Endoplasmic Reticulum Stress in Metabolic Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. ER Functions and Homeostasis
1.1. The Multiple Functions of ER in Protein, Lipid, and Calcium Homeostasis
1.2. ER Interconnections with Other Organelles
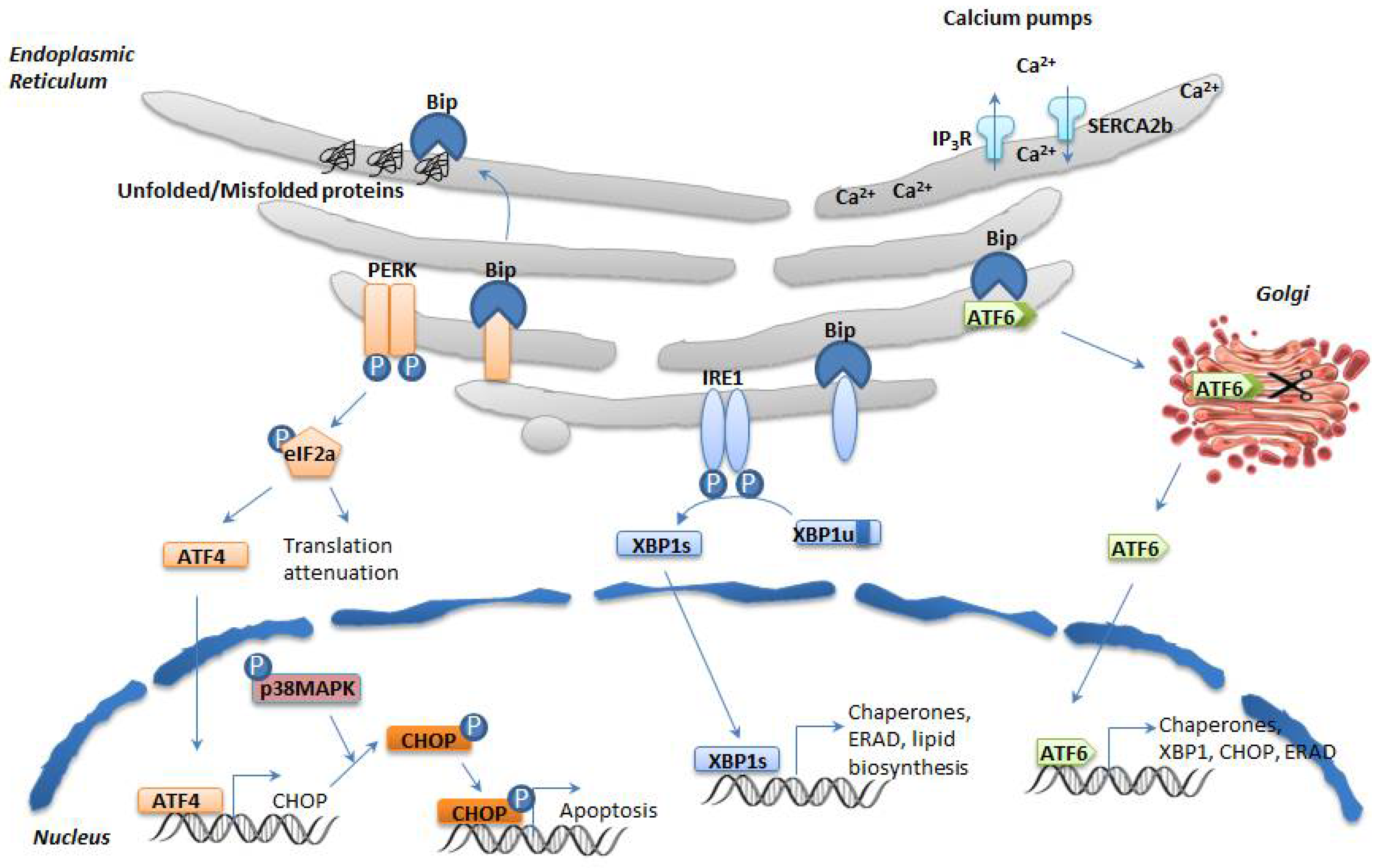
2. ER Stress and the Unfolded Protein Response (UPR)
2.1. The UPR
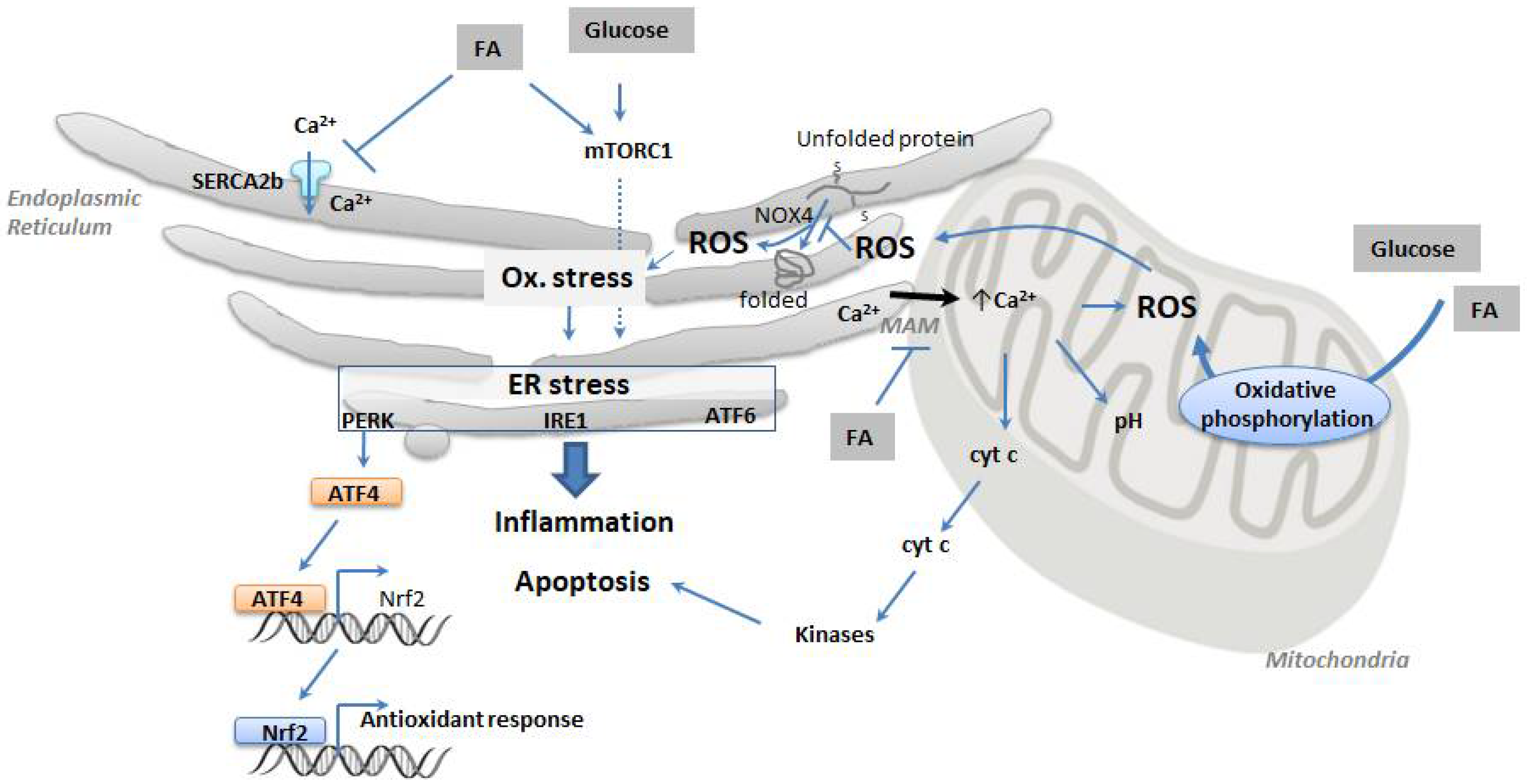
2.2. ER Senses Cell Stress and Integrates Metabolic Stress Signals
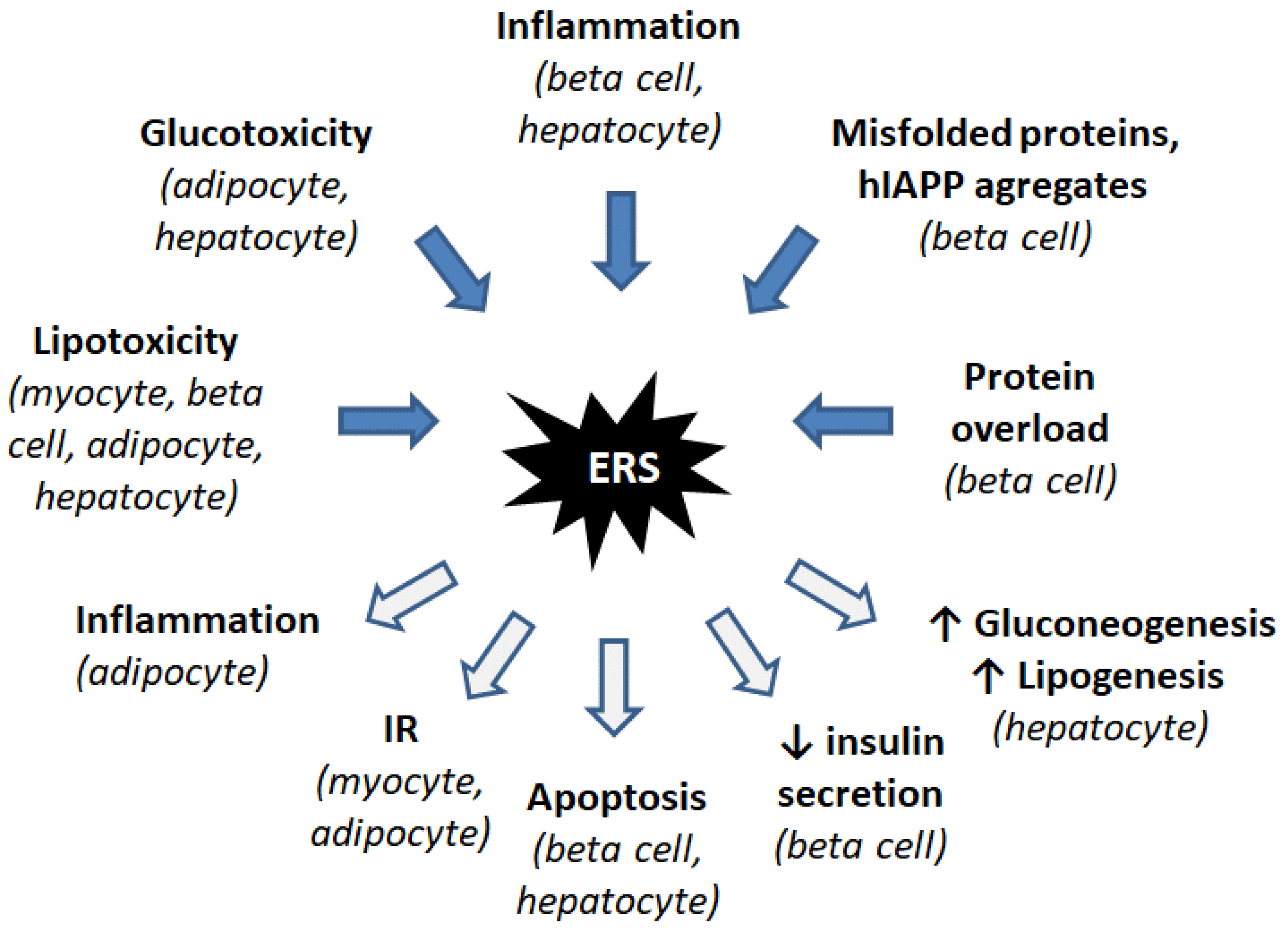
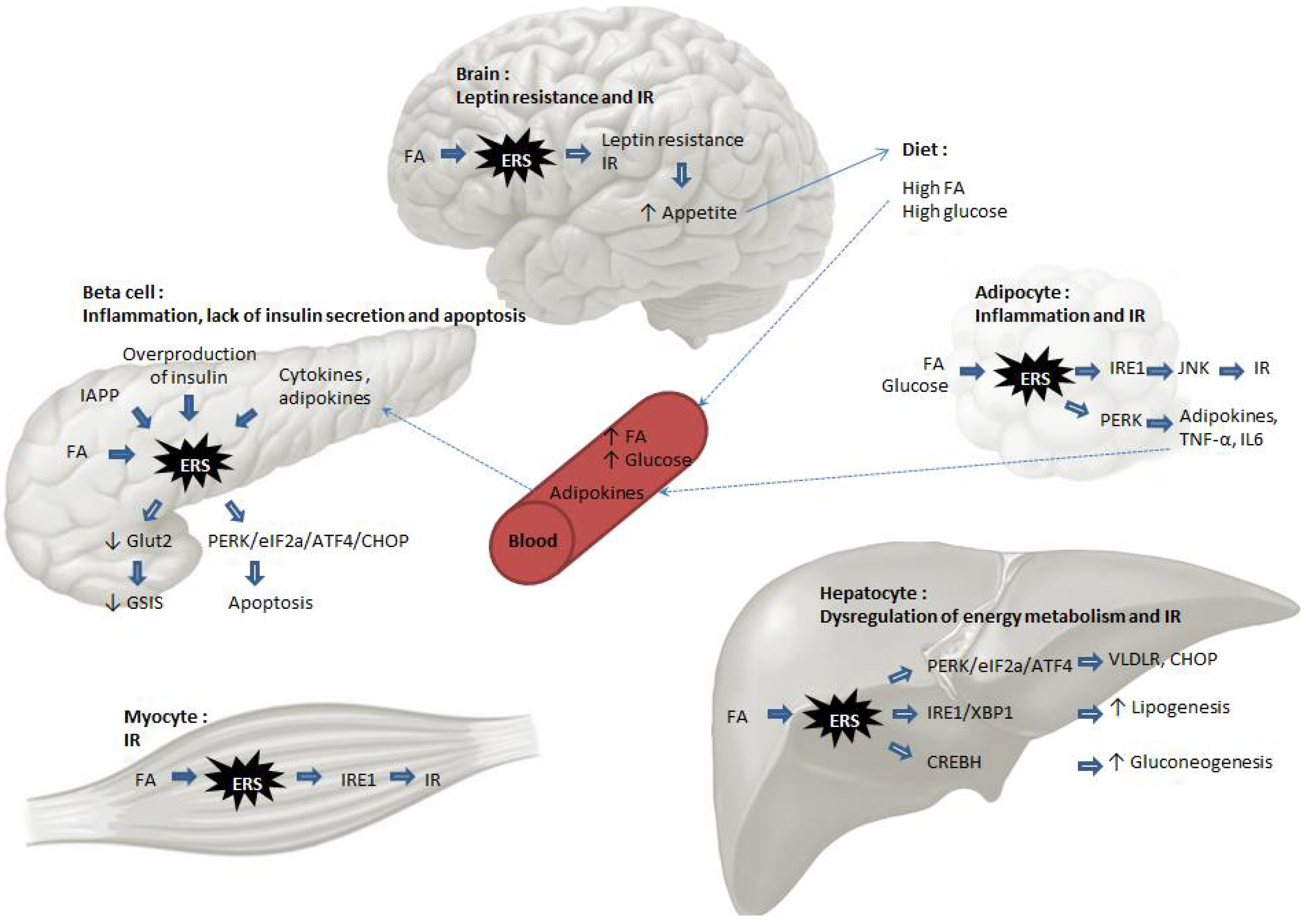
3. ER Stress Induces IR and Diabetes
3.1. Importance of the ER in IR and Diabetes
3.2. Inflammatory Response
3.3. Lipotoxicity
3.4. Glucotoxicity
3.5. Insulin Receptor Modulation
3.6. Mechanisms Specific to Liver Cells: Activation of Gluconeogenesis and Lipogenesis
3.7. Mechanisms Specific to Beta-Cells: Accumulation of Proinsulin and Amyloid Deposits in the ER
3.8. Brain-Specific Mechanisms: Leptin Resistance
4. ER and Fatty Liver Disease
4.1. Oxidative Stress
4.2. Disturbance of Lipid Metabolism
4.3. Inflammation
4.4. Apoptosis
4.5. Autophagy
5. ER Stress and Atherosclerosis
5.1. ER Stress in Macrophages
5.2. ER Stress in Endothelial Cells (ECs)
5.3. ER Stress in Vascular Smooth Muscle Cells
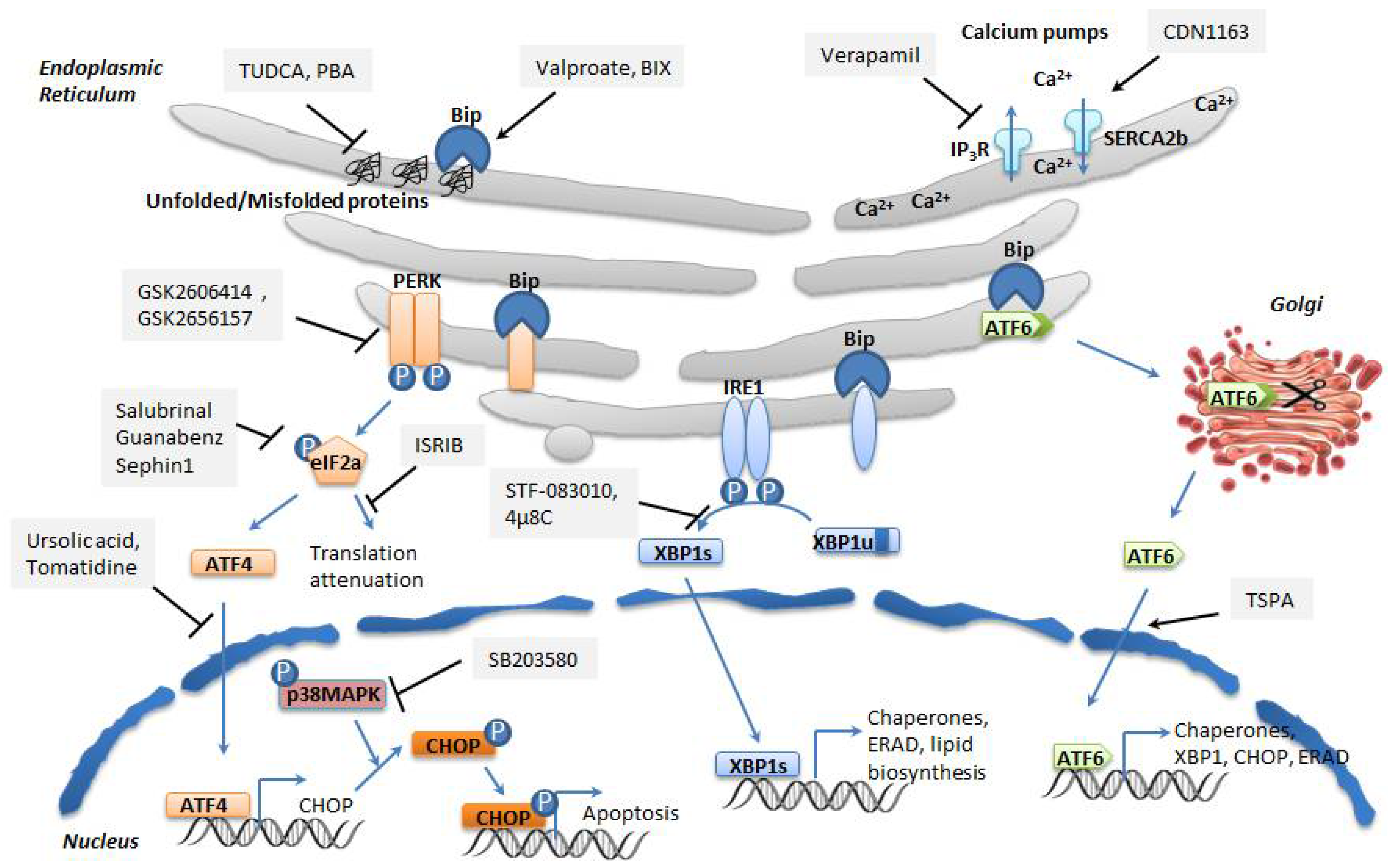
6. Therapeutic Intervention on ER Stress in Metabolic Diseases
6.1. Regulators of ER Calcium Homeostasis
6.2. Non-Specific Protein Misfolding Inhibitors
6.3. Bip Activators
6.4. Regulators of the PERK/eIF2α/ATF4/CHOP Branch
6.5. Regulators of the IRE1α/XBP1 Branch
6.6. Regulators of the ATF6 Branch
7. General Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenylbutyrate |
| ALDH | Aldehyde dehydrogenase |
| AMPK | AMP-activated protein kinase |
| AP1 | Activator protein 1 |
| ApoE | Apolipoprotein E |
| ASK1 | Apoptosis signal-regulating kinase |
| ATF | Activating transcription factor |
| ATP | Adenosine triphosphate |
| BCL | B-cell lymphoma |
| BI-1 | Bax inhibitor-1 (BI-1) |
| C/EBP | CCAAT/enhancer-binding protein |
| CD36 | cluster of differentiation 36 |
| CNPY2 | Canopy homolog 2 |
| CREBH | Cyclic AMP-responsive element binding protein, hepatocyte-specific |
| Cyt c | Cytochrome c |
| DKK1 | Dickkopf1 |
| ECs | Endothelial cells |
| eIF2α | Eukaryotic initiation factor 2α |
| ER | Endoplasmic Reticulum |
| ERAD | ER-associated degradation |
| FA | Fatty acids |
| FDA | Food and Drug Administration |
| FGF21 | Fibroblast growth factor 21 |
| FOXO1 | Forkhead box protein O1 |
| GABA | Gamma-Aminobutyric acid |
| GADD153 or CHOP | C/EBPα-homologous protein |
| GLUT | glucose transporter |
| GM-CSF | Granulocyte macrophage colony stimulating factor |
| GRP78 | 78 KDa glucose-regulated proteins |
| GSIS | Glucose-stimulated insulin secretion |
| GSK | Glycogen synthase kinase |
| HDAC | Histone deacetylases |
| Herpud1 | Homocysteine inducible ER protein with ubiquitin like domain 1 |
| HFD | High fat diet |
| hIAPP | Human islet amyloid polypeptide |
| HNAs | Hydroxynaphthoic acids |
| HNF4α | Hepatocyte nuclear factor 4 |
| HSPA5 | Heat shock 70 kDa protein 5 |
| HUVECs | Human umbilical vein endothelial cells |
| IAPP | Islet amyloid polypeptide |
| IFN-γ | Interferon gamma |
| IGF1 | Insulin-like growth factor 1 |
| IKKβ | IκB kinase β |
| IL | interleukin |
| iNOS | Inducible nitric oxide synthase |
| INS1 cells | Insulin-secreting cell lines |
| IP3Rs | Inositol trisphosphate receptors |
| IR | Insulin resistance |
| IRE1α | Inositol- requiring enzyme 1α |
| IRS-1 | Insulin receptor substrate-1 |
| ISRIB | Integrated stress response inhibitor |
| JAK | Janus kinase |
| JNK | JUN N-terminal kinase |
| LDL | Low density lipoprotein |
| LPS | Lipopolysaccharide |
| MAM | Mitochondria-associated ER membrane |
| MAPK | Mitogen-activated protein kinase |
| MIDY | Mutant insulin INS gene-induced diabetes of youth |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| Nfe2L1 | Nuclear factor erythroid 2 like 1 |
| NFκB | Nuclear factor κB |
| NLRP | NOD-like receptor family and pyrin domain |
| NO | Nitric oxide |
| NOX4 | NADPH oxidase 4 |
| Nrf1 | Nuclear respiratory factor 1 |
| PBMCs | Peripheral blood monocytes |
| PC | Phosphatidylcholine |
| PDI | Protein disulfide isomerase |
| PE | Phosphatidylethanolamine |
| PERK | RNA-dependent protein kinase (PKR)-like ER kinase |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PKA | Protein kinase A |
| PKC | protein kinase C |
| PM | Plasma membrane |
| PMCA | Plasma membrane Ca2+ ATPase |
| POMC | Pro-opiomelanocortin |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PVAT | Perivascular adipose tissue |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| RORα | receptor-related orphan receptor α |
| ROS | Reactive oxygen species |
| SERCA | Sarco-endoplasmic reticulum Ca(2+)-ATPase |
| SIRT3 | Sirtuin-3 |
| SPCA | Secretory Pathway Calcium ATPase |
| SREBP | Sterol regulatory element-binding protein |
| STAT | Signal transducer and activator of transcription |
| T1D | Type 1 diabetes |
| T2DM | Type 2 diabetes mellitus |
| TA | Transactivation domain |
| TCA cycle | Tricarboxylic acid cycle |
| TLR2 | Toll-like Receptor 2 |
| TMAO | Trimethylamine N-oxide |
| TNFα | Tumor necrosis factor alpha |
| TRAF2 | TNF receptor–associated factor 2 |
| TUDCA | Tauroursodeoxycholic acid |
| uORF | Upstream Open Reading Frame |
| UPR | Unfolded protein response |
| VLDL | Very low-density lipoprotein |
| VLDLR | VLDL receptor |
| VSMCs | Vascular smooth muscle cells |
| XBP1 | X-box binding protein 1 |
| YAP1 | Yes-associated protein 1 |
References
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 2015, 21, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Autophagy: An ER protein quality control process. Autophagy 2006, 2, 135–137. [Google Scholar] [CrossRef] [PubMed]
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3d analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Prinz, W.A. Atp-binding cassette (abc) transporters mediate nonvesicular, raft-modulated sterol movement from the plasma membrane to the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 45226–45234. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Meyer, T. Stim proteins and the endoplasmic reticulum-plasma membrane junctions. Annu. Rev. Biochem. 2011, 80, 973–1000. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (mam) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J. Role of endoplasmic reticulum-mitochondria communication in type 2 diabetes. Adv. Exp. Med. Biol. 2017, 997, 171–186. [Google Scholar] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.K.; Choudhary, V.; Toulmay, A.; Prinz, W.A. An inducible ER-Golgi tether facilitates ceramide transport to alleviate lipotoxicity. J. Cell Biol. 2017, 216, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, K.; Shibata, M.A.; Ito, Y.; Sohma, Y.; Azuma, H.; Otsuki, Y. Riluzole induces apoptotic cell death in human prostate cancer cells via endoplasmic reticulum stress. Anticancer Res. 2009, 29, 2195–2204. [Google Scholar] [PubMed]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. Nrf1 is an ER membrane sensor that is central to cholesterol homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of beta-cells by Ca2+/Calpain-2 pathways. PLoS ONE 2013, 8, e59921. [Google Scholar]
- Zhang, J.; Li, Y.; Jiang, S.; Yu, H.; An, W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am. J. Physiol. Cell Physiol. 2014, 306, C279–C290. [Google Scholar] [CrossRef] [PubMed]
- Thresher, J.S.; Podolin, D.A.; Wei, Y.; Mazzeo, R.S.; Pagliassotti, M.J. Comparison of the effects of sucrose and fructose on insulin action and glucose tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1334–R1340. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontes, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Haas, M.J. Glucose-induced endoplasmic reticulum stress is independent of oxidative stress: A mechanistic explanation for the failure of antioxidant therapy in diabetes. Free Radic. Biol. Med. 2011, 50, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002, 51 (Suppl. 3), S405–S413. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Kim, H.J.; Kim, J.Y.; Lee, I.K. Guards and culprits in the endoplasmic reticulum: Glucolipotoxicity and beta-cell failure in type II diabetes. Exp. Diabetes Res. 2012, 2012, 639762. [Google Scholar] [CrossRef] [PubMed]
- Bachar, E.; Ariav, Y.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 2009, 4, e4954. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Luo, K.L.; Shi, L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J. Cell Physiol. 2016, 231, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Stoss, H.; Pesch, H.J.; Pontz, B.; Otten, A.; Spranger, J. Wolcott-Rallison syndrome: Diabetes mellitus and spondyloepiphyseal dysplasia. Eur. J. Pediatr. 1982, 138, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Maris, M.; Overbergh, L.; Gysemans, C.; Waget, A.; Cardozo, A.K.; Verdrengh, E.; Cunha, J.P.; Gotoh, T.; Cnop, M.; Eizirik, D.L.; et al. Deletion of C/EBP homologous protein (CHOP) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 2012, 55, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. Atf6alpha-null mice are glucose intolerant due to pancreatic beta-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 2012, 61, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Z.; Zhao, S.; Xiang, R. Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci. Rep. 2016, 6, 27486. [Google Scholar] [CrossRef] [PubMed]
- Qiang, G.; Whang Kong, H.; Gil, V.; Liew, C.W. Transcription regulator TRIP-Br2 mediates ER stress-induced brown adipocytes dysfunction. Sci. Rep. 2017, 7, 40215. [Google Scholar] [CrossRef] [PubMed]
- Qiang, G.; Kong, H.W.; Fang, D.; McCann, M.; Yang, X.; Du, G.; Bluher, M.; Zhu, J.; Liew, C.W. The obesity-induced transcriptional regulator TRIP-Br2 mediates visceral fat endoplasmic reticulum stress-induced inflammation. Nat. Commun. 2016, 7, 11378. [Google Scholar] [CrossRef] [PubMed]
- Alcala, M.; Calderon-Dominguez, M.; Bustos, E.; Ramos, P.; Casals, N.; Serra, D.; Viana, M.; Herrero, L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep. 2017, 7, 16082. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgul, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Zhang, J.; Liu, Y.; Zhang, Y.; Su, Z. Retinoic acid receptor-related orphan receptor alpha stimulates adipose tissue inflammation by modulating endoplasmic reticulum stress. J. Biol. Chem. 2017, 292, 13959–13969. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Borg, D.J.; Harcourt, B.E.; Tong, H.; Sheng, Y.H.; Ng, C.P.; Das, I.; Wang, R.; Chen, A.C.; Loudovaris, T.; et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 2014, 20, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yang, S.; Yang, L.; Cheng, Y.; Zhang, H. Interleukin-22 alleviated palmitate-induced endoplasmic reticulum stress in INS-1 cells through activation of autophagy. PLoS ONE 2016, 11, e0146818. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ehses, J.A.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.M.; Reinecke, M.; Halban, P.A.; Donath, M.Y. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Boni-Schnetzler, M.; Ellingsgaard, H.; Ehses, J.A. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology (Bethesda) 2009, 24, 325–331. [Google Scholar] [PubMed]
- Miani, M.; Barthson, J.; Colli, M.L.; Brozzi, F.; Cnop, M.; Eizirik, D.L. Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1beta-induced apoptosis via Bim/A1 imbalance. Cell Death Dis. 2013, 4, e701. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladriere, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Salvado, L.; Palomer, X.; Barroso, E.; Vazquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ibi, D.; Taniguchi, K.; Lee, J.; Herrema, H.; Akosman, B.; Mucka, P.; Salazar Hernandez, M.A.; Uyar, M.F.; Park, S.W.; et al. Inflammation improves glucose homeostasis through IKKbeta-XBP1s interaction. Cell 2016, 167, 1052–1066.e18. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Kawai, T.; Hirose, H.; Tanaka, K.; Kurosawa, H.; Fujii, C.; Fujita, H.; Seto, Y.; Matsumoto, H.; Itoh, H. Palmitic acid-rich diet suppresses glucose-stimulated insulin secretion (GSIS) and induces endoplasmic reticulum (ER) stress in pancreatic islets in mice. Endocr. Res. 2016, 41, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Luzuriaga, J.; Maxwell, E.L.; West, P.K.; Bensellam, M.; Laybutt, D.R. The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol. Cell Endocrinol. 2015, 413, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.S.; Oh, J.E.; Nan, J.; Lee, H.; Jung, H.S.; Chung, S.S.; Park, K.S. SIRT3 overexpression attenuates palmitate-induced pancreatic beta-cell dysfunction. PLoS ONE 2015, 10, e0124744. [Google Scholar]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, P.; Wang, T.; Chen, K.; Zhu, W.; Wang, H. Inhibition of calcium influx reduces dysfunction and apoptosis in lipotoxic pancreatic beta-cells via regulation of endoplasmic reticulum stress. PLoS ONE 2015, 10, e0132411. [Google Scholar]
- Peng, G.; Li, L.; Liu, Y.; Pu, J.; Zhang, S.; Yu, J.; Zhao, J.; Liu, P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 2011, 152, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Salvado, L.; Coll, T.; Gomez-Foix, A.M.; Salmeron, E.; Barroso, E.; Palomer, X.; Vazquez-Carrera, M. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2013, 56, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, S.; Zou, L.; Xu, C.; Geng, B.; Xu, G. Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 2012, 287, 6240–6249. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Claycombe, K.; Schommer, J.; Collins, D.; Ghribi, O. Palmitate-induced endoplasmic reticulum stress and subsequent C/EBPalpha homologous protein activation attenuates leptin and insulin-like growth factor 1 expression in the brain. Cell Signal. 2016, 28, 1789–1805. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic beta-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [PubMed]
- Manuel, A.M.; Walla, M.D.; Faccenda, A.; Martin, S.L.; Tanis, R.M.; Piroli, G.G.; Adam, J.; Kantor, B.; Mutus, B.; Townsend, D.M.; et al. Succination of protein disulfide isomerase links mitochondrial stress and endoplasmic reticulum stress in the adipocyte during diabetes. Antioxid. Redox Signal. 2017, 27, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. A switch from life to death in endoplasmic reticulum stressed beta-cells. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 58–65. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: Recent findings and future research directions. Mol. Cell Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.Y.; Kim, Y.D.; Lee, K.M.; Min, A.K.; Kim, M.K.; Kim, H.S.; Won, K.C.; Park, J.Y.; Lee, K.U.; Choi, H.S.; et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 2008, 149, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Christulia, F.; Ortis, F.; Pirot, P.; Lortz, S.; Lenzen, S.; Eizirik, D.L.; Cardozo, A.K. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010, 53, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Homko, C.; Barrero, C.A.; Stein, T.P.; Chen, X.; Cheung, P.; Fecchio, C.; Koller, S.; Merali, S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 2015, 7, 304re7. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Takatani, T.; Shirakawa, J.; Roe, M.W.; Leech, C.A.; Maier, B.F.; Mirmira, R.G.; Kulkarni, R.N. IRS1 deficiency protects beta-cells against ER stress-induced apoptosis by modulating sXBP-1 stability and protein translation. Sci. Rep. 2016, 6, 28177. [Google Scholar] [CrossRef] [PubMed]
- Simon-Szabo, L.; Kokas, M.; Mandl, J.; Keri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Lee, J.; Zhou, Y.; Copps, K.D.; White, M.F.; Ozcan, U. IRS1Ser307 phosphorylation does not mediate mTORC1-induced insulin resistance. Biochem. Biophys. Res. Commun. 2014, 443, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Choi, J.; Nam, T.G.; Ku, J.M.; Jeong, K. Anti-diabetic effect of 3-hydroxy-2-naphthoic acid, an endoplasmic reticulum stress-reducing chemical chaperone. Eur. J. Pharmacol. 2016, 779, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Makinen, S.; Nguyen, Y.H.; Skrobuk, P.; Koistinen, H.A. Palmitate and oleate exert differential effects on insulin signalling and glucose uptake in human skeletal muscle cells. Endocr. Connect. 2017, 6, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Zanotto, T.M.; Quaresma, P.G.; Guadagnini, D.; Weissmann, L.; Santos, A.C.; Vecina, J.F.; Calisto, K.; Santos, A.; Prada, P.O.; Saad, M.J. Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab. 2017, 6, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH couples circadian clock with hepatic lipid metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, Z.; Walker, P.D.; Kapatos, G.; Zhang, K. CREBH maintains circadian glucose homeostasis by regulating hepatic glycogenolysis and gluconeogenesis. Mol. Cell Biol. 2017, 37, e00048-17. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Salazar Hernandez, M.A.; Auen, T.; Mucka, P.; Lee, J.; Ozcan, U. PGC-1alpha functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol. Metab. 2018, 7, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Zhou, Y.; Zhang, D.; Lee, J.; Salazar Hernandez, M.A.; Shulman, G.I.; Ozcan, U. XBP1s is an anti-lipogenic protein. J. Biol. Chem. 2016, 291, 17394–17404. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Wang, B.; Palladino, E.N.; de Aguiar Vallim, T.Q.; Ford, D.A.; Tontonoz, P. ER phospholipid composition modulates lipogenesis during feeding and in obesity. J. Clin. Investig. 2017, 127, 3640–3651. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hodish, I.; Haataja, L.; Lara-Lemus, R.; Rajpal, G.; Wright, J.; Arvan, P. Proinsulin misfolding and diabetes: Mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 2010, 21, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Kayo, T.; Ikeda, T.; Koizumi, A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997, 46, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; He, K.; Arunagiri, A.; Paton, A.W.; Paton, J.C.; Arvan, P.; Tsai, B. Chaperone-driven degradation of a misfolded proinsulin mutant in parallel with restoration of wild-type insulin secretion. Diabetes 2017, 66, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Opie, E.L. The relation Oe diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands Oe langerhans. J. Exp. Med. 1901, 5, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Gurlo, T.; Rivera, J.F.; Butler, A.E.; Cory, M.; Hoang, J.; Costes, S.; Butler, P.C. CHOP contributes to, but is not the only mediator of, IAPP induced beta-cell apoptosis. Mol. Endocrinol. 2016, 30, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cadavez, L.; Montane, J.; Alcarraz-Vizan, G.; Visa, M.; Vidal-Fabrega, L.; Servitja, J.M.; Novials, A. Chaperones ameliorate beta cell dysfunction associated with human islet amyloid polypeptide overexpression. PLoS ONE 2014, 9, e101797. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Camargo, D.C.; Tripsianes, K.; Buday, K.; Franko, A.; Gobl, C.; Hartlmuller, C.; Sarkar, R.; Aichler, M.; Mettenleiter, G.; Schulz, M.; et al. The redox environment triggers conformational changes and aggregation of hiAPP in Type II Diabetes. Sci. Rep. 2017, 7, 44041. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational switching within dynamic oligomers underpins toxic gain-of-function by diabetes-associated amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic beta cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Colburn, W.A.; Gottlieb, A.B.; Koda, J.; Kolterman, O.G. Pharmacokinetics and pharmacodynamics of AC137 (25,28,29 tripro-amylin, human) after intravenous bolus and infusion doses in patients with insulin-dependent diabetes. J. Clin. Pharmacol. 1996, 36, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.; Claret, M. Hypothalamic ER stress: A bridge between leptin resistance and obesity. FEBS Lett. 2015, 589, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Liu, G.; Guo, J.; Su, Z. Hypothalamic endoplasmic reticulum stress as a key mediator of obesity-induced leptin resistance. Obes. Rev. 2018, 19, 770–785. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Deng, Z.; Gao, Y.; Sun, J.; Kong, X.; Huang, Y.; He, Z.; Xu, Y.; Chang, Y.; Yu, K.J.; et al. Ire1alpha in Pomc neurons is required for thermogenesis and glycemia. Diabetes 2017, 66, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Gonzalez-Garcia, I.; Seoane-Collazo, P.; Martinez-Sanchez, N.; Linares-Pose, L.; Rial-Pensado, E.; Ferno, J.; Tena-Sempere, M.; Casals, N.; Dieguez, C.; et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes 2017, 66, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.W.; Liu, T.; Kong, X.; Fukuda, M.; Deng, Y.; Berglund, E.D.; Deng, Z.; Gao, Y.; Liu, T.; Sohn, J.W.; et al. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 2014, 20, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Spinas, G.A.; Niessen, M. ER stress in adipocytes inhibits insulin signaling, represses lipolysis, and alters the secretion of adipokines without inhibiting glucose transport. Horm. Metab. Res. 2010, 42, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental nonalcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, eaah4477. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-Lopez, T.; Escola-Gil, J.C.; Cedo, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Shan, B.; Liu, Y.; Deng, Y.; Yan, C.; Wu, Y.; Mao, T.; Qiu, Y.; Zhou, Y.; Jiang, S.; et al. Hepatic IRE1alpha regulates fasting-induced metabolic adaptive programs through the XBP1s-PPARalpha axis signalling. Nat. Commun. 2014, 5, 3528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Imarisio, C.; Alchera, E.; Bangalore Revanna, C.; Valente, G.; Follenzi, A.; Trisolini, E.; Boldorini, R.; Carini, R. Oxidative and ER stress-dependent ASK1 activation in steatotic hepatocytes and Kupffer cells sensitizes mice fatty liver to ischemia/reperfusion injury. Free Radic. Biol. Med. 2017, 112, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Porteiro, B.; Fondevila, M.F.; Delgado, T.C.; Iglesias, C.; Imbernon, M.; Iruzubieta, P.; Crespo, J.; Zabala-Letona, A.; Ferno, J.; Gonzalez-Teran, B.; et al. Hepatic p63 regulates steatosis via IKKbeta/ER stress. Nat. Commun. 2017, 8, 15111. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.Q.; Wang, H.; Zeng, X.Y.; Chan, S.M.; Li, S.P.; Jo, E.; Leung, S.L.; Molero, J.C.; Ye, J.M. IRE1 impairs insulin signaling transduction of fructose-fed mice via JNK independent of excess lipid. Biochim. Biophys. Acta 2015, 1852, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Hwang, S.; Cherrington, N.J.; Ryu, D.Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 2017, 8, 63370–63381. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell Physiol. 2012, 227, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Liu, Y.; Kumar, P.; Smith, T.; Thorn, N.E.; Farris, A.B.; Anania, F.A. C/EBP homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab. Investig. 2016, 96, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallee, D.; Rousseau, D.; Patouraux, S.; Bonnafous, S.; Adam, G.; Luciano, F.; Luci, C.; Anty, R.; Iannelli, A.; et al. Bax inhibitor-1 protects from non-alcoholic steatohepatitis by limiting ire1alpha signaling. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Liu, B.; Wu, B.X.; Morreall, J.; Roth, B.; Davies, C.; Sun, S.; Diehl, J.A.; Li, Z. CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat. Struct. Mol. Biol. 2017, 24, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yoo, W.H.; Chae, H.J. ER stress and autophagy. Curr. Mol. Med. 2015, 15, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Hong, H.C.; Hwang, H.J.; Yoo, H.J.; Baik, S.H.; Choi, K.M. C1q/TNF-related protein 9 (CTRP9) attenuates hepatic steatosis via the autophagy-mediated inhibition of endoplasmic reticulum stress. Mol. Cell Endocrinol. 2015, 417, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Garbin, U.; Mozzini, C.; Stranieri, C.; Pasini, A.; Solani, E.; Tinelli, I.A.; Pasini, A.F. The atherosclerotic plaque vulnerability: Focus on the oxidative and endoplasmic reticulum stress in orchestrating the macrophage apoptosis in the formation of the necrotic core. Curr. Med. Chem. 2015, 22, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, D.; Nystrom, T. Hypoglycemic pharmacological treatment of type 2 diabetes: Targeting the endothelium. Mol. Cell Endocrinol. 2009, 297, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Young, T.L.; Dang, V.T.; Shi, Y.; McAlpine, C.S.; Werstuck, G.H. 4-phenylbutyrate and valproate treatment attenuates the progression of atherosclerosis and stabilizes existing plaques. Atherosclerosis 2017, 266, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Banko, N.S.; McAlpine, C.S.; Venegas-Pino, D.E.; Raja, P.; Shi, Y.; Khan, M.I.; Werstuck, G.H. Glycogen synthase kinase 3alpha deficiency attenuates atherosclerosis and hepatic steatosis in high fat diet-fed low density lipoprotein receptor-deficient mice. Am. J. Pathol. 2014, 184, 3394–3404. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Lipoprotein metabolism in the macrophage: Implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 1983, 52, 223–261. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Werstuck, G.H. Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3alpha/beta regulate foam cell formation. J. Lipid Res. 2014, 55, 2320–2333. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guo, Y.; Sun, S.; Jiang, X.; Tang, B.; Wang, Q.; Wang, L. Free cholesterol-induced macrophage apoptosis is mediated by inositol-requiring enzyme 1 alpha-regulated activation of Jun N-terminal kinase. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Tsukano, H.; Gotoh, T.; Endo, M.; Miyata, K.; Tazume, H.; Kadomatsu, T.; Yano, M.; Iwawaki, T.; Kohno, K.; Araki, K.; et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ruas, J.L.; Estall, J.L.; Rasbach, K.A.; Choi, J.H.; Ye, L.; Bostrom, P.; Tyra, H.M.; Crawford, R.W.; Campbell, K.P.; et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab. 2011, 13, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Pivotal role of calcium/calmodulin-dependent protein kinase II in ER stress-induced apoptosis. Cell Cycle 2010, 9, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Li, Y.; Bao, L.; Yao, P.M.; Kuriakose, G.; Rong, J.; Fisher, E.A.; Tabas, I. Brief report: Increased apoptosis in advanced atherosclerotic lesions of Apoe−/− mice lacking macrophage Bcl-2. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Ying, R.; Li, S.W.; Chen, J.Y.; Zhang, H.F.; Yang, Y.; Gu, Z.J.; Chen, Y.X.; Wang, J.F. Endoplasmic reticulum stress in perivascular adipose tissue promotes destabilization of atherosclerotic plaque by regulating GM-CSF paracrine. J. Transl. Med. 2018, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.; Kim, M.; Noh, J.S.; Park, C.H.; Song, Y.O. Kimchi attenuates fatty streak formation in the aorta of low-density lipoprotein receptor knockout mice via inhibition of endoplasmic reticulum stress and apoptosis. Nutr. Res. Pract. 2017, 11, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.C.; Granville, D.J.; Hunt, D.W.; McManus, B.M. Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. J. Mol. Cell Cardiol. 2001, 33, 1673–1690. [Google Scholar] [CrossRef] [PubMed]
- Bombeli, T.; Schwartz, B.R.; Harlan, J.M. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood 1999, 93, 3831–3838. [Google Scholar] [PubMed]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Feaver, R.E.; Hastings, N.E.; Pryor, A.; Blackman, B.R. GRP78 upregulation by atheroprone shear stress via p38-, alpha2beta1-dependent mechanism in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.S.; Gharavi, N.M.; Clark, M.J.; Pagnon, J.; Yang, W.P.; He, A.; Truong, A.; Baruch-Oren, T.; Berliner, J.A.; Kirchgessner, T.G.; et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Gora, S.; Maouche, S.; Atout, R.; Wanherdrick, K.; Lambeau, G.; Cambien, F.; Ninio, E.; Karabina, S.A. Phospholipolyzed LDL induces an inflammatory response in endothelial cells through endoplasmic reticulum stress signaling. FASEB J. 2010, 24, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Outinen, P.A.; Sood, S.K.; Pfeifer, S.I.; Pamidi, S.; Podor, T.J.; Li, J.; Weitz, J.I.; Austin, R.C. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 1999, 94, 959–967. [Google Scholar] [PubMed]
- Ushio-Fukai, M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 2009, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Willecke, F.; Yuan, C.; Oka, K.; Chan, L.; Hu, Y.; Barnhart, S.; Bornfeldt, K.E.; Goldberg, I.J.; Fisher, E.A. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in LDL receptor-deficient mice. PLoS ONE 2015, 10, e0128996. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Di, M.; Wang, L.; Li, M.; Zhang, Y.; Liu, X.; Zeng, R.; Wang, H.; Chen, Y.; Chen, W.; Zhang, Y.; et al. Dickkopf1 destabilizes atherosclerotic plaques and promotes plaque formation by inducing apoptosis of endothelial cells through activation of ER stress. Cell Death Dis. 2017, 8, e2917. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Pan, S.; Meng, L.; Zhou, C.; Jiang, C.; Ji, Z.; Chi, J.; Guo, H. MicroRNA-384-mediated Herpud1 upregulation promotes angiotensin II-induced endothelial cell apoptosis. Biochem. Biophys. Res. Commun. 2017, 488, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Takaguri, A.; Kubo, T.; Mori, M.; Satoh, K. The protective role of YAP1 on ER stress-induced cell death in vascular smooth muscle cells. Eur. J. Pharmacol. 2017, 815, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.K.; Yu, P.L.; Bai, Y.P.; Yan, S.T.; Zhao, S.P.; Zhang, G.Q. Role of PERK/eiF2alpha/CHOP endoplasmic reticulum stress pathway in oxidized low-density lipoprotein mediated induction of endothelial apoptosis. Biomed. Environ. Sci. 2016, 29, 868–876. [Google Scholar] [PubMed]
- Clarke, M.C.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.P.; Hung, H.F.; Wang, B.W.; Shyu, K.G. The molecular regulation of GADD153 in apoptosis of cultured vascular smooth muscle cells by cyclic mechanical stretch. Cardiovasc. Res. 2008, 77, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [PubMed]
- Kedi, X.; Ming, Y.; Yongping, W.; Yi, Y.; Xiaoxiang, Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis 2009, 207, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Werstuck, G.H.; Lhotak, S.; de Koning, A.B.; Sood, S.K.; Hossain, G.S.; Moller, J.; Ritskes-Hoitinga, M.; Falk, E.; Dayal, S.; et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation 2004, 110, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Austin, R.C. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. Biofactors 2009, 35, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, J.G.; Sood, S.K.; Austin, R.C. Role of endoplasmic reticulum calcium disequilibria in the mechanism of homocysteine-induced ER stress. Antioxid. Redox Signal. 2007, 9, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Wang, Y.B.; Han, B.; Yang, B.; Qiang, Y.W.; Zhang, Y.; Wang, Z.; Huang, X.; Liu, J.; Chen, Y.D.; et al. Activation of aldehyde dehydrogenase 2 slows down the progression of atherosclerosis via attenuation of ER stress and apoptosis in smooth muscle cells. Acta Pharmacol. Sin. 2018, 39, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Calcium pumps: Why so many? Compr. Physiol. 2012, 2, 1045–1060. [Google Scholar] [PubMed]
- Guerrero-Hernandez, A.; Verkhratsky, A. Calcium signalling in diabetes. Cell Calcium 2014, 56, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin. Ther. Targets 2013, 17, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 2012, 61, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhou, Y.; Lee, J.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J. Biol. Chem. 2018, 293, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Li, Y.; Kuang, Y.; Cui, H.; Yang, Y.; Sun, W.; Liu, K.; Chen, D.; Yan, Q.; Wen, L. PKCdelta silencing alleviates saturated fatty acid induced ER stress by enhancing SERCA activity. Biosci. Rep. 2017, 37, BSR20170869. [Google Scholar] [CrossRef] [PubMed]
- Leandro, P.; Gomes, C.M. Protein misfolding in conformational disorders: Rescue of folding defects and chemical chaperoning. Mini Rev. Med. Chem. 2008, 8, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.M. Protein misfolding in disease and small molecule therapies. Curr. Top. Med. Chem. 2012, 12, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Shepshelovich, J.; Goldstein-Magal, L.; Globerson, A.; Yen, P.M.; Rotman-Pikielny, P.; Hirschberg, K. Protein synthesis inhibitors and the chemical chaperone TMAO reverse endoplasmic reticulum perturbation induced by overexpression of the iodide transporter pendrin. J. Cell Sci. 2005, 118, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhu, M.; Wu, W.; Rashid, A.; Liang, Y.; Hou, L.; Ning, Q.; Luo, X. Valproate pretreatment protects pancreatic beta-cells from palmitate-induced ER stress and apoptosis by inhibiting glycogen synthase kinase-3beta. J. Biomed. Sci. 2014, 21, 38. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Zyryanova, A.; Crespillo-Casado, A.; Fischer, P.M.; Harding, H.P.; Ron, D. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015, 348, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.N.; Abiraman, K.; Plyler, E.S.; Sprenkle, N.T.; Gibson, S.A.; McFarland, B.C.; Rajbhandari, R.; Rowse, A.L.; Benveniste, E.N.; Meares, G.P. Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J. Biol. Chem. 2016, 291, 15830–15840. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Dyle, M.C.; Bullard, S.A.; Dierdorff, J.M.; Murry, D.J.; Fox, D.K.; Bongers, K.S.; Lira, V.A.; Meyerholz, D.K.; Talley, J.J.; et al. Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. J. Biol. Chem. 2015, 290, 25497–25511. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Ron, D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Bouchecareilh, M.; Higa, A.; Fribourg, S.; Moenner, M.; Chevet, E. Peptides derived from the bifunctional kinase/RNase enzyme IRE1alpha modulate IRE1alpha activity and protect cells from endoplasmic reticulum stress. FASEB J. 2011, 25, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Cheng, Y.; Yan, W.; Shi, X.; Xu, X.; Zhou, J.; Li, J.; Chen, J.; Shen, X. TSPA as a novel ATF6alpha translocation inducer efficiently ameliorates insulin sensitivity restoration and glucose homeostasis in db/db mice. Biochem. Biophys. Res. Commun. 2018, 499, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xu, F.; Liang, H.; Cao, H.; Cai, M.; Xu, W.; Weng, J. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology 2017, 66, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2alpha deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Silva-Veiga, F.M.; Rachid, T.L.; de Oliveira, L.; Graus-Nunes, F.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. GW0742 (PPAR-beta agonist) attenuates hepatic endoplasmic reticulum stress by improving hepatic energy metabolism in high-fat diet fed mice. Mol. Cell Endocrinol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. https://doi.org/10.3390/cells7060063
Ghemrawi R, Battaglia-Hsu S-F, Arnold C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells. 2018; 7(6):63. https://doi.org/10.3390/cells7060063
Chicago/Turabian StyleGhemrawi, Rose, Shyue-Fang Battaglia-Hsu, and Carole Arnold. 2018. "Endoplasmic Reticulum Stress in Metabolic Disorders" Cells 7, no. 6: 63. https://doi.org/10.3390/cells7060063
APA StyleGhemrawi, R., Battaglia-Hsu, S.-F., & Arnold, C. (2018). Endoplasmic Reticulum Stress in Metabolic Disorders. Cells, 7(6), 63. https://doi.org/10.3390/cells7060063