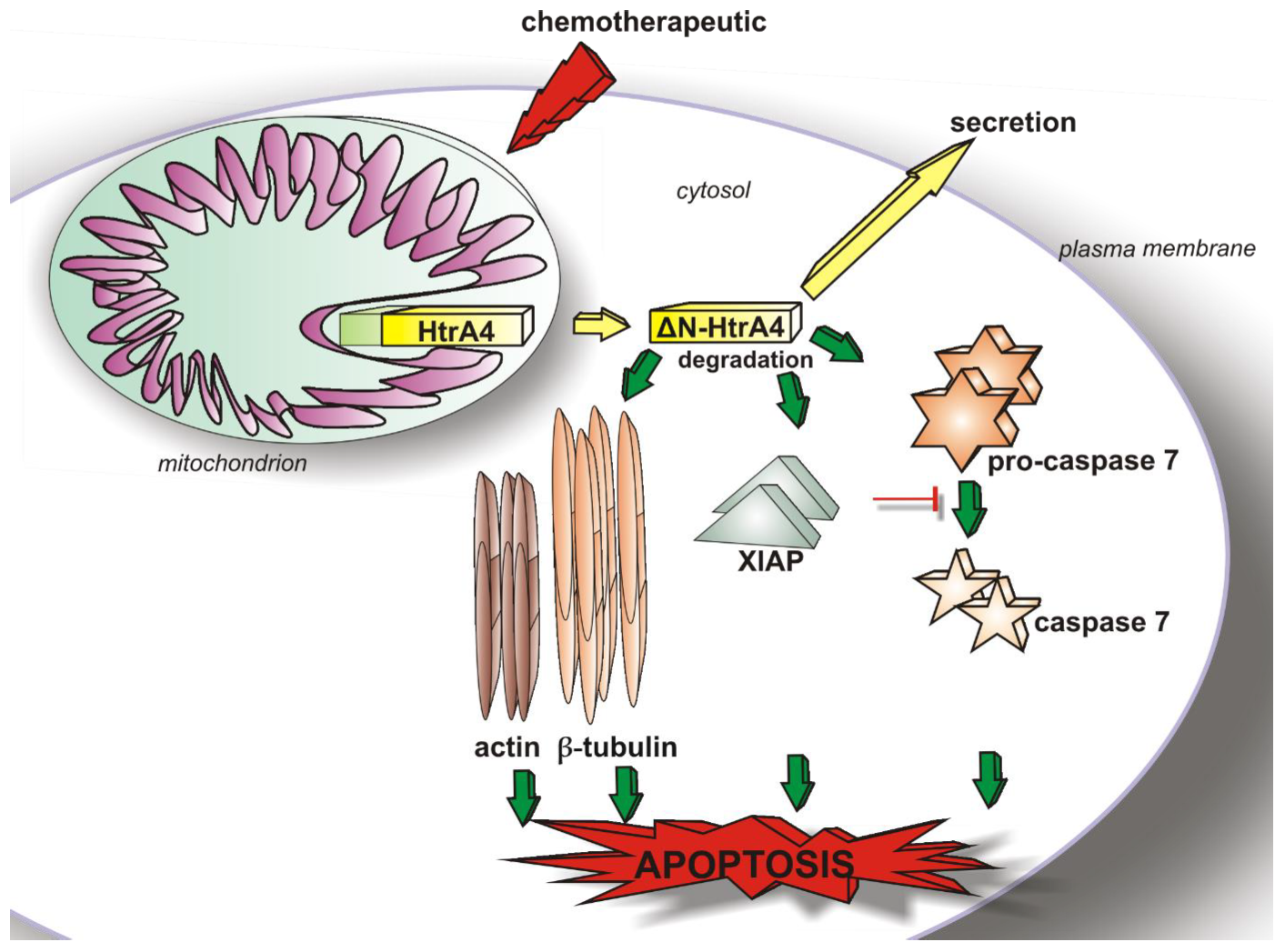
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmid Construction
2.3. Cell Lines and Cell Culture
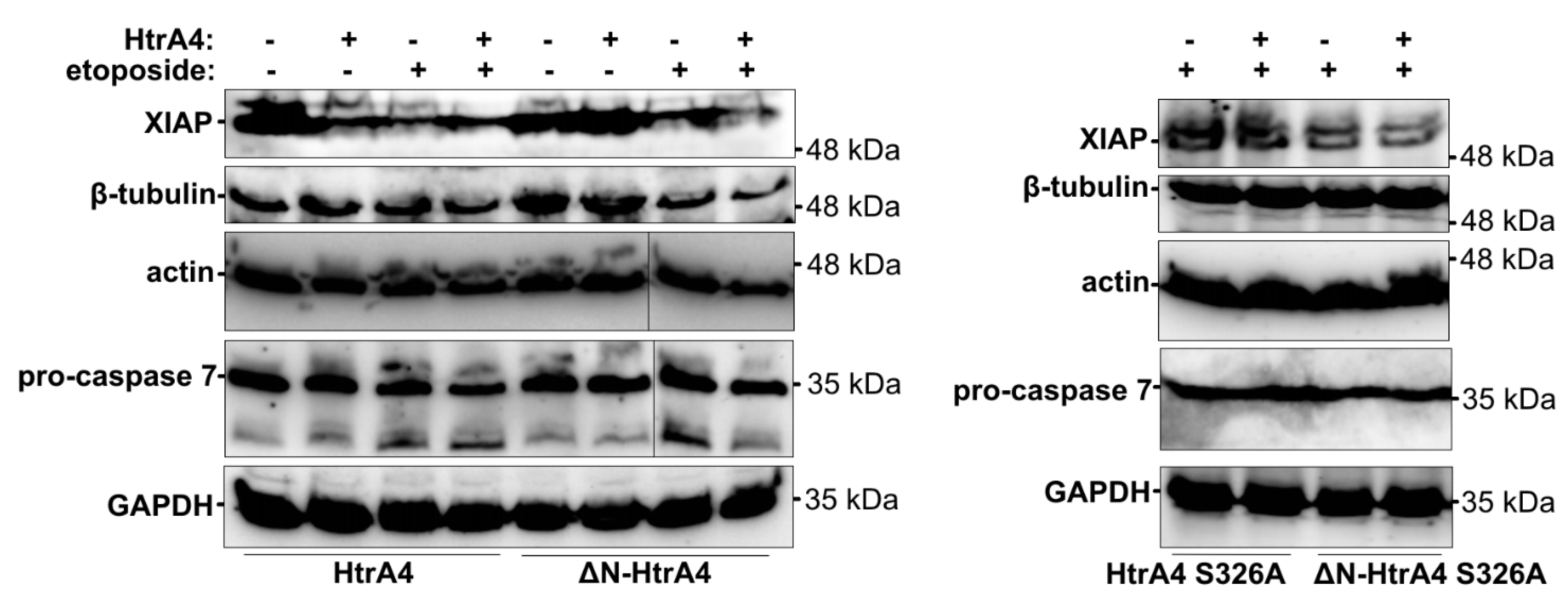
2.4. Western Blotting
2.5. Retroviral Transduction
2.6. Fluorescence Microscopy
2.7. Cell Viability Assay
2.8. EB/AO Staining
2.9. Cell Death Analysis
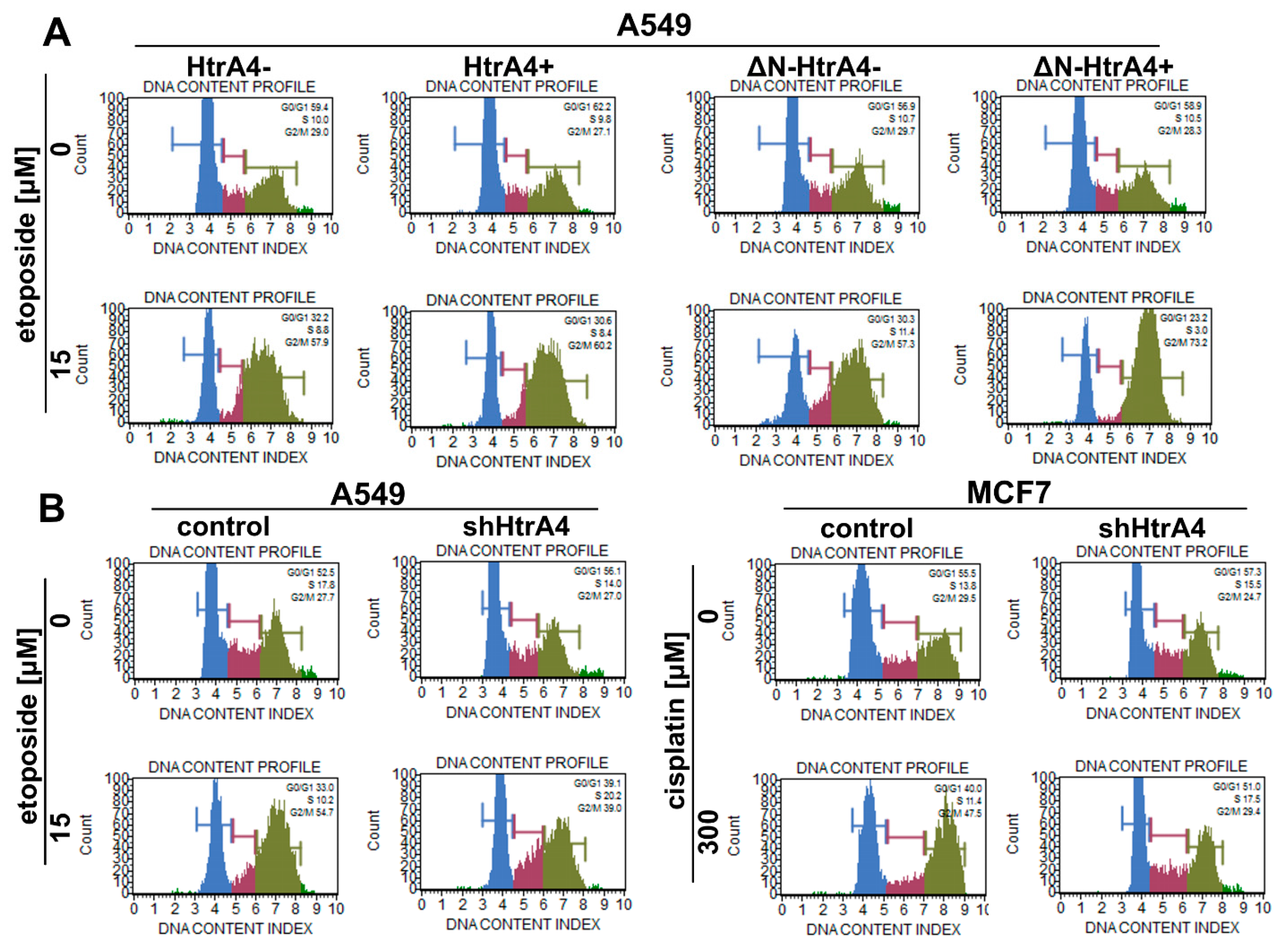
2.10. Cell Cycle Analysis
2.11. Clonogenic Assay
2.12. Wound Healing Assay
2.13. Statistical Analyses
3. Results
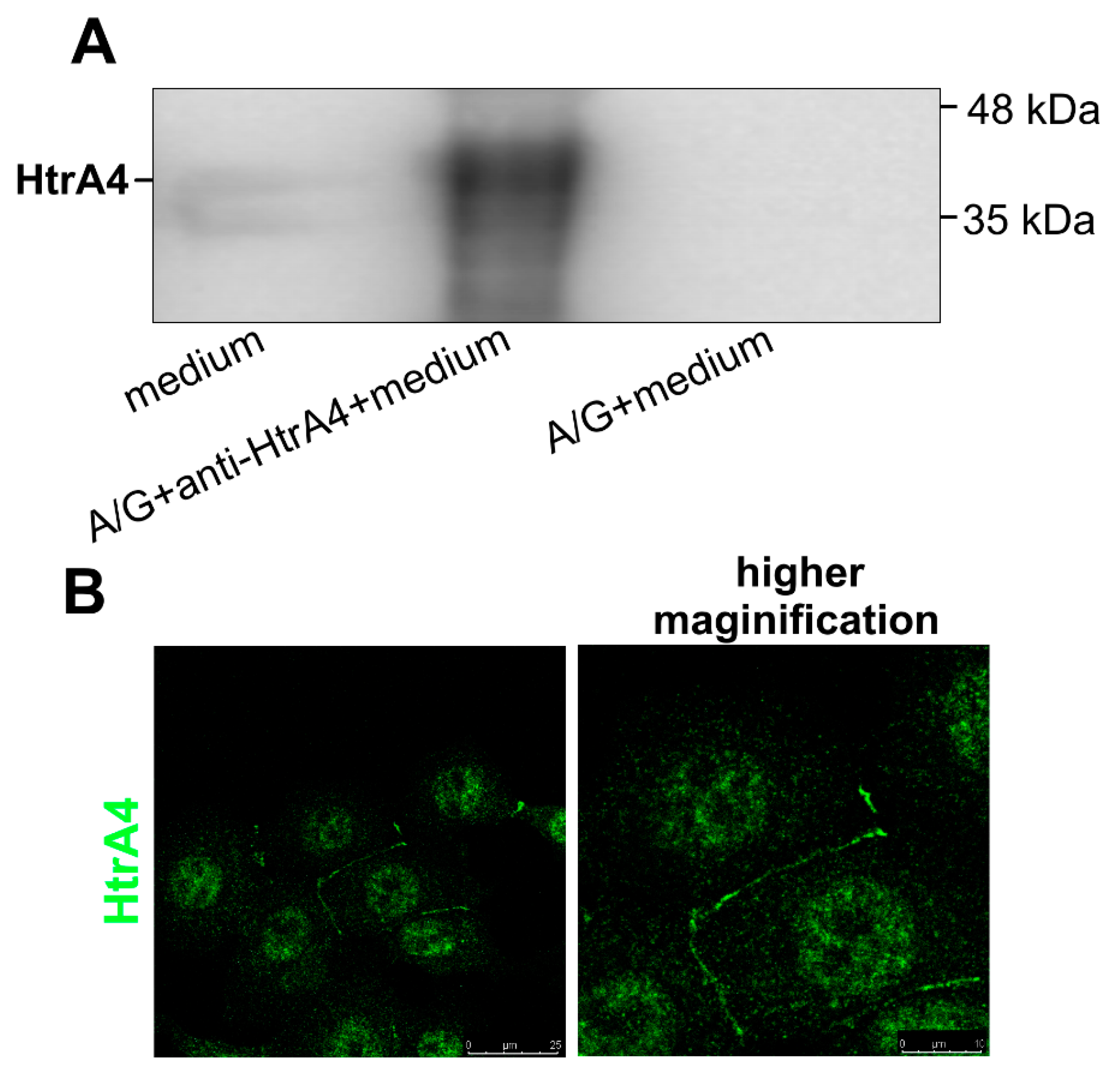
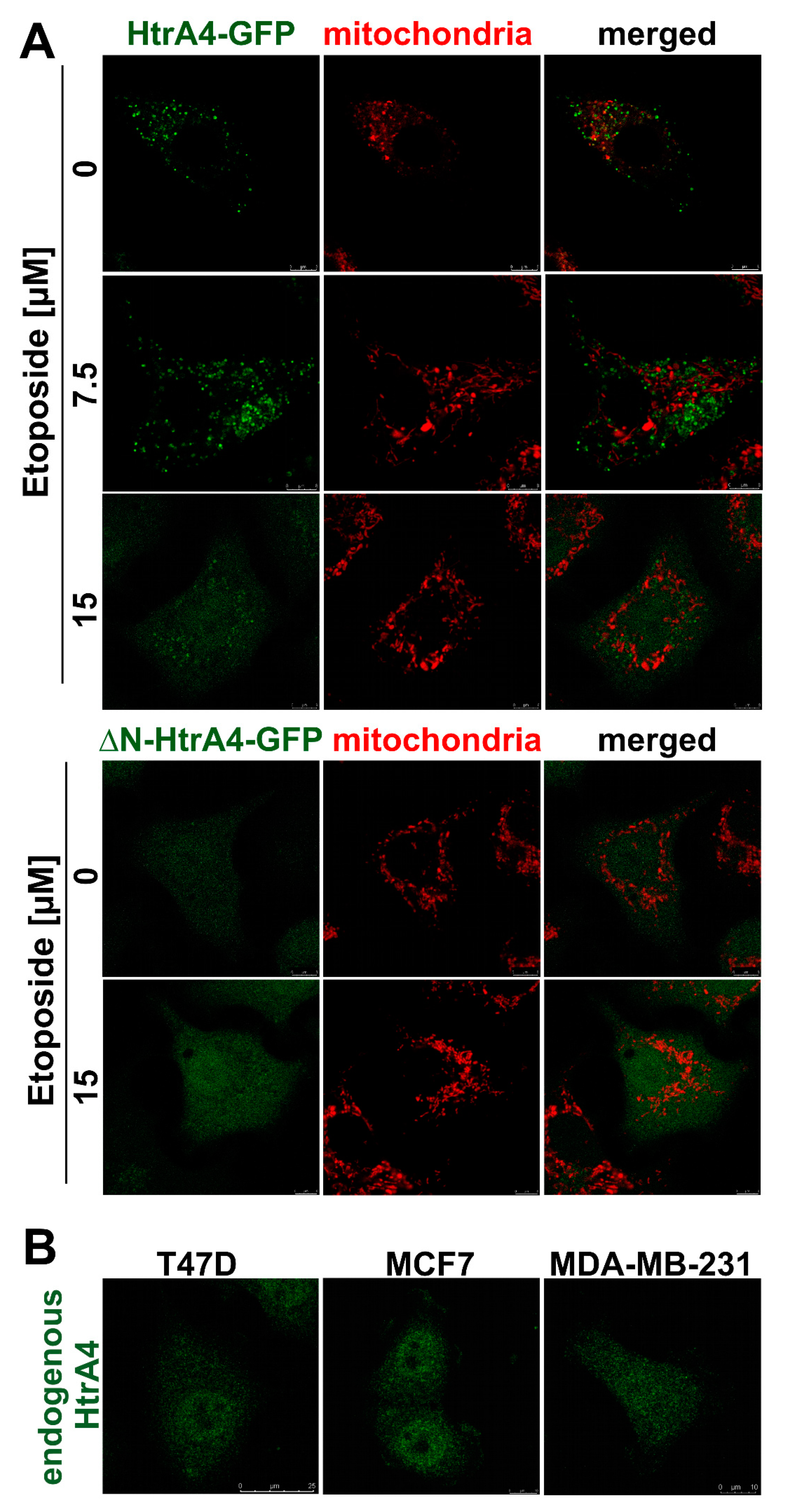
3.1. HtrA4 Is a Secreted Protein but Also Co-Localizes with Mitochondria and Is Present in the Cytoplasm
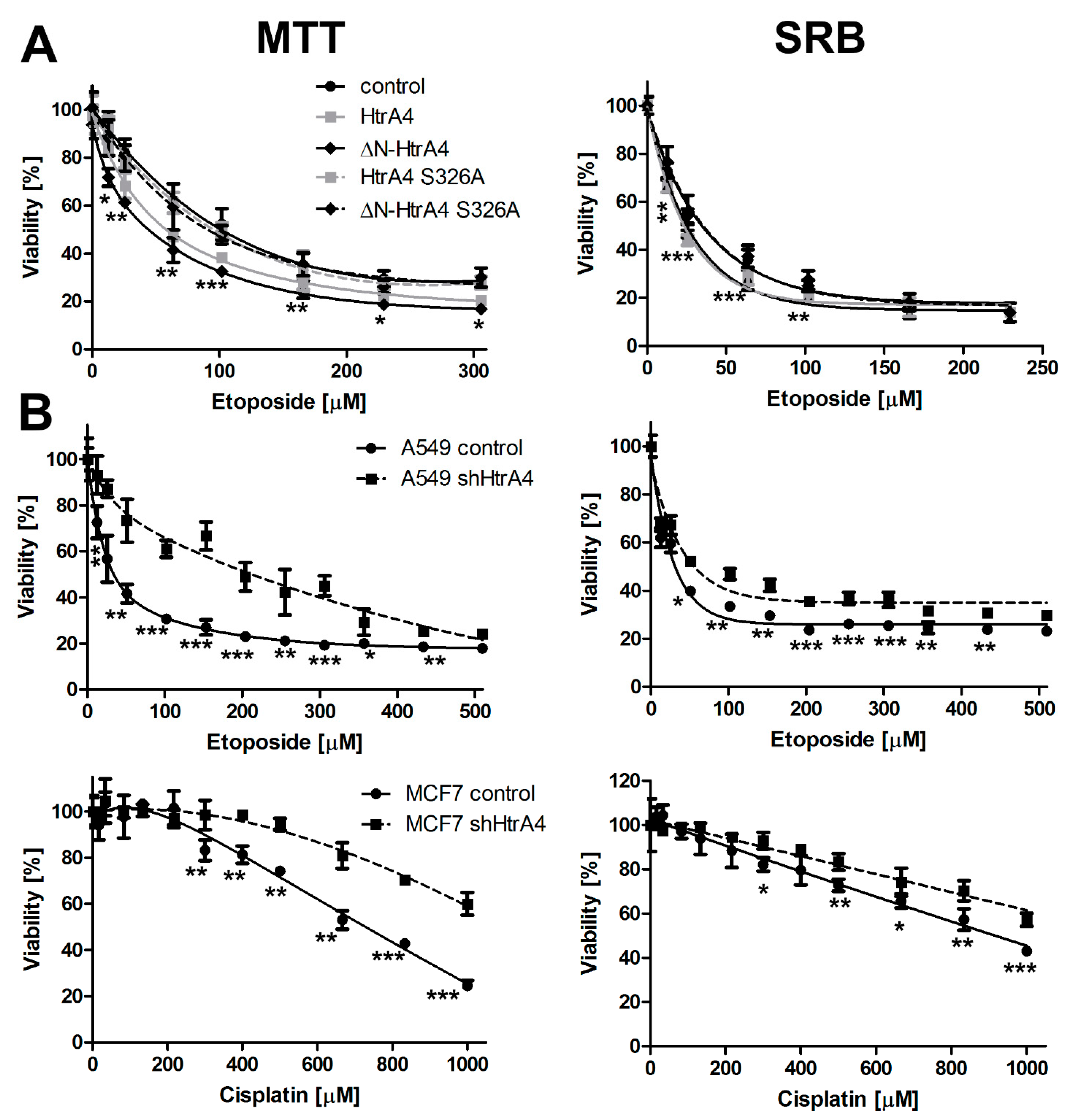
3.2. HtrA4 Decreases Survival of Cancer Cells
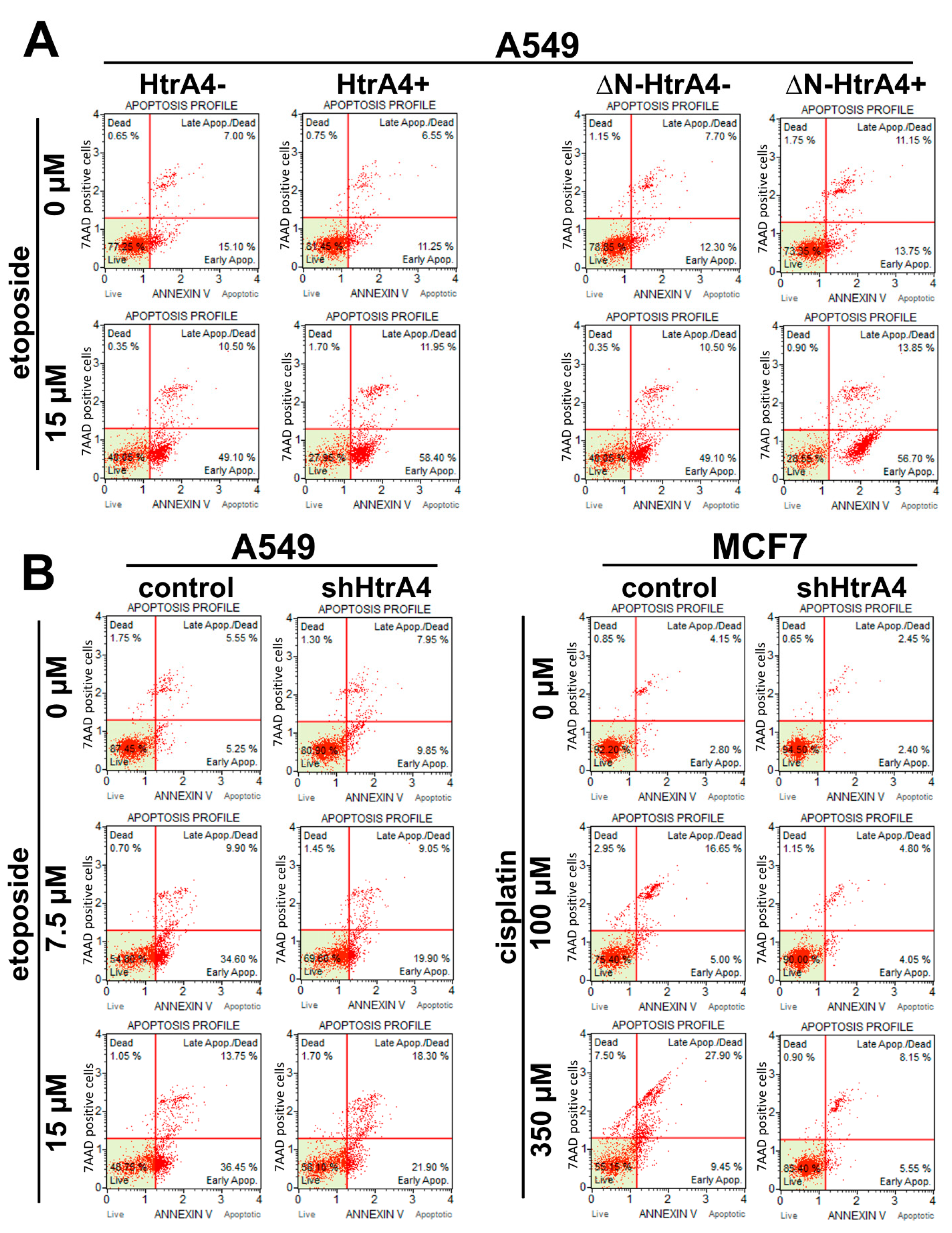
3.3. HtrA4 Promotes Cell Death by Enhancing Apoptosis
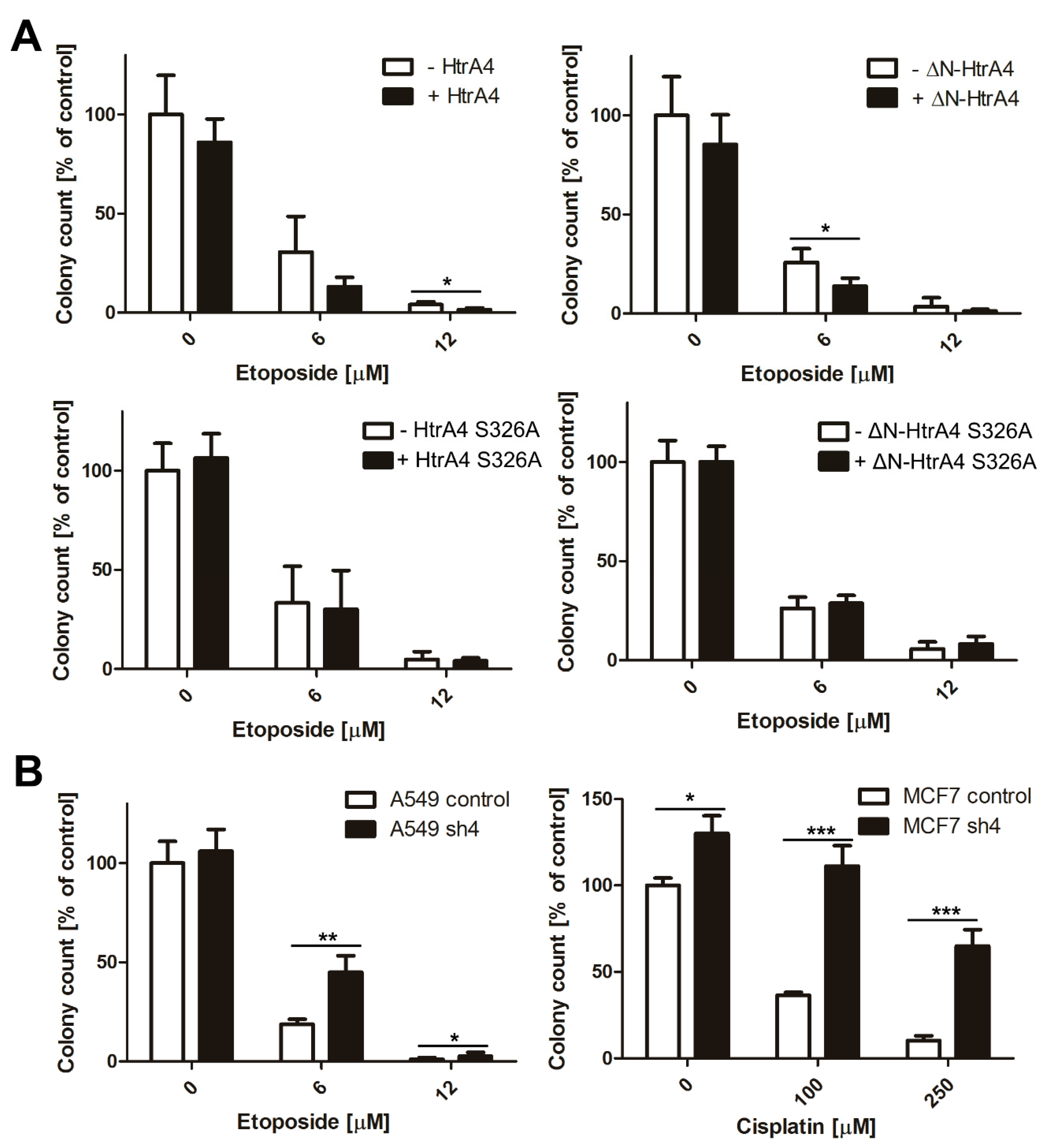
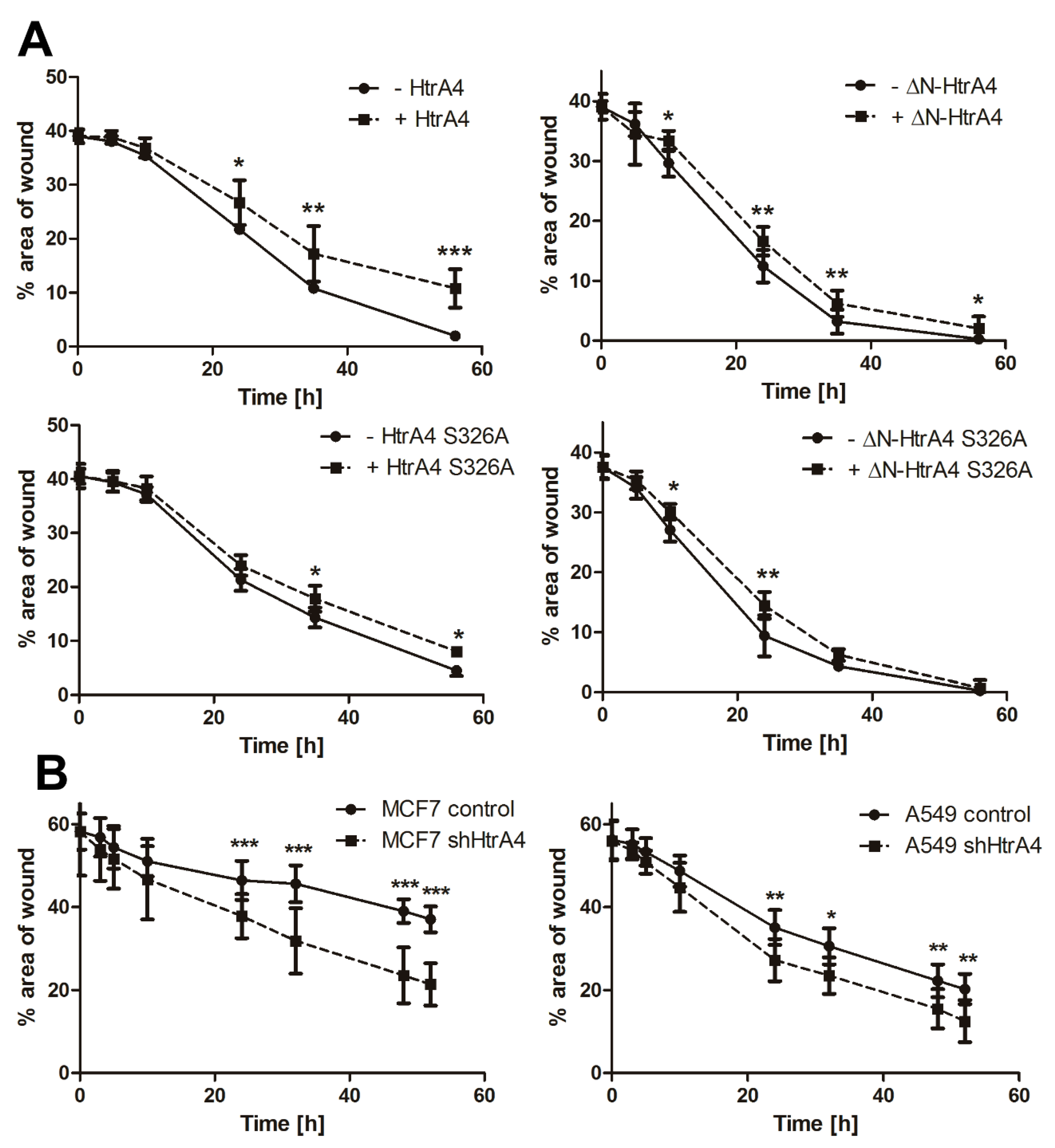
3.4. HtrA4 Enhances the Effect of Chemotherapeutic Agents on Clonogenic Potential and Motility of Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HtrA | High-temperature requirement A |
| PD | protease domain |
| PDZ domain | Postsynaptic density protein 95, Drosophila disc large tumor suppressor and Zonula occludens-1 protein domain |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Skorko-Glonek, J.; Zurawa-Janicka, D.; Koper, T.; Jarzab, M.; Figaj, D.; Glaza, P.; Lipinska, B. HtrA protease family as therapeutic targets. Curr. Pharm. Des. 2013, 19, 977–1009. [Google Scholar] [CrossRef] [PubMed]
- Zurawa-Janicka, D.; Wenta, T.; Jarzab, M.; Skorko-Glonek, J.; Glaza, P.; Gieldon, A.; Ciarkowski, J.; Lipinska, B. Structural insights into the activation mechanisms of human HtrA serine proteases. Arch. Biochem. Biophys. 2017, 621, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Rand, M.L.; Schmugge, M.; Speer, O. Omi/HtrA2 and XIAP are components of platelet apoptosis signalling. Thromb. Haemost. 2013, 109, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.; Siegelin, M.D.; Dehghani, F.; Cilenti, L.; Zervos, A.S.; Rami, A. The serine protease Omi/HtrA2 is involved in XIAP cleavage and in neuronal cell death following focal cerebral ischemia/reperfusion. Neurochem. Int. 2007, 50, 172–180. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Khurana, A.; Maguire, J.L.; Chien, J.; Shridhar, V. HtrA1 sensitizes ovarian cancer cells to cisplatin-induced cytotoxicity by targeting XIAP for degradation. Int. J. Cancer. 2012, 130, 1029–1035. [Google Scholar] [CrossRef]
- Bao, W.; Zhu, F.; Duan, Y.; Yang, Y.; Cai, H. HtrA1 resensitizes multidrug-resistant hepatocellular carcinoma cells by targeting XIAP. Biomed. Pharmacother. 2015, 70, 97–102. [Google Scholar] [CrossRef]
- Xiong, Z.; Fu, Z.; Shi, J.; Jiang, X.; Wan, H. HtrA1 Down-regulation Induces Cisplatin Resistance in Colon Cancer by Increasing XIAP and Activating PI3K/Akt Pathway. Ann. Clin. Lab. Sci. 2017, 47, 264–270. [Google Scholar]
- Wenta, T.; Rychlowski, M.; Jurewicz, E.; Jarzab, M.; Zurawa-Janicka, D.; Filipek, A.; Lipinska, B. The HtrA3 protease promotes drug-induced death of lung cancer cells by cleavage of the X-linked inhibitor of apoptosis protein (XIAP). FEBS J. 2019. [Google Scholar] [CrossRef]
- Wenta, T.; Glaza, P.; Jarząb, M.; Zarzecka, U.; Żurawa-Janicka, D.; Lesner, A.; Skórko-Glonek, J.; Lipińska, B. The role of the LB structural loop and its interactions with the PDZ domain of the human HtrA3 protease. Biochim. Biophys. Acta Proteins Proteomics 2017, 1865, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Kummari, R.; Dutta, S.; Chaganti, L.K.; Bose, K. Discerning the Mechanism of Action of HtrA4: A Serine Protease Implicated in the Cell Death Pathway. Biochem. J. 2019, 476, 1445–1463. [Google Scholar] [CrossRef] [PubMed]
- Glaza, P.; Osipiuk, J.; Wenta, T.; Zurawa-Janicka, D.; Jarzab, M.; Lesner, A.; Banecki, B.; Skorko-Glonek, J.; Joachimiak, A.; Lipinska, B. Structural and Functional Analysis of Human HtrA3 Protease and Its Subdomains. PLoS ONE 2015, 10, e0131142. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Srinivasula, S.M.; Chai, J.; Li, P.; Wu, J.-W.; Zhang, Z.; Alnemri, E.S.; Shi, Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 2002, 9, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Truebestein, L.; Tennstaedt, A.; Mönig, T.; Krojer, T.; Canellas, F.; Kaiser, M.; Clausen, T.; Ehrmann, M. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 2011, 18, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, A.; Nishizawa, H.; Ota, S.; Suzuki, M.; Inuzuka, H.; Miyamura, H.; Sekiya, T.; Kurahashi, H.; Udagawa, Y. Upregulation of HtrA4 in the placentas of patients with severe pre-eclampsia. Placenta 2012, 33, 919–926. [Google Scholar] [CrossRef]
- Singh, H.; Zhao, M.; Chen, Q.; Wang, Y.; Li, Y.; Kaitu’u-Lino, T.J.; Tong, S.; Nie, G. Human HtrA4 Expression is Restricted to the Placenta, is Significantly Up-Regulated in Early-Onset Preeclampsia, and High Levels of HtrA4 Cause Endothelial Dysfunction. J. Clin. Endocrinol. Metab. 2015, 100, E936–E945. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Chuang, P.-Y.; Chen, C.-P.; Chiu, Y.-H.; Lo, H.-F.; Cheong, M.-L.; Huang, J.-Y.; Kuo, P.-L.; Chen, H. Functional antagonism between high temperature requirement protein A (HtrA) family members regulates trophoblast invasion. J. Biol. Chem. 2014, 289, 22958–22968. [Google Scholar] [CrossRef]
- Dynon, K.; Heng, S.; Puryer, M.; Li, Y.; Walton, K.; Endo, Y.; Nie, G. HtrA3 as an early marker for preeclampsia: Specific monoclonal antibodies and sensitive high-throughput assays for serum screening. PLoS ONE 2012, 7, e45956. [Google Scholar] [CrossRef]
- Liu, C.; Xing, F.; He, Y.; Zong, S.; Luo, C.; Li, C.; Duan, T.; Wang, K.; Zhou, Q. Elevated HTRA1 and HTRA4 in severe preeclampsia and their roles in trophoblast functions. Mol. Med. Rep. 2018, 18, 2937–2944. [Google Scholar] [CrossRef]
- Chien, J.; Campioni, M.; Shridhar, V.; Baldi, A. HtrA serine proteases as potential therapeutic targets in cancer. Curr. Cancer Drug Targets 2009, 9, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hui, A.M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.L.; Wang, Z.C.; de Nicolo, A.; Lu, X.; Brown, M.; Miron, A.; Liao, X.; Iglehart, J.D.; Livingston, D.M.; Ganesan, S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006, 9, 121–132. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Choudhury, B.; Campbell, P.J.; Feng, B.; Wong, K.K.; Protopopov, A.; O’Neil, J.; Gutierrez, A.; Ivanova, E.; Perna, I.; et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007, 447, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; La, M.; Pham, T.; Lovrecz, G.O.; Nie, G. High levels of HtrA4 detected in preeclamptic circulation may disrupt endothelial cell function by cleaving the main VEGFA receptor KDR. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5058–5066. [Google Scholar] [CrossRef] [PubMed]
- Wenta, T.; Zurawa-Janicka, D.; Rychlowski, M.; Jarzab, M.; Glaza, P.; Lipinska, A.; Bienkowska-Szewczyk, K.; Herman-Antosiewicz, A.; Skorko-Glonek, J.; Lipinska, B. HtrA3 is a cellular partner of cytoskeleton proteins and TCP1α chaperonin. J. Proteomics 2018, 177, 88–111. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.Y.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Chien, J.; Ota, T.; Aletti, G.; Shridhar, R.; Boccellino, M.; Quagliuolo, L.; Baldi, A.; Shridhar, V. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol. Cell Biol. 2009, 29, 4177–4187. [Google Scholar] [CrossRef]
- Beleford, D.; Rattan, R.; Chien, J.; Shridhar, V. High temperature requirement A3 (HtrA3) promotes etoposide- and cisplatin-induced cytotoxicity in lung cancer cell lines. J. Biol. Chem. 2010, 285, 12011–12027. [Google Scholar] [CrossRef]
- Chien, J.; Aletti, G.; Baldi, A.; Catalano, V.; Muretto, P.; Keeney, G.L.; Kalli, K.R.; Staub, J.; Ehrmann, M.; Cliby, W.A.; et al. Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J. Clin. Invest. 2006, 116, 1994–2004. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clawson, G.A.; Bui, V.; Xin, P.; Wang, N.; Pan, W. Intracellular localization of the tumor suppressor HtrA1/Prss11 and its association with HPV16 E6 and E7 proteins. J. Cell Biochem. 2008, 105, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, T.; Mori, M.; Terada, K. Mitochondrial import of Omi: The definitive role of the putative transmembrane region and multiple processing sites in the amino-terminal segment. Biochem. Biophys. Res. Commun. 2007, 361, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Wenta, T.; Jarzab, M.; Rychlowski, M.; Borysiak, M.; Latala, A.; Zurawa-Janicka, D.; Filipek, A.; Lipinska, B. Cellular substrates and pro-apoptotic function of the human HtrA4 protease. J. Proteomics 2019, 209, 103505. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, Z.; Zhang, Z.; Sun, N.; Zhang, M.; Xie, J.; Li, T.; Hou, Y.; Wu, D. HtrA1 downregulation induces cisplatin resistance in lung adenocarcinoma by promoting cancer stem cell-like properties. J. Cell Biochem. 2014, 115, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Lin, Y.S.; Su, W.C.; Chen, C.L.; Lin, C.F. Glycogen synthase kinase-3 and Omi/HtrA2 induce annexin A2 cleavage followed by cell cycle inhibition and apoptosis. Mol. Biol. Cell 2009, 20, 4153–4161. [Google Scholar] [CrossRef][Green Version]
- Martins, L.M.; Iaccarino, I.; Tenev, T.; Gschmeissner, S.; Totty, N.F.; Lemoine, N.R.; Savopoulos, J.; Gray, C.W.; Creasy, C.L.; Dingwall, C.; et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 2002, 277, 439–444. [Google Scholar] [CrossRef]
- Berthelet, J.; Dubrez, L. Regulation of Apoptosis by Inhibitors of Apoptosis (IAPs). Cells 2013, 2, 163–187. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Zhang, X.; Feng, M.; Qu, J. High temperature requirement A3 (HTRA3) expression predicts postoperative recurrence and survival in patients with non-small-cell lung cancer. Oncotarget 2016, 7, 40725–40734. [Google Scholar] [CrossRef]
- Melo, E.; Oertle, P.; Trepp, C.; Meistermann, H.; Burgoyne, T.; Sborgi, L.; Cabrera, A.C.; Chen, C.-Y.; Hoflack, J.-C.; Kam-Thong, T.; et al. HtrA1 Mediated Intracellular Effects on Tubulin Using a Polarized RPE Disease Model. EBioMedicine 2018, 27, 258–274. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HtrA4 Variant | Etoposide [µM] | Pearson’s Correlation | Overlap Coefficient |
|---|---|---|---|
| HtrA4-GFP | 0 | 0.52 ± 0.09 | 0.58 ± 0.08 |
| 7.5 | 0.17 ± 0.04 | 0.26 ± 0.03 | |
| 15 | 0.13 ± 0.03 | 0.23 ± 0.03 | |
| ∆N-HtrA4-GFP | 0 | 0.12 ± 0.02 | 0.23 ± 0.03 |
| 15 | 0.11 ± 0.02 | 0.19 ± 0.03 |
| Cell Line | Chemotherapeutic Drug [µM] | HtrA4 Variant | Live Cells [%±SD] | Early Apoptotic Cells [%±SD] | Late Apoptotic/Dead Cells [%±SD] | Total Apoptotic Cells | ||
|---|---|---|---|---|---|---|---|---|
| [%] | Statistical Analysis | |||||||
| A549 | Etoposide | 0 | − HtrA4 | 80.6 ± 4.7 | 12.9 ± 3.1 | 6.4 ± 0.8 | 19.3 | ns |
| + HtrA4 | 80.9 ± 0.8 | 13.4 ± 3.0 | 5.3 ± 1.1 | 18.7 | ||||
| 15 | − HtrA4 | 39.0 ± 1.5 | 46.5 ± 3.7 | 12.7 ± 0.3 | 59.2 | * | ||
| + HtrA4 | 29.4 ± 2.1 | 55.4 ± 4.3 | 14.4 ± 3.4 | 69.7 | ||||
| 0 | − ∆N-HtrA4 | 80.5 ± 2.3 | 10.7 ± 2.3 | 8.4 ± 0.9 | 19 | ns | ||
| + ∆N-HtrA4 | 77.0 ± 5.1 | 12.2 ± 0.7 | 10.1 ± 1.4 | 22.4 | ||||
| 15 | − ∆N-HtrA4 | 37.9 ± 2.8 | 47.8 ± 3.5 | 13.0 ± 1.5 | 60.8 | ** | ||
| + ∆N-HtrA4 | 26.7 ± 2.6 | 57.5 ± 1.1 | 15.3 ± 2.1 | 72.8 | ||||
| A549 | Etoposide | 0 | control | 86.8 ± 0.9 | 5.9 ± 0.9 | 6.7 ± 1.2 | 12.6 | ns |
| shHtrA4 | 85.1 ± 5.8 | 8.2 ± 2.3 | 6.4 ± 1.1 | 14.7 | ||||
| 7.5 | control | 61.6 ± 7.1 | 26.65 ± 3.0 | 10.7 ± 0.5 | 37.3 | ns | ||
| shHtrA4 | 67.9 ± 3.8 | 19.0 ± 0.1 | 11.4 ± 2.0 | 30.4 | ||||
| 15 | control | 51.8 ± 4.3 | 35.5 ± 1.3 | 11.8 ± 2.1 | 47.4 | ** | ||
| shHtrA4 | 63.9 ± 3.1 | 20.9 ± 1.4 | 13.7 ± 2.0 | 34.6 | ||||
| MCF7 | Cisplatin | 0 | control | 89.2 ± 4.2 | 3.3 ± 0.7 | 6.8 ± 0.2 | 10.1 | ns |
| shHtrA4 | 92.7 ± 2.5 | 2.8 ± 0.5 | 4.2 ± 0.4 | 7 | ||||
| 100 | control | 73.9 ± 2.1 | 4.8 ± 0.2 | 18.0 ± 1.8 | 22.8 | *** | ||
| shHtrA4 | 88.4 ± 2.3 | 4.0 ± 0.1 | 6.4 ± 0.5 | 10.3 | ||||
| 350 | control | 46.8 ± 5.4 | 16.8 ± 2.6 | 30.9 ± 3.2 | 47.7 | ** | ||
| shHtrA4 | 80.3 ± 7.3 | 6.3 ± 0.8 | 12.1 ± 1.5 | 18.4 | ||||
| Cell Line | Chemotherapeutic Drug [µM] | HtrA4 Variant | Cell Cycle Phase | ||||||
|---|---|---|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | |||||||
| [% ± SD] | Statistical Analysis | [% ± SD] | Statistical Analysis | [% ± SD] | Statistical Analysis | ||||
| A549 | Etoposide | 0 | − HtrA4 | 63.6 ± 3.8 | ns | 9.3 ± 1.2 | ns | 26.9 ± 2.2 | ns |
| + HtrA4 | 64.1 ± 1.9 | 11.0 ± 1.9 | 24.7 ± 2.7 | ||||||
| 15 | − HtrA4 | 32.5 ± 3.6 | ns | 10.0 ± 1.5 | ns | 53.3 ± 5.4 | * | ||
| + HtrA4 | 29.6 ± 6.0 | 10.6 ± 1.9 | 58.6 ± 6.5 | ||||||
| A549 | Etoposide | 0 | − ∆N-HtrA4 | 55.6 ± 2.1 | ns | 11.1 ± 2.7 | ns | 31.0 ± 1.4 | ns |
| + ∆N-HtrA4 | 58.1 ± 2.9 | 11.5 ± 0.9 | 31.4 ± 2.5 | ||||||
| 15 | − ∆N-HtrA4 | 29.2 ± 1.8 | ** | 14.2 ± 2.3 | ** | 54.5 ± 2.9 | ** | ||
| + ∆N-HtrA4 | 21.5 ± 2.4 | 4.4 ± 2.5 | 73.7 ± 5.1 | ||||||
| A549 | Etoposide | 0 | control | 56.1 ± 3.4 | ns | 15.8 ± 1.5 | * | 27.3 ± 1.4 | ns |
| shHtrA4 | 57.2 ± 1.2 | 13.6 ± 0.3 | 27.9 ± 1.0 | ||||||
| 15 | control | 32.4 ± 1.6 | *** | 12.8 ± 1.9 | * | 55.7 ± 1.7 | *** | ||
| shHtrA4 | 41.6 ± 2.4 | 16.2 ± 2.3 | 35.8 ± 2.0 | ||||||
| MCF7 | Cisplatin | 0 | control | 56.8 ± 3.3 | ns | 15.5 ± 1.9 | ns | 31.4 ± 2.9 | ns |
| shHtrA4 | 55.2 ± 4.1 | 17.9 ± 2.5 | 26.5 ± 3.2 | ||||||
| 300 | control | 41.5 ± 1.1 | *** | 7.2 ± 2.5 | ** | 48.5 ± 1.3 | *** | ||
| shHtrA4 | 56.5 ± 3.5 | 13.4 ± 2.4 | 28.7 ± 1.2 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenta, T.; Rychlowski, M.; Jarzab, M.; Lipinska, B. HtrA4 Protease Promotes Chemotherapeutic-Dependent Cancer Cell Death. Cells 2019, 8, 1112. https://doi.org/10.3390/cells8101112
Wenta T, Rychlowski M, Jarzab M, Lipinska B. HtrA4 Protease Promotes Chemotherapeutic-Dependent Cancer Cell Death. Cells. 2019; 8(10):1112. https://doi.org/10.3390/cells8101112
Chicago/Turabian StyleWenta, Tomasz, Michal Rychlowski, Miroslaw Jarzab, and Barbara Lipinska. 2019. "HtrA4 Protease Promotes Chemotherapeutic-Dependent Cancer Cell Death" Cells 8, no. 10: 1112. https://doi.org/10.3390/cells8101112
APA StyleWenta, T., Rychlowski, M., Jarzab, M., & Lipinska, B. (2019). HtrA4 Protease Promotes Chemotherapeutic-Dependent Cancer Cell Death. Cells, 8(10), 1112. https://doi.org/10.3390/cells8101112