The ER Stress Inducer l-Azetidine-2-Carboxylic Acid Elevates the Levels of Phospho-eIF2α and of LC3-II in a Ca2+-Dependent Manner


 ,
,  and
and 
Abstract
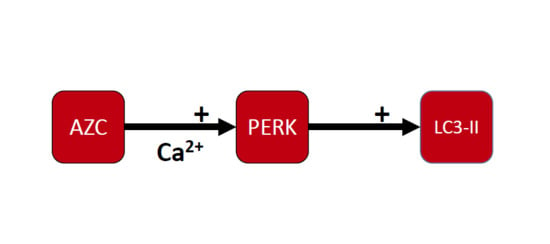
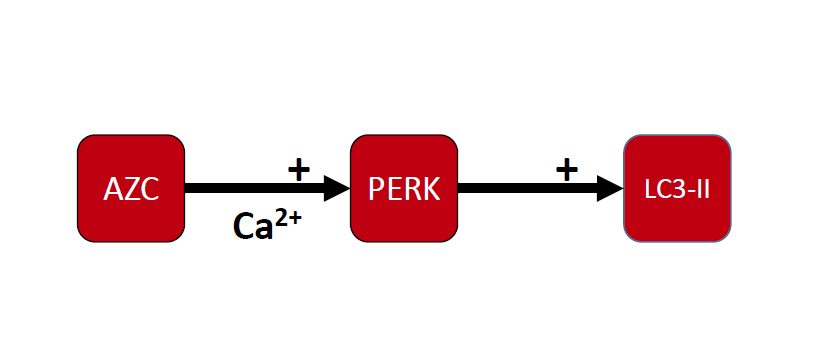{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents and Antibodies
2.3. Sodium Dodecyl Sulfate (SDS) Polyacrylamide Gel Electrophoresis and Western Blotting
2.4. XBP1 Splicing
2.5. Cell Death Assays
2.6. Ca2+ Measurements at the Population Level
2.7. Single-Cell Ca2+ Measurements
2.8. Statistics
3. Results
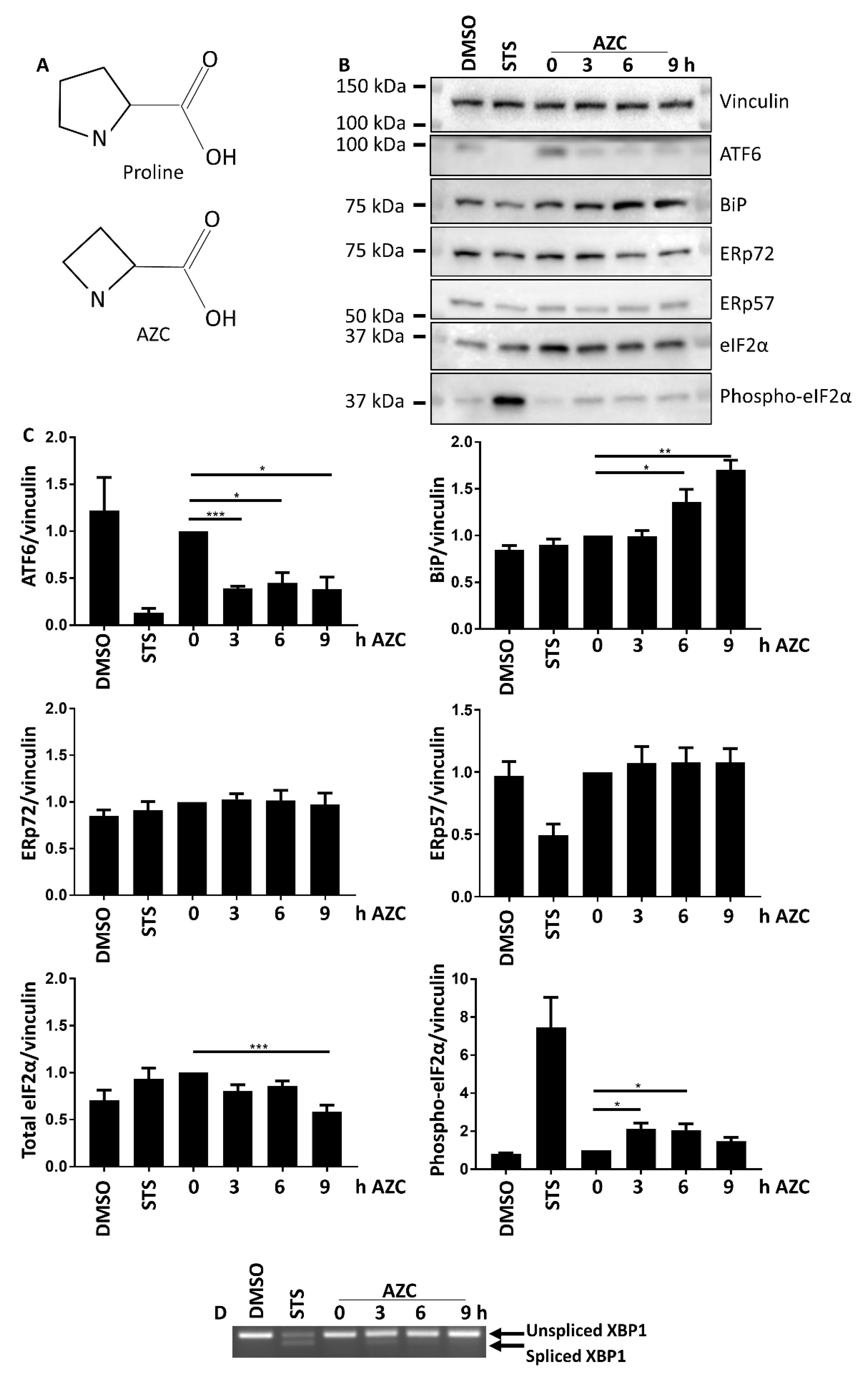
3.1. AZC Upregulates the Levels of BiP and Phospho-eIF2α while Decreasing the Level of Full-Length ATF6
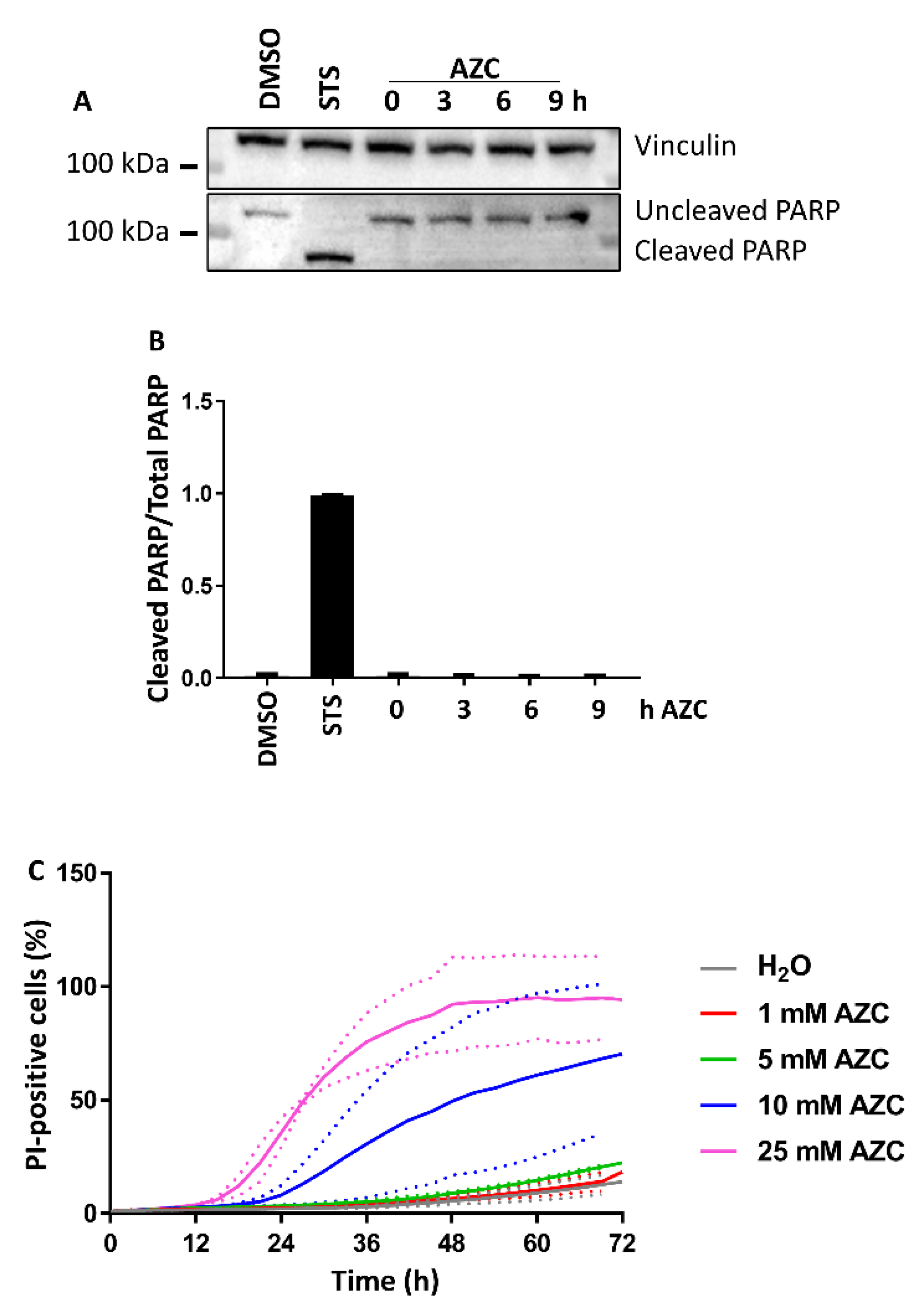
3.2. AZC Does Not Induce Cell Death within 6 h of Treatment
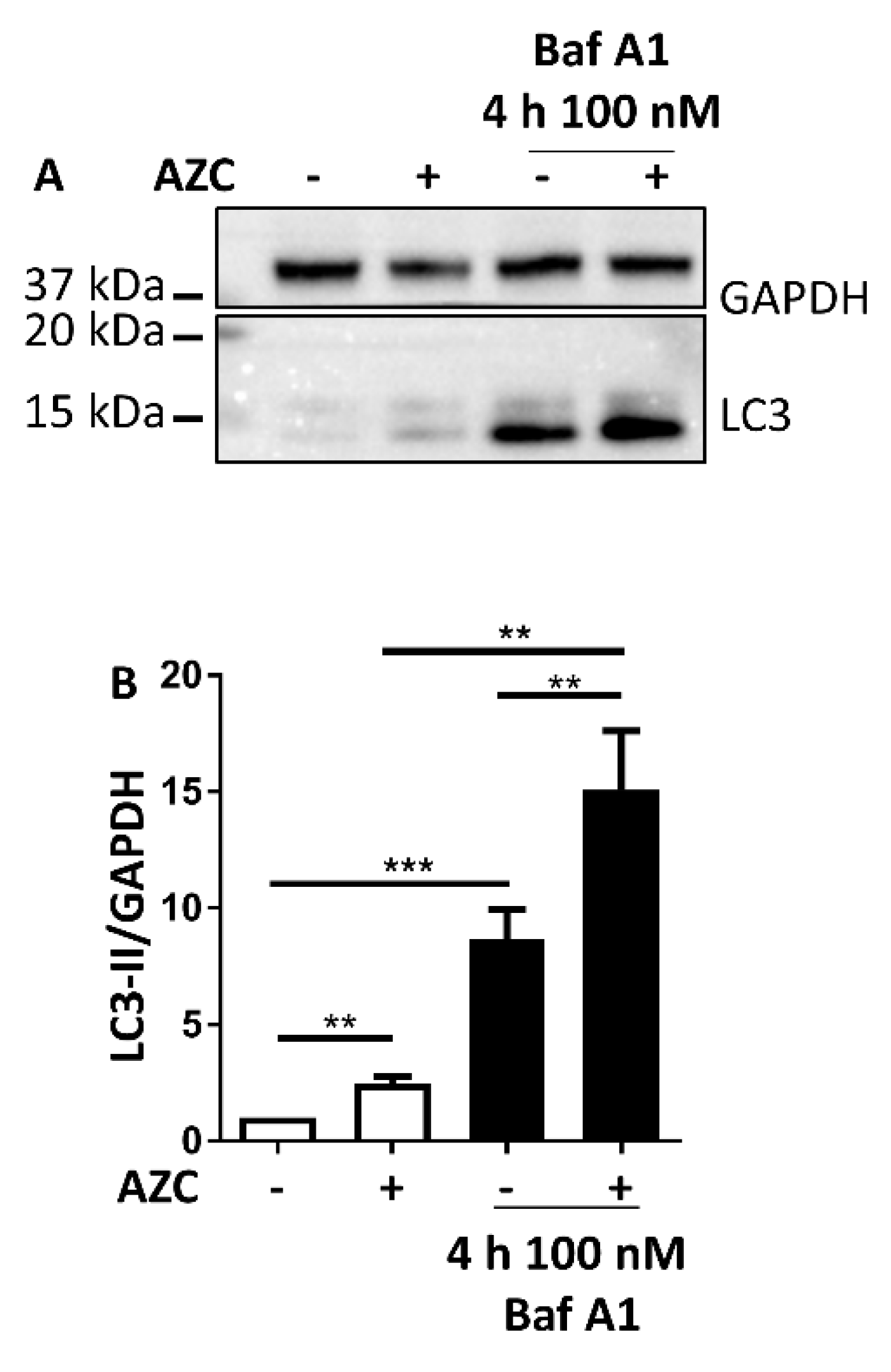
3.3. AZC Increases the Levels of Lipidated Autophagy Marker LC3
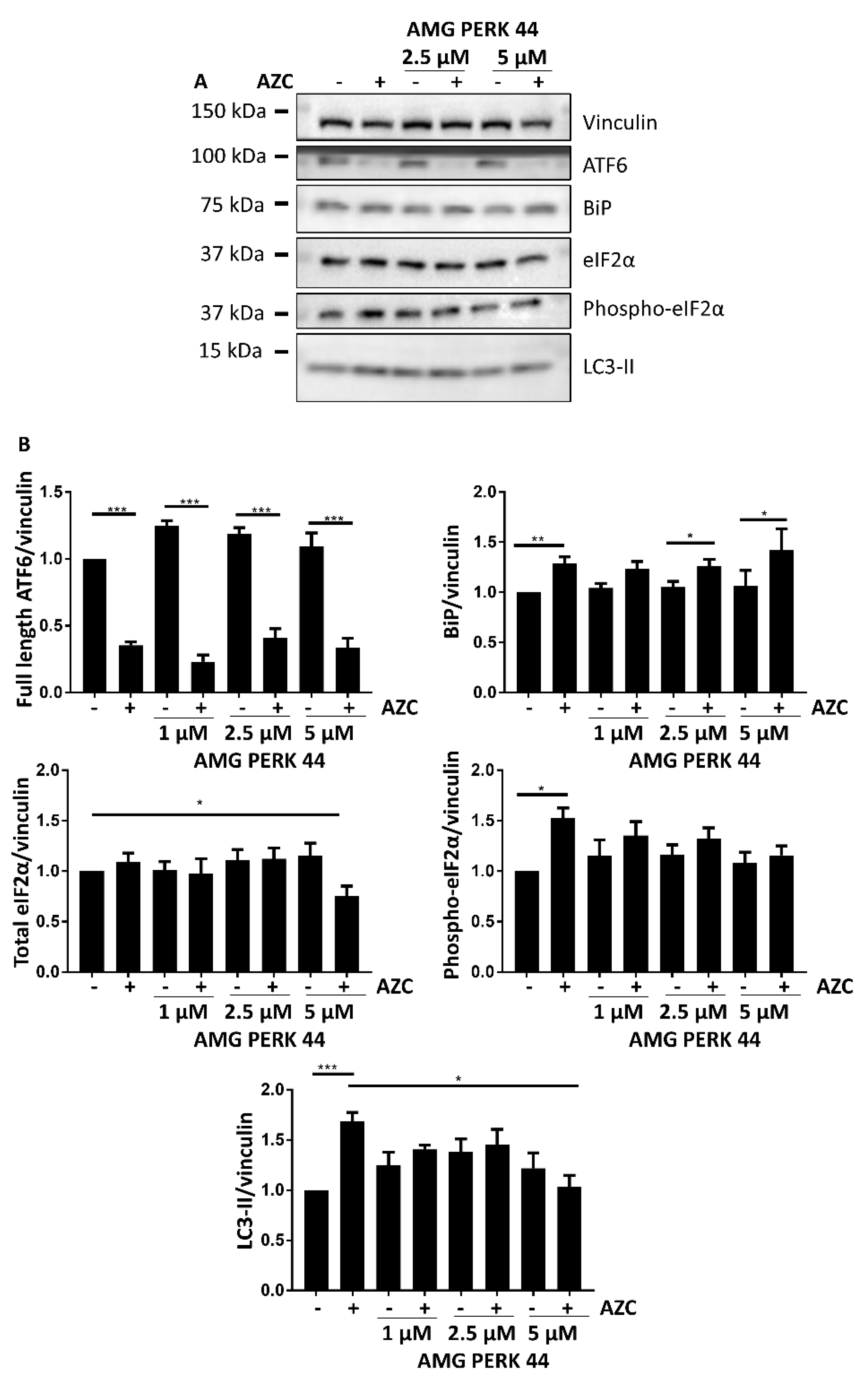
3.4. AZC Upregulates the Levels of LC3-II Subsequently to Activation of the PERK Pathway
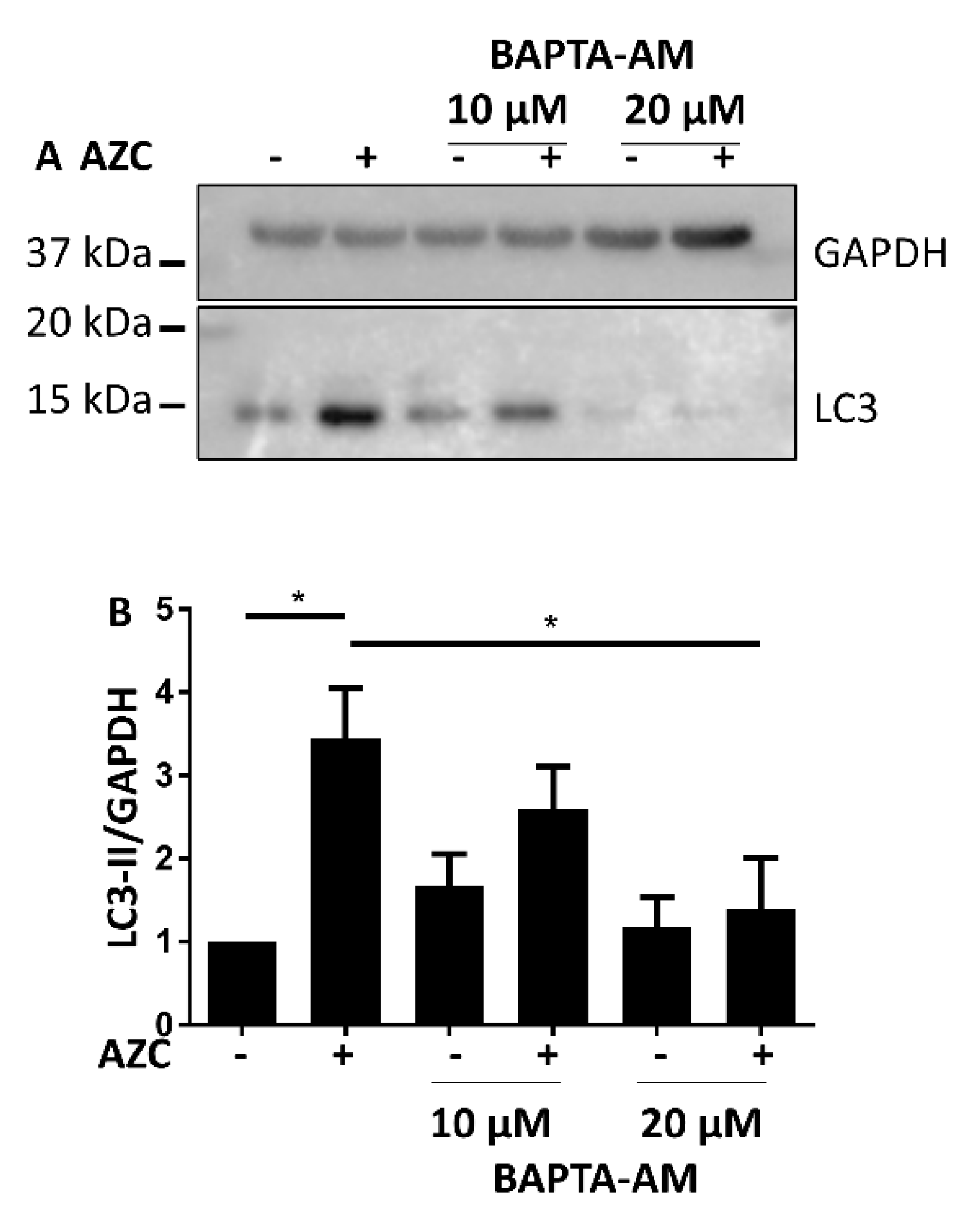
3.5. AZC Upregulates the Levels of LC3-II in a Ca2+-Dependent Manner
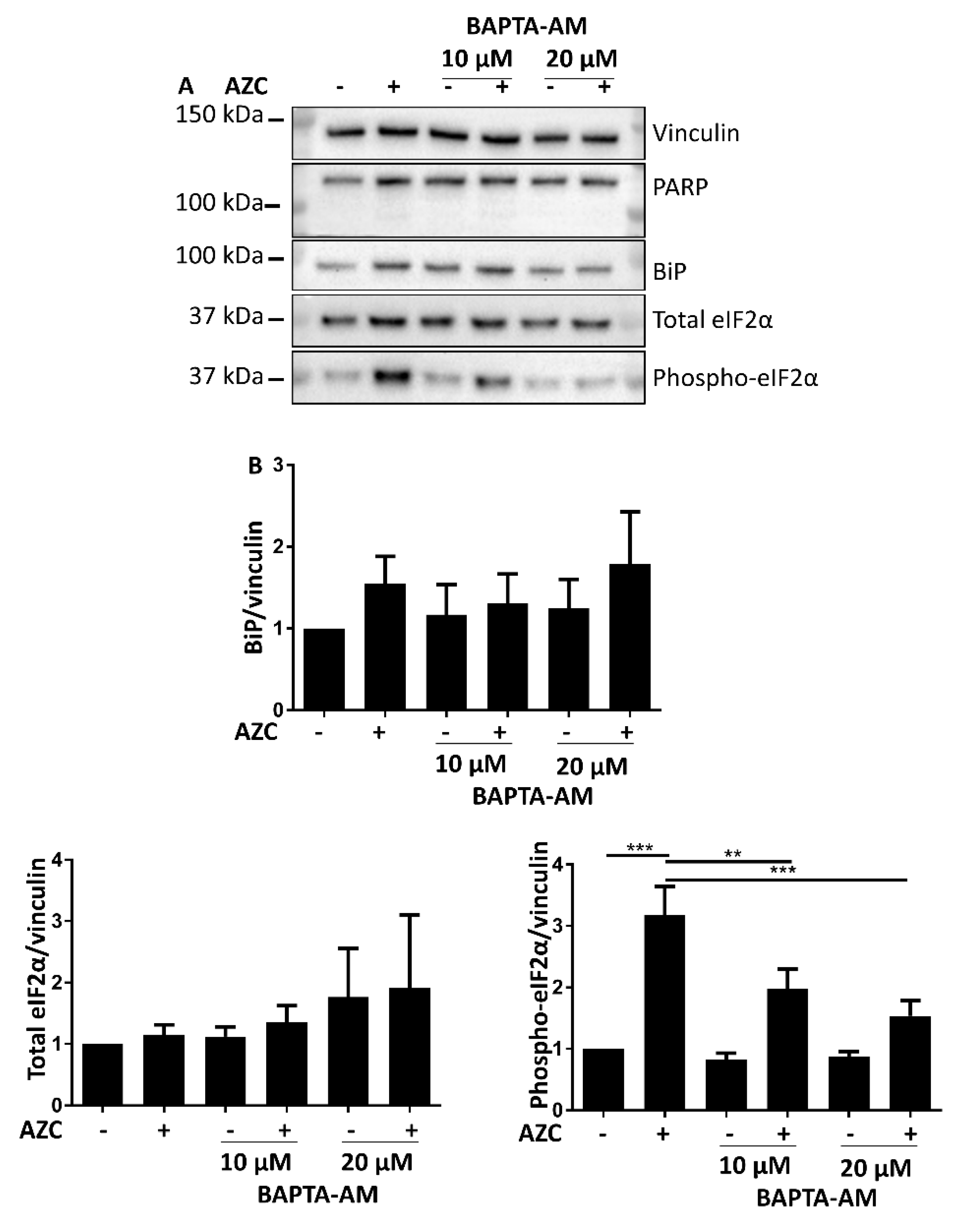
3.6. AZC-Induced Elevation of Phospho-eIF2α Levels Is Ca2+ Dependent
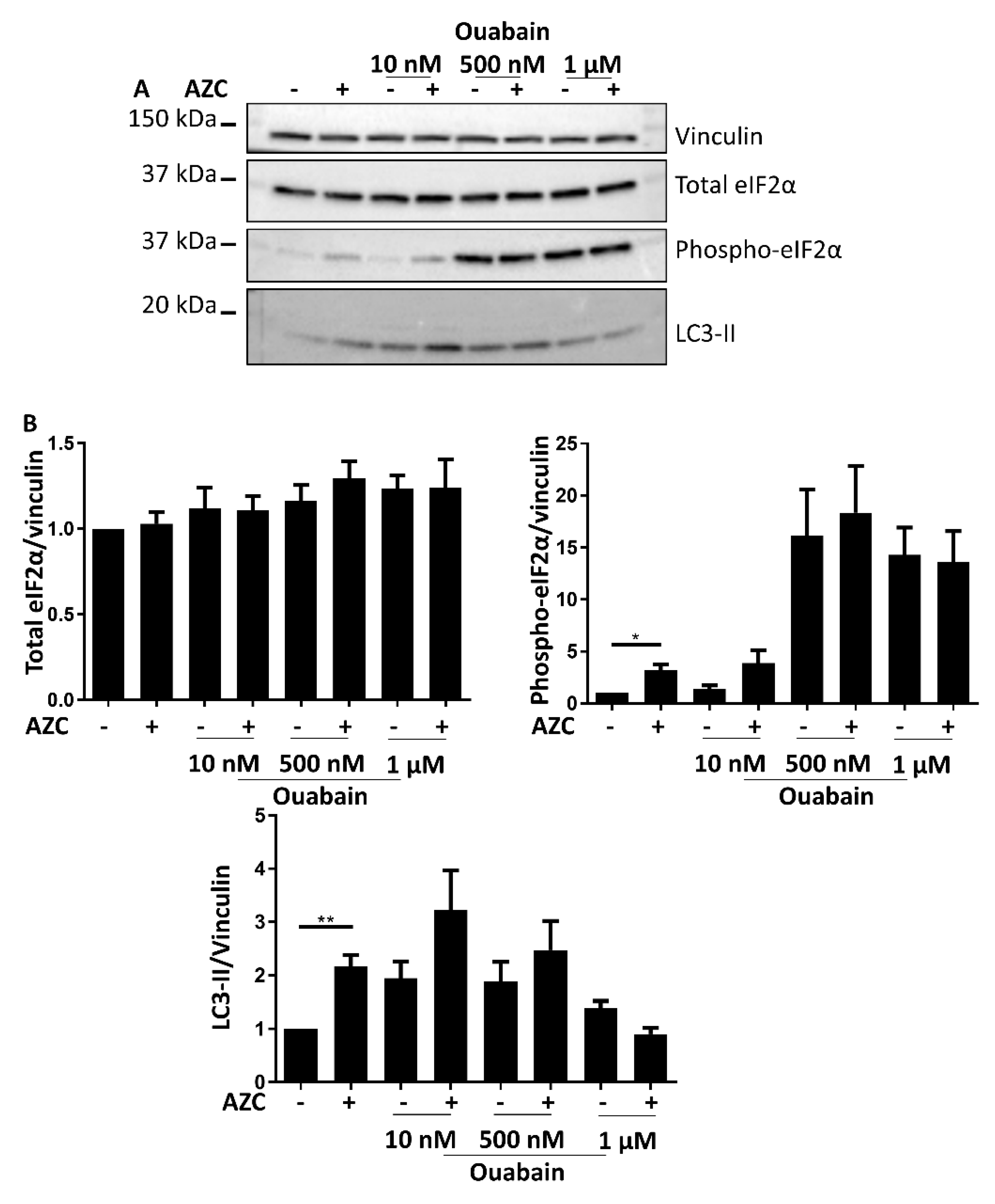
3.7. Effects of BAPTA-AM Treatment Are Not Related to Na+/K+ ATPase Inhibition
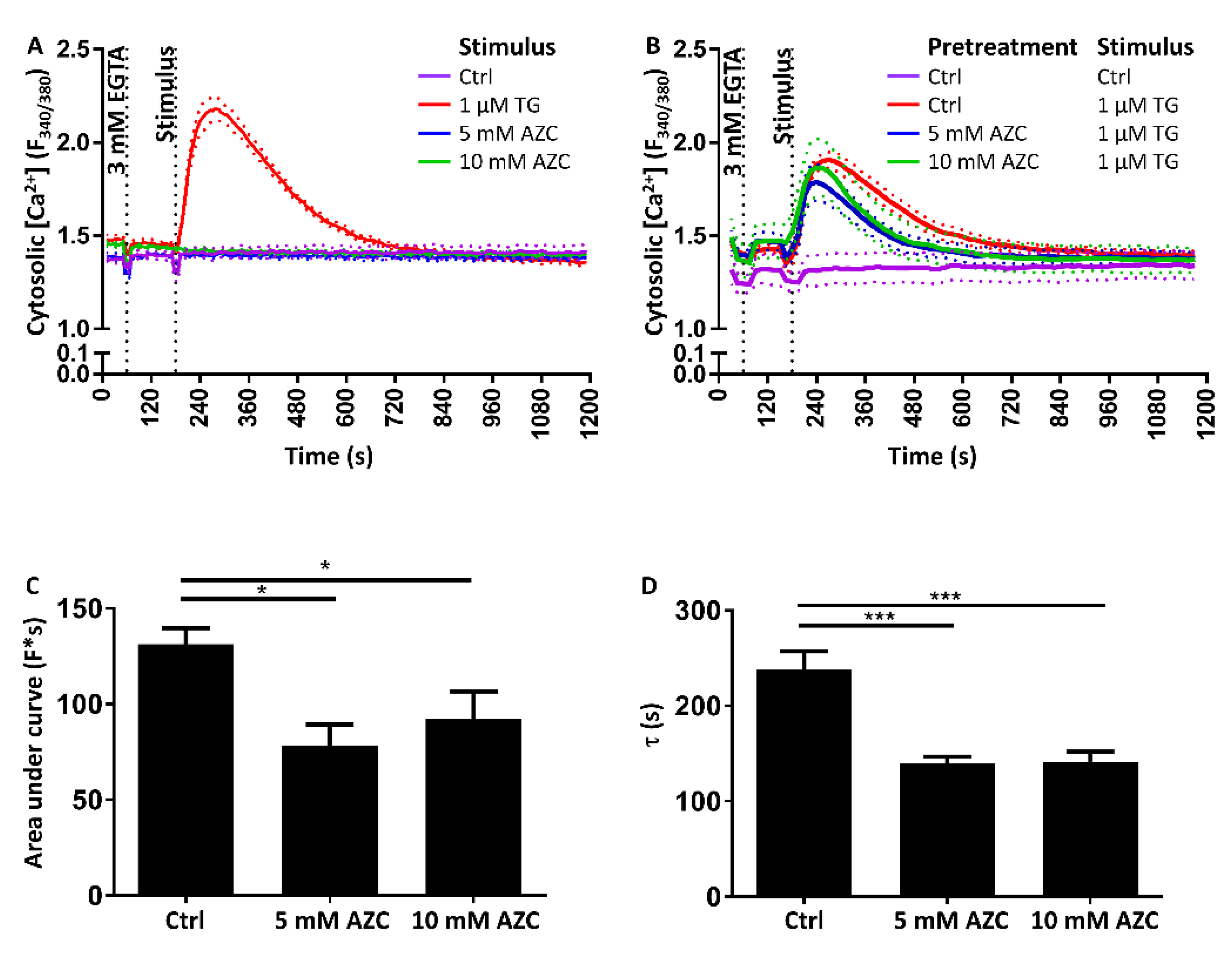
3.8. Pretreatment with AZC Reduces the Cytosolic Amount of Ca2+ after ER Store Release
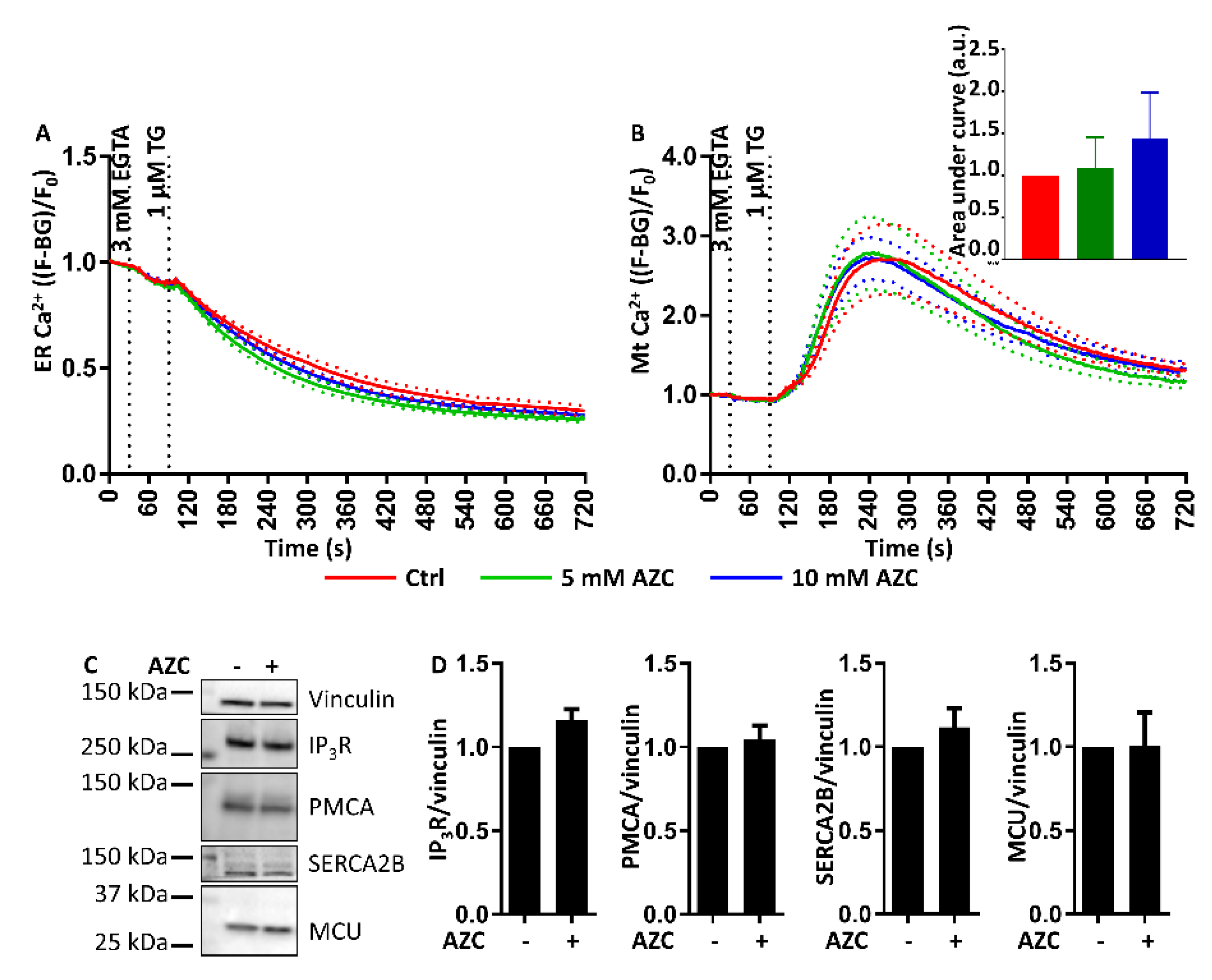
3.9. AZC Does Not Affect the ER Ca2+ Store Content or the ER-Mitochondrial Ca2+ Transfer
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF | Activating transcription factor |
| ATG | Autophagy-related |
| AZC | l-azetidine-2-carboxylic acid |
| Baf A1 | Bafilomycin A1 |
| BAPTA-AM | 1,2-bis(O-aminophenoxy)ethane-N,N,N′,N′-tetraaceticacid tetra(acetoxy-methyl) ester |
| BiP | Binding immunoglobulin protein |
| eIF2α | Eukaryotic translation initiation factor 2α |
| ECL | Enhanced chemiluminescence |
| EDTA | Ethylene diamine tetraacetic acid |
| EGTA | Ethylene glycol tetraacetic acid |
| ER | Endoplasmic reticulum |
| ERp | Endoplasmic reticulum protein |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IP3R | Inositol trisphosphate receptor |
| IRE1 | Inositol-requiring enzyme 1 |
| LC3 | Microtubule-associated protein light chain 3 |
| MCU | Mitochondrial Ca2+ uniporter |
| mTOR | Mechanistic target of rapamycin |
| PARP | Poly adenosine diphosphate ribose polymerase |
| PBS | Phosphate-buffered saline |
| PERK | Protein kinase RNA-like ER kinase |
| PI | Propidium iodide |
| PMCA | Plasma membrane Ca2+ ATPase |
| SDS | Sodium dodecyl sulfate |
| SEM | Standard error of the mean |
| SERCA | Sarco-/endoplasmic reticulum Ca2+ ATPase |
| STS | Staurosporine |
| TBS | Tris-buffered saline |
| TG | Thapsigargin |
| UPR | Unfolded protein response |
| XBP | X-box protein |
References
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 2013, 1833, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Persp. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, B.; Wang, M.; Wang, W.; Liao, P.; Sun, G.B.; Sun, X.B. Calcium homeostasis and endoplasmic reticulum stress are involved in Salvianolic acid B-offered protection against cardiac toxicity of arsenic trioxide. Oncotarget 2017, 8, 97384–97393. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Z.; Wu, Y.; Shi, Q.; Yan, H.; Mei, N.; Tolleson, W.H.; Guo, L. Endoplasmic reticulum stress and store-operated calcium entry contribute to usnic acid-induced toxicity in hepatic cells. Toxicol. Sci. 2015, 146, 116–126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hammadi, M.; Oulidi, A.; Gackiere, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Demaurex, N.; Lew, D.P.; Krause, K.H. Cyclopiazonic acid depletes intracellular Ca2+ stores and activates an influx pathway for divalent cations in HL-60 cells. J. Biol. Chem. 1992, 267, 2318–2324. [Google Scholar] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Armstrong, J.L.; Flockhart, R.; Veal, G.J.; Lovat, P.E.; Redfern, C.P. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J. Biol. Chem. 2010, 285, 6091–6100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.B.; et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. mTOR-controlled autophagy requires intracellular Ca2+ signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef] [PubMed]
- Luyten, T.; Welkenhuyzen, K.; Roest, G.; Kania, E.; Wang, L.; Bittremieux, M.; Yule, D.I.; Parys, J.B.; Bultynck, G. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2+. Biochim. Biophys. Acta 2017, 1864, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgó, J.; Diaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Joseph, S.K. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 2010, 285, 16912–16920. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Grotemeier, A.; Alers, S.; Pfisterer, S.G.; Paasch, F.; Daubrawa, M.; Dieterle, A.; Viollet, B.; Wesselborg, S.; Proikas-Cezanne, T.; Stork, B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010, 22, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Saetre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Mauthe, M.; Codogno, P.; Proikas-Cezanne, T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol. Pharmacol. 2011, 80, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.E.; Dyck, L.; Laussmann, M.A.; Rehm, M. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis. 2014, 5, e1011. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.K.; Hajnoczky, G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 2007, 12, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 2011, 1813, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Persp. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Ibstedt, S.; Tamas, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.P.; Hetz, C.; Atkin, J.D.; et al. NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell 2016, 27, 1712–1727. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.B.; Zhang, X.; Sun, J.; Bennink, J.R.; Yewdell, J.W.; Patterson, C. mTORC1 links protein quality and quantity control by sensing chaperone availability. J. Biol. Chem. 2010, 285, 27385–27395. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lee, A.S. Requirement of the p38 mitogen-activated protein kinase signalling pathway for the induction of the 78 kDa glucose-regulated protein/immunoglobulin heavy-chain binding protein by azetidine stress: Activating transcription factor 6 as a target for stress-induced phosphorylation. Biochem. J. 2002, 366, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Lehrman, M.A. Discordance of UPR signaling by ATF6 and Ire1p-XBP1 with levels of target transcripts. Biochem. Biophys. Res. Commun. 2004, 317, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Szlufcik, K.; Kasri, N.N.; Assefa, Z.; Callewaert, G.; Missiaen, L.; Parys, J.B.; De Smedt, H. Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem. J. 2004, 381, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Szalai, P.; Engedal, N. An image-based assay for high-throughput analysis of cell proliferation and cell death of adherent cells. Bio-protocol 2018, 8, e2835. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Short, D.M.; Heron, I.D.; Birse-Archbold, J.L.; Kerr, L.E.; Sharkey, J.; McCulloch, J. Apoptosis induced by staurosporine alters chaperone and endoplasmic reticulum proteins: Identification by quantitative proteomics. Proteomics 2007, 7, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Simenc, J.; Lipnik-Stangelj, M. Staurosporine induces different cell death forms in cultured rat astrocytes. Radiol. Oncol. 2012, 46, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Persson, J.L. Induction of apoptosis by staurosporine involves the inhibition of expression of the major cell cycle proteins at the G2/M checkpoint accompanied by alterations in ERK and AKT kinase activities. Anticancer Res. 2009, 29, 2893–2898. [Google Scholar] [PubMed]
- Kabir, J.; Lobo, M.; Zachary, I. Staurosporine induces endothelial cell apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis. Biochem. J. 2002, 367, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Kress, B.T.; Lu, Y.; Chandler-Militello, D.; Benraiss, A.; Nedergaard, M. Fluorescent Ca2+ indicators directly inhibit the Na+,K+-ATPase and disrupt cellular functions. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Bach, T.M.; Takagi, H. Properties, metabolisms, and applications of l-proline analogues. Appl. Microbiol. Biotechnol. 2013, 97, 6623–6634. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Viegas, M.S.; Thome, M.P.; Buffon, A.; Wink, M.R.; Lenz, G. Modulation of autophagy by calcium signalosome in human disease. Mol. Pharmacol. 2016, 90, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Allman, S.; Rietdorf, K.; Bultynck, G. Deleterious effects of calcium indicators within cells; an inconvenient truth. Cell Calcium 2018, 73, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Crambert, G.; Hasler, U.; Beggah, A.T.; Yu, C.; Modyanov, N.N.; Horisberger, J.D.; Lelièvre, L.; Geering, K. Transport and pharmacological properties of nine different human Na+, K+-ATPase isozymes. J. Biol. Chem. 2000, 275, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 2017, 65, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Pedersen, P.S.; Treiman, M. The sarco/endoplasmic reticulum calcium-ATPase 2B is an endoplasmic reticulum stress-inducible protein. J. Biol. Chem. 2000, 275, 22363–22372. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roest, G.; Hesemans, E.; Welkenhuyzen, K.; Luyten, T.; Engedal, N.; Bultynck, G.; Parys, J.B. The ER Stress Inducer l-Azetidine-2-Carboxylic Acid Elevates the Levels of Phospho-eIF2α and of LC3-II in a Ca2+-Dependent Manner. Cells 2018, 7, 239. https://doi.org/10.3390/cells7120239
Roest G, Hesemans E, Welkenhuyzen K, Luyten T, Engedal N, Bultynck G, Parys JB. The ER Stress Inducer l-Azetidine-2-Carboxylic Acid Elevates the Levels of Phospho-eIF2α and of LC3-II in a Ca2+-Dependent Manner. Cells. 2018; 7(12):239. https://doi.org/10.3390/cells7120239
Chicago/Turabian StyleRoest, Gemma, Evelien Hesemans, Kirsten Welkenhuyzen, Tomas Luyten, Nikolai Engedal, Geert Bultynck, and Jan B. Parys. 2018. "The ER Stress Inducer l-Azetidine-2-Carboxylic Acid Elevates the Levels of Phospho-eIF2α and of LC3-II in a Ca2+-Dependent Manner" Cells 7, no. 12: 239. https://doi.org/10.3390/cells7120239
APA StyleRoest, G., Hesemans, E., Welkenhuyzen, K., Luyten, T., Engedal, N., Bultynck, G., & Parys, J. B. (2018). The ER Stress Inducer l-Azetidine-2-Carboxylic Acid Elevates the Levels of Phospho-eIF2α and of LC3-II in a Ca2+-Dependent Manner. Cells, 7(12), 239. https://doi.org/10.3390/cells7120239