Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells

 ,
,  , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals
2.3. Isolation of Goblet Cells
2.4. RNA Extraction and cDNA Conversion
2.5. qRTPCR
2.6. The Assessment of Cytokines by Bioplex
2.7. Toxicity Assay Lactate Dehydrogenase (LDH)
2.8. Western Blot
2.9. Apoptosis Assays
2.9.1. Caspase-3 Assay
2.9.2. Annexin V Assay
2.10. Proliferation Assay
2.11. Statistical Analysis
3. Results
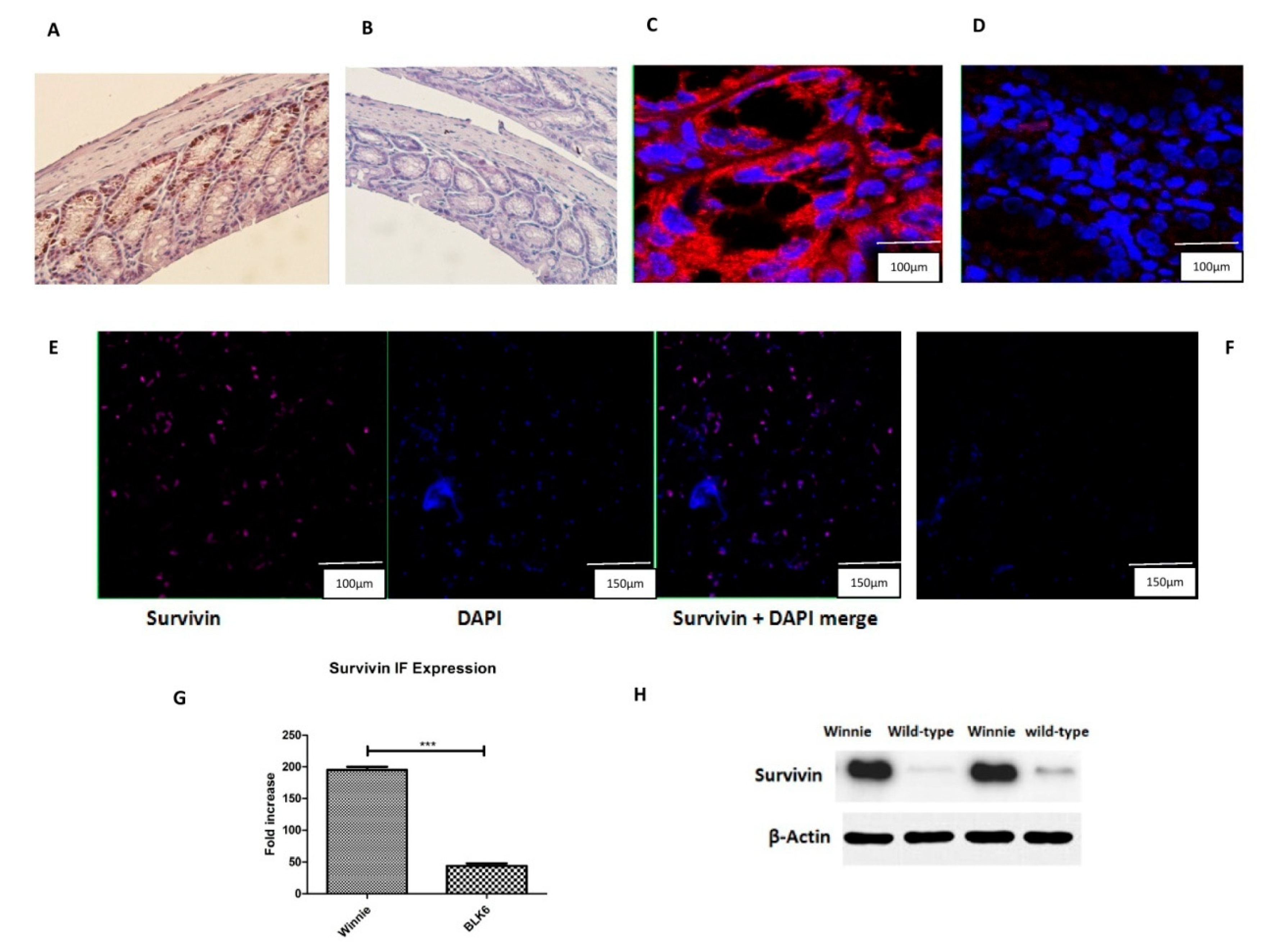
3.1. Increased Survivin Expression in the Colon of Winnie
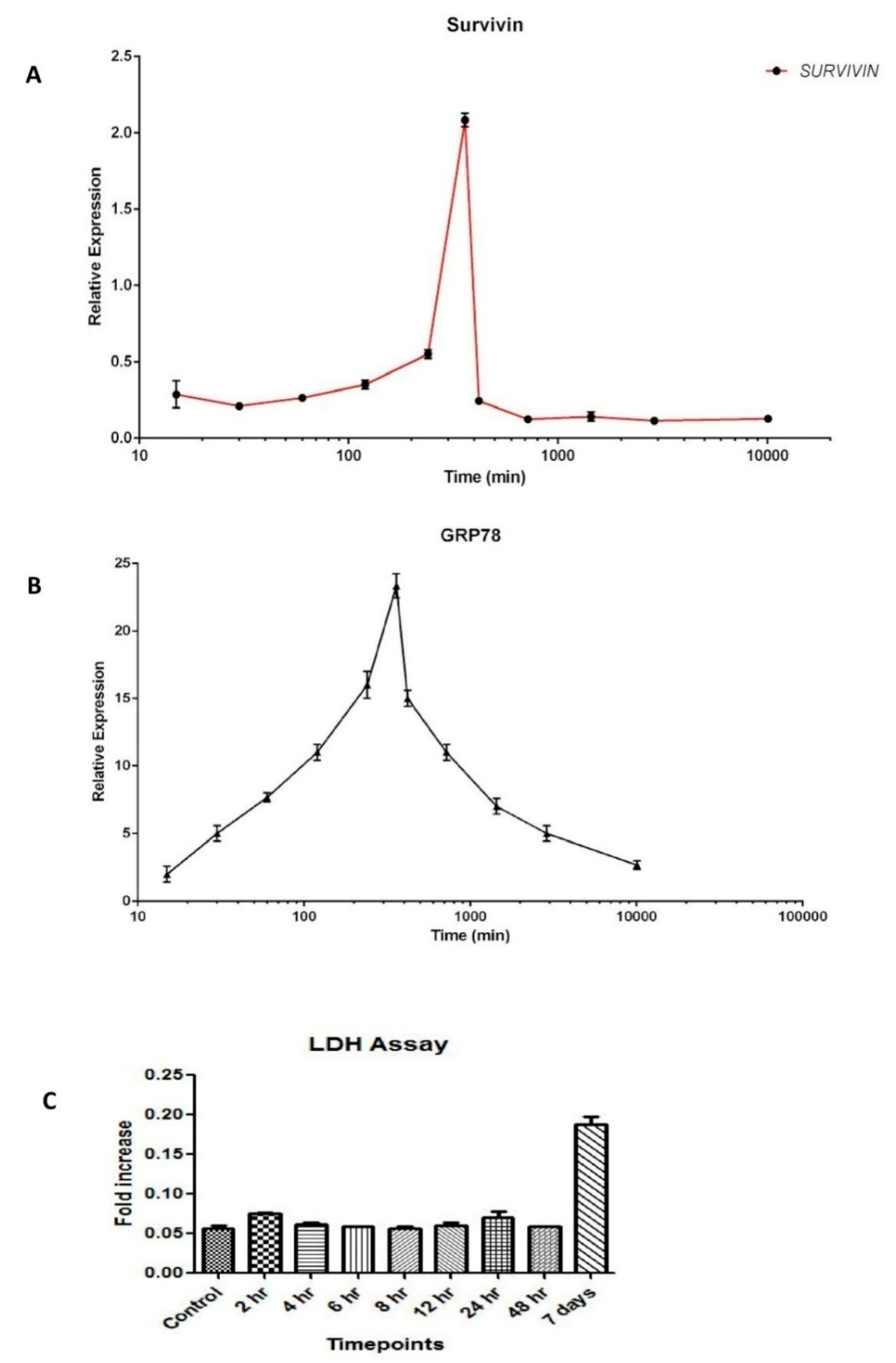
3.2. Dynamics of Survivin in Line with Endoplasmic Reticular Stress (ERS)
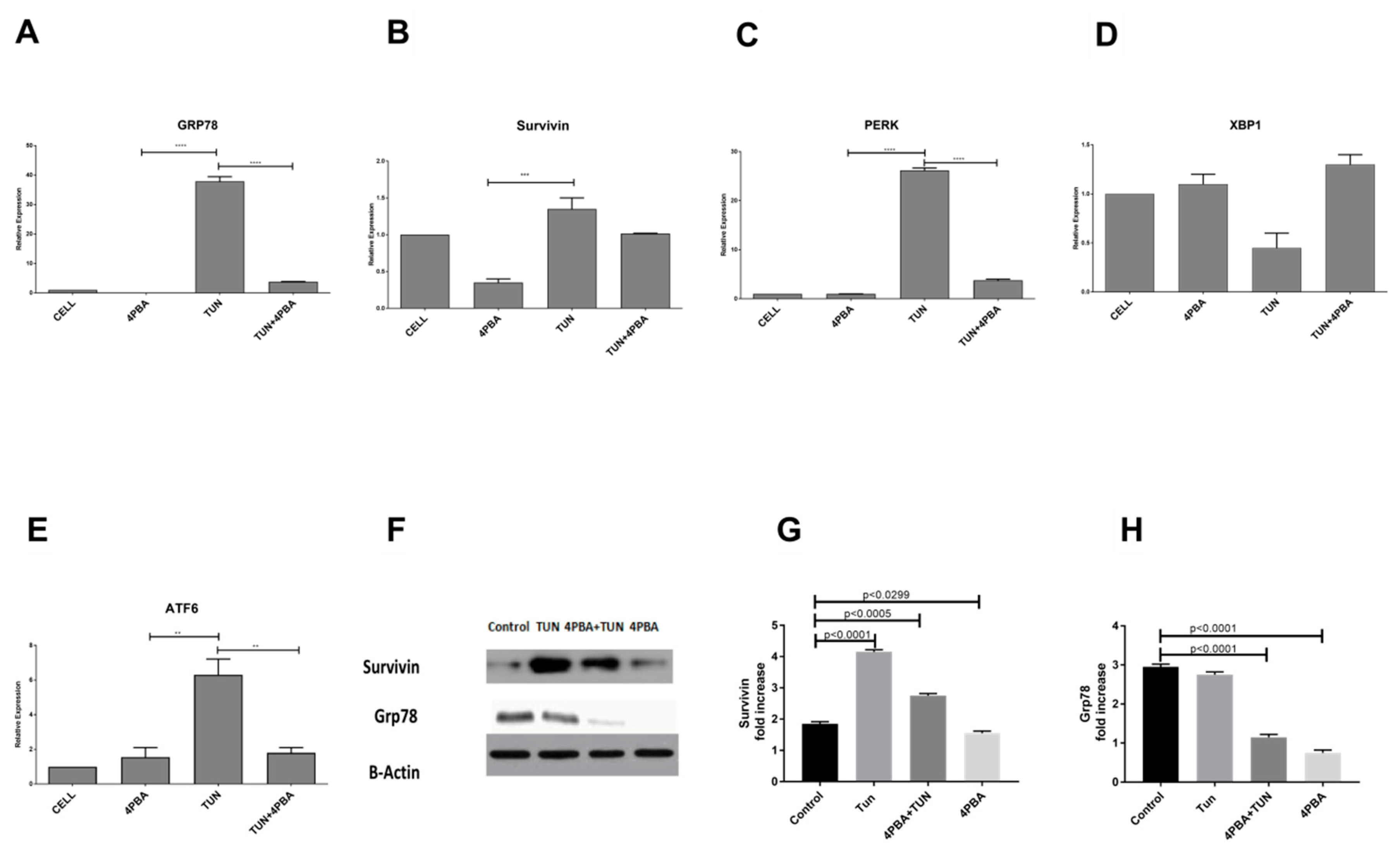
3.3. Inhibition of Endoplasmic Reticular (ER) Stress Is Correlated to Survivin Expression in the LS174T Cell Line
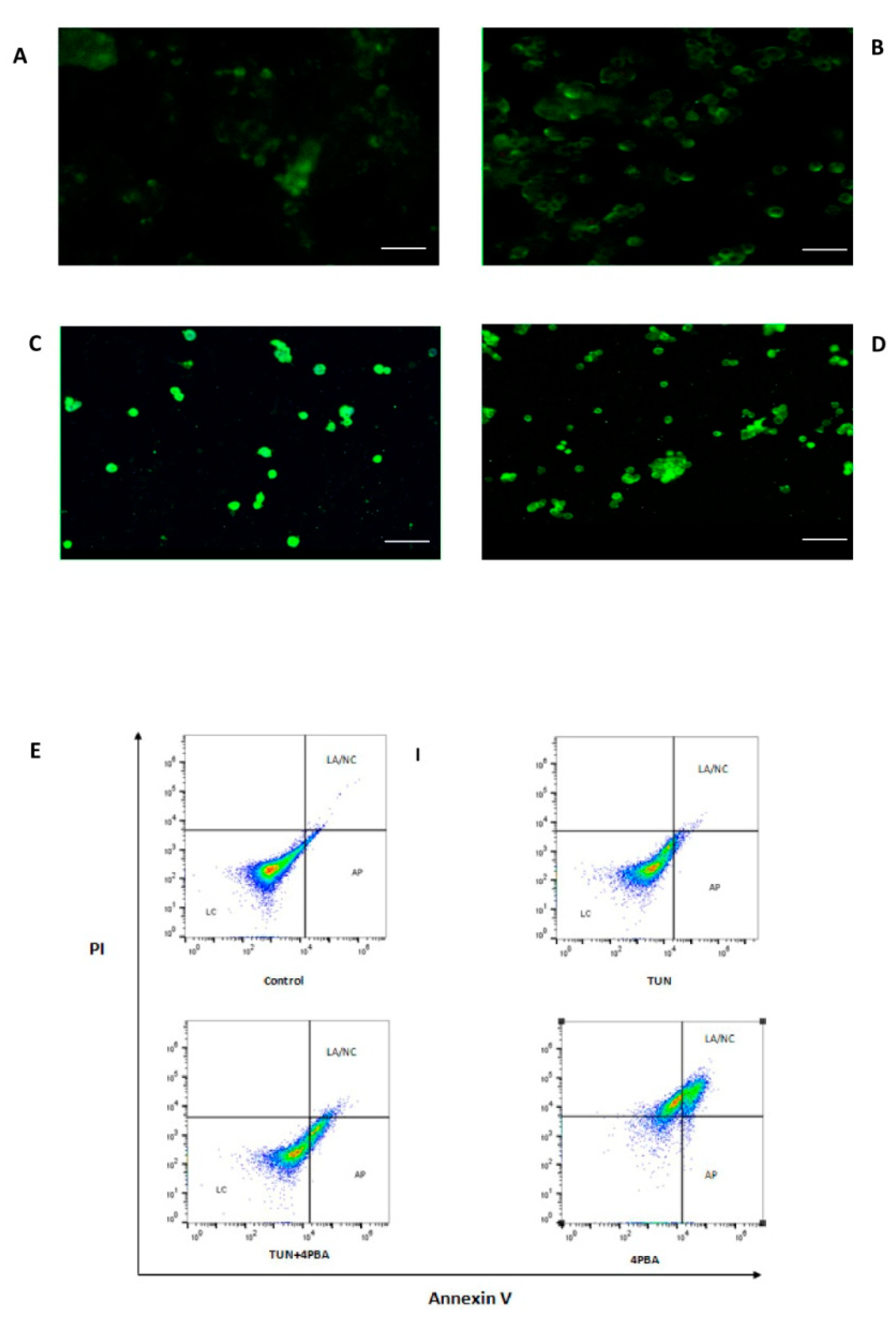
3.4. Inhibition of ERS Escalates Apoptosis in Colon Cancer Cells
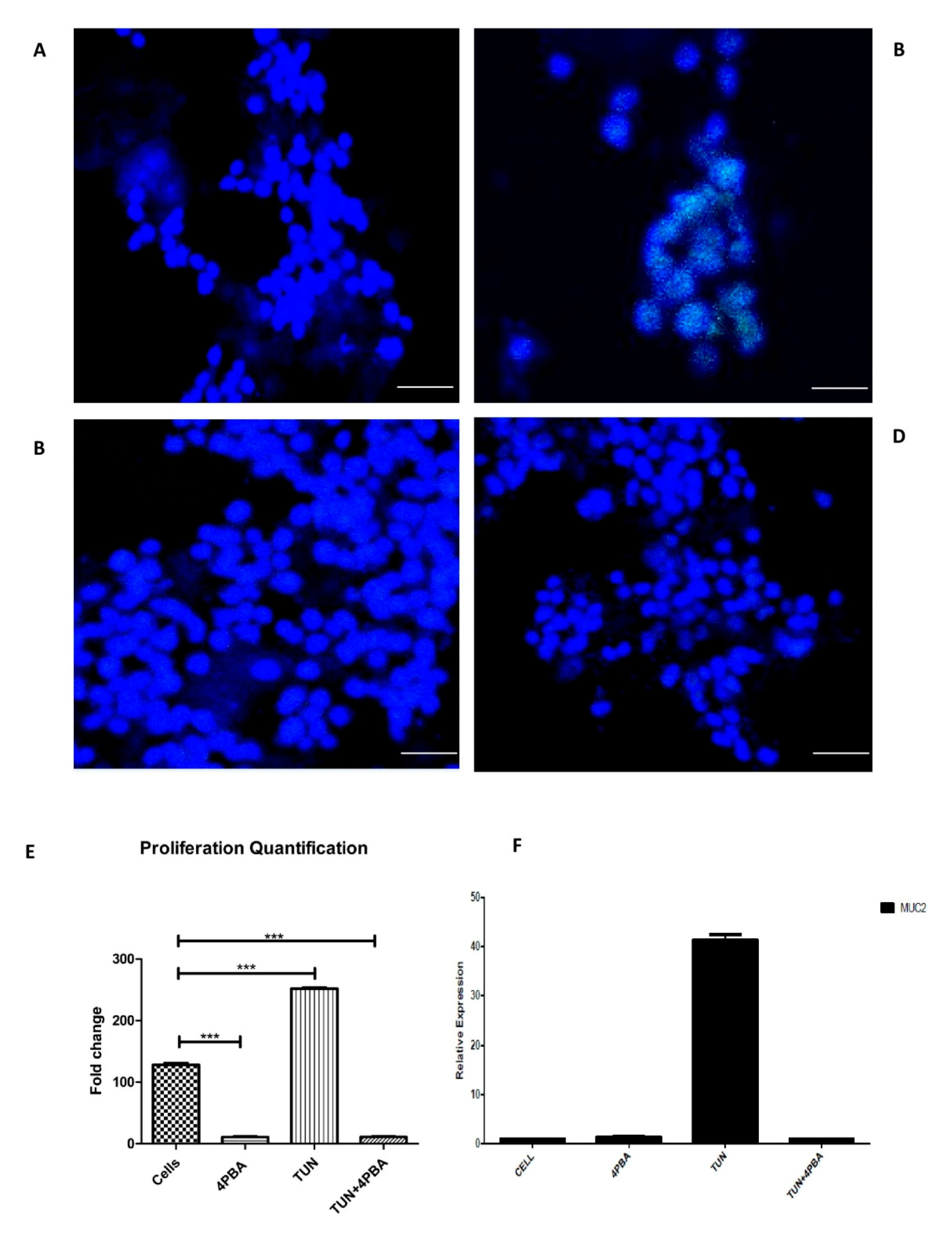
3.5. ERS Exhibits Direct Relationship to Proliferation in the LS174T Cell Line
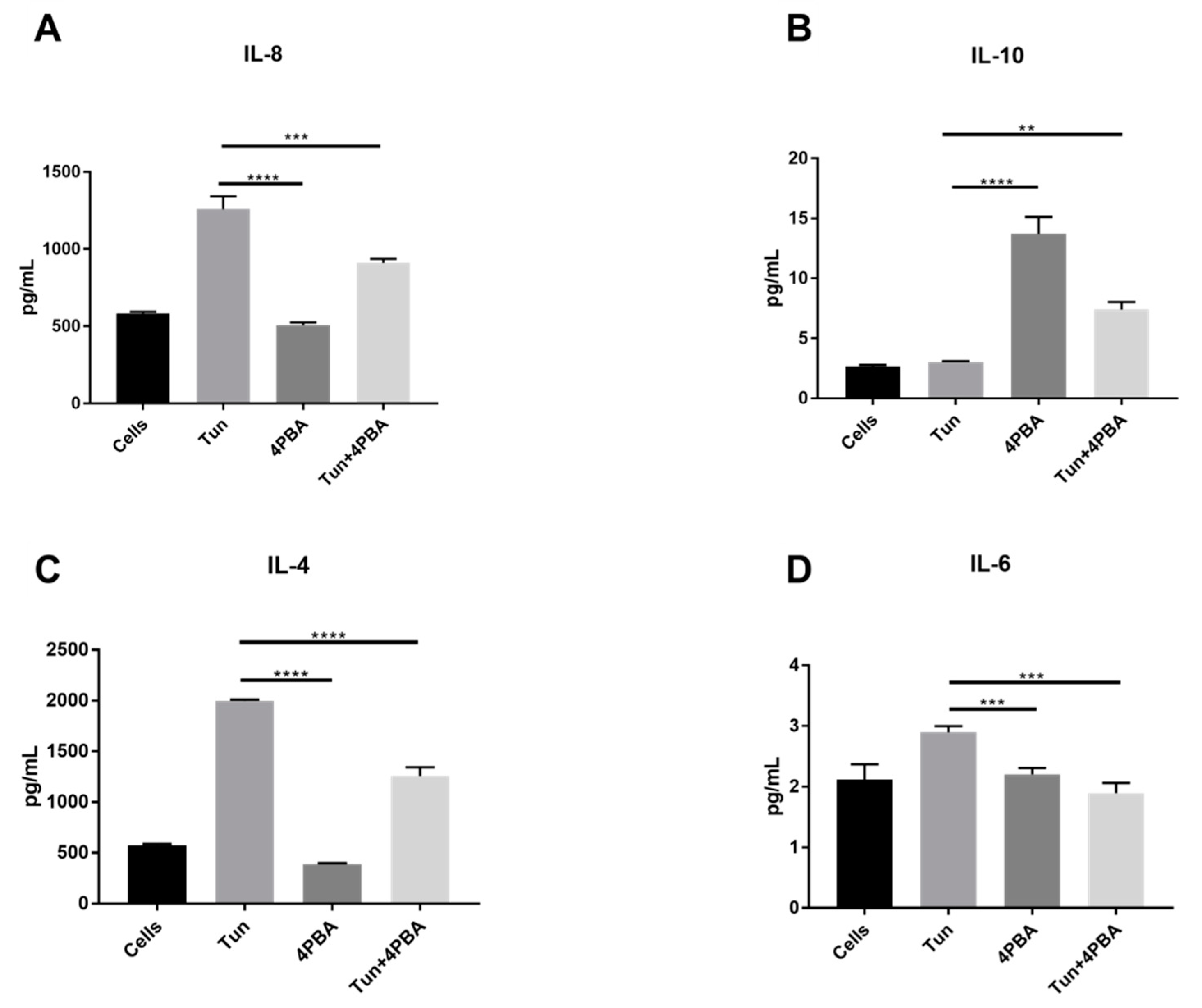
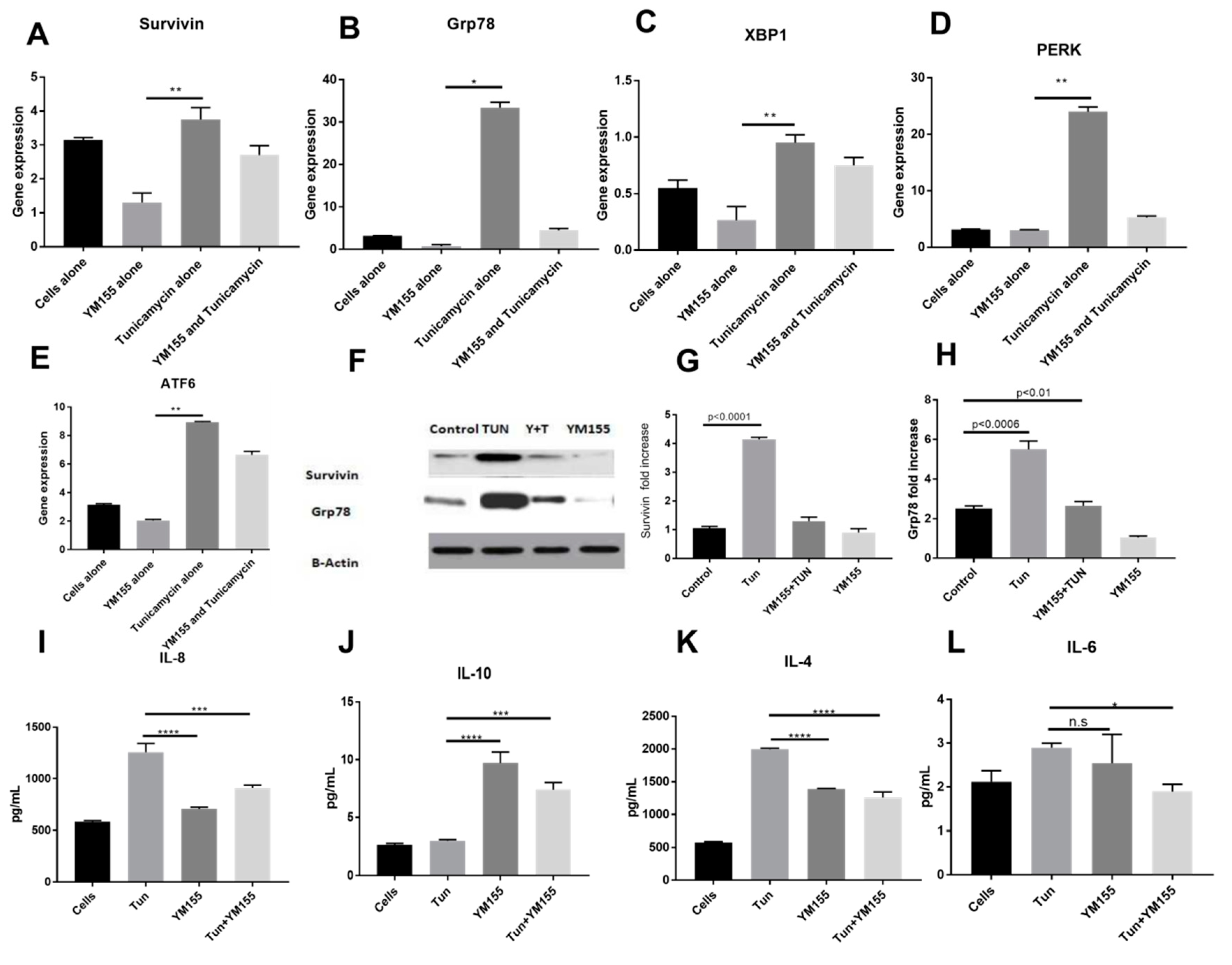
3.6. YM155, a Survivin Inhibitor, Subsides ERS and ERS Mediated Inflammation
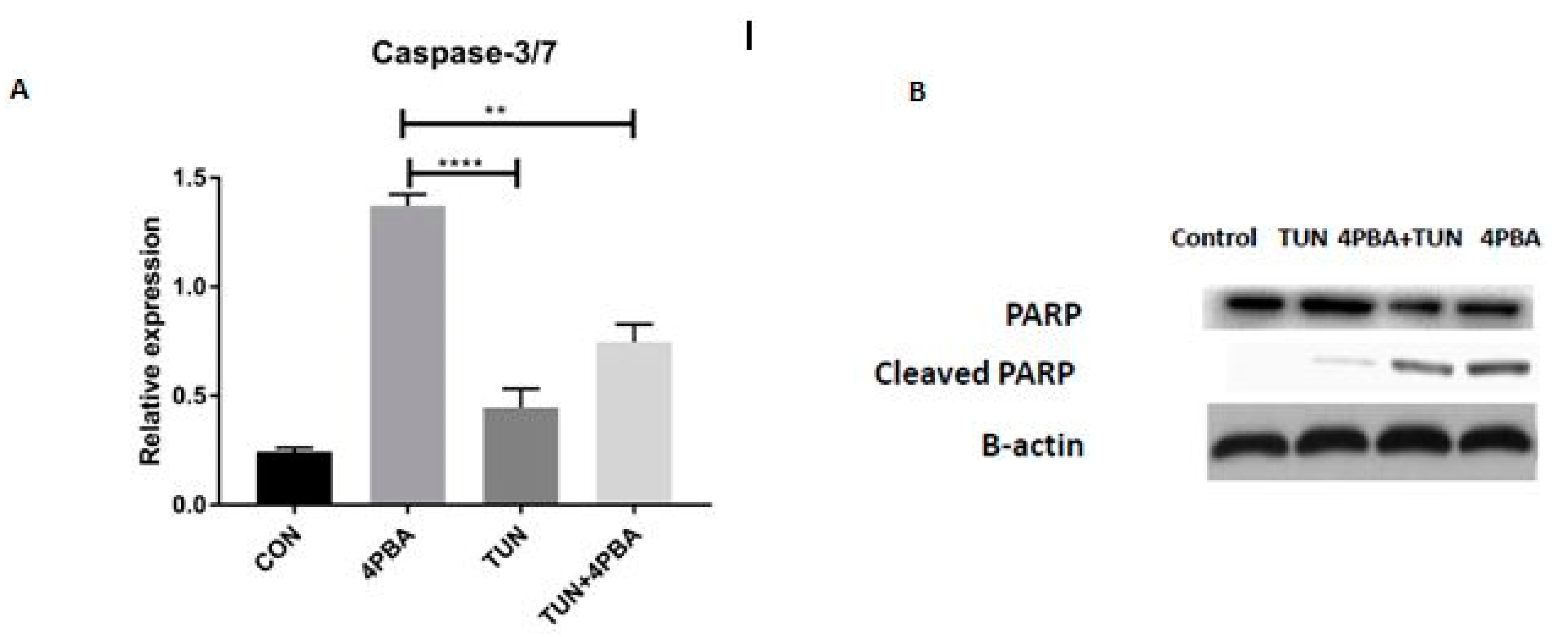
3.7. YM155 Initiates Cell Death in LS174T Cells via Caspase3/7
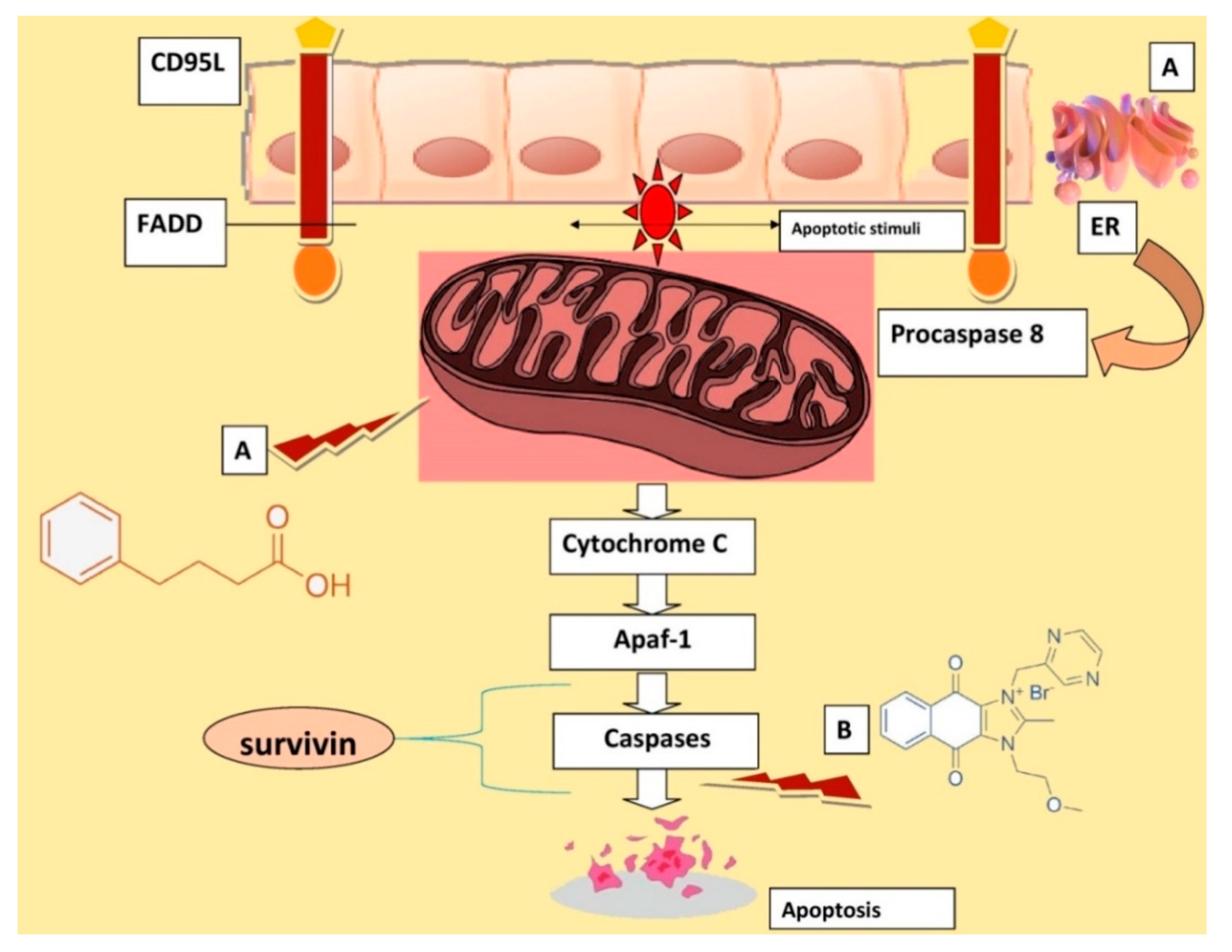
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2009, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta 2014, 1843, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- Mc Guckin, M.A.; Eri, R.D.; Das, I.; Lourie, R.; Florin, T.H. ER stress and the unfolded protein response in intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Conti, S.; Facchiano, A.; Filippini, A.; Ziparo, E. Cancer microenvironment and endoplasmic reticulum stress response. Mediators Inflamm. 2015, 2015, 417281. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Meier, P. Inhibitor of Apoptosis (IAP) Proteins–Modulators of Cell Death and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef] [PubMed]
- Altznauer, F.; Martinelli, S.; Yousefi, S.; Thürig, C.; Schmid, I.; Conway, E.M.; Schöni, M.H.; Vogt, P.; Mueller, C.; Fey, M.F.; et al. Inflammation-associated cell cycle-independent block of apoptosis by survivin in terminally differentiated neutrophils. J. Exp. Med. 2004, 199, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Wittkopf, N.; Günther, C.; Leppkes, M.; Okada, H.; Watson, A.J.; Podstawa, E.; Backert, I.; Amann, K.; Neurath, M.F.; et al. Loss of Survivin in intestinal epithelial progenitor cells leads to mitotic catastrophe and breakdown of gut immune homeostasis. Cell. Rep. 2016, 14, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Kudo, Y.; Tang, B.; Liu, T.; Jin, S.; Liu, J.; Zuo, X.; Mi, S.; Shao, W.; Ma, X.; et al. PARP6 acts as a tumor suppressor via downregulating Survivin expression in colorectal cancer. Oncotarget 2016, 7, 18812–18824. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Li, K.T.; Fayyaz, S.; Chang, Y.T.; Ismail, M.; Liaw, C.C.; Yuan, S.S.; Tang, J.Y.; Chang, H.W. Anticancer drugs for the modulation of endoplasmic reticulum stress and oxidative stress. Tumour Biol. 2015, 36, 5743–5752. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS. Med. 2008, 5, e54. [Google Scholar] [CrossRef] [PubMed]
- Tawiah, A.; Cornick, S.; Moreau, F. High Muc2 mucin expression and misfolding induce cellular stress, reactive oxygen production, and apoptosis in goblet cells. Am. J. Pathol. 2018, 188, 1354–1373. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.J.; Cho, M.Y. Cyclooxygenase-2 expression and cell proliferation are increased in Muc2-positive area of columnar-lined esophagus. Pathol. Int. 2005, 55, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Farma, J.M.; Coppola, D.; Hakam, A.; Fulp, W.J.; Chen, D.T.; Siegel, E.M.; Yeatman, T.J.; Shibata, D. Expression of the antiapoptotic protein survivin in colon cancer. Clin. Colorectal. Cancer 2011, 10, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Liu, Q.; Fu, J.X.; Kang, S.Y. Expression of survivin and its significance in colorectal cancer. World J. Gastroenterol. 2004, 10, 2886–2889. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, J.; Anishetty, S. Molecular dynamics simulations of inhibitor of apoptosis proteins and identification of potential small molecule inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jeon, J.S.; Kim, H.R.; Park, S.Y.; Chae, H.J.; Lee, Y.C. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-κB/HIF-1α signaling pathway. Sci. Rep. 2013, 3, 1142. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef] [PubMed]
- Koller, F.L.; Hwang, D.G.; Dozier, E.A.; Fingleton, B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis 2010, 31, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Perez Alea, M.; Scopelliti, A.; Medema, J.P.; Stassi, G. IL4-mediated drug resistance in colon cancer stem cells. Cell. Cycle 2008, 7, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Francipane, M.G.; Alea, M.P.; Lombardo, Y.; Todaro, M.; Medema, J.P.; Stassi, G. Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res. 2008, 68, 4022–4025. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.B.; Iovino, F.; Lombardo, Y.; Eterno, V.; Höger, T.; Dieli, F.; Stassi, G.; Todaro, M. Survivin is regulated by interleukin-4 in colon cancer stem cells. J. Cell. Physiol. 2010, 225, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. IL10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Purushottam, L.; Lavakumar, K.; Barath, S.; Keith, K. PD-1 and IL-10: Partners in crime against anti-tumor immunity in ovarian cancer. J. Immunol. 2015, 194, 13. [Google Scholar]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Gerl, R.; Vaux, D.L. Apoptosis in the development and treatment of cancer. Carcinogenesis 2005, 26, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L.; Ni, M.; Schontal, A.H.; Chen, T.C.; Hofman, F.M.; Lee, A.S. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 2008, 6, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Badmus, J.A.; Ekpo, O.E.; Hussein, A.A.; Meyer, M.; Hiss, D.C. Antiproliferative and apoptosis induction potential of the methanolic leaf extract of Holarrhena floribunda (G. Don). Evid. Based Complement. Altern. Med. 2015, 2015, 756482. [Google Scholar] [CrossRef] [PubMed]
- Kwan, Y.P.; Saito, T.; Ibrahim, D.; Al-Hassan, F.M.; Ein Oon, C.; Chen, J.; Jothy, S.L.; Kanwar, J.L.; Sasidharan, S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by Euphorbia hirta in MCF-7 breast cancer cells. Pharm. Biol. 2016, 54, 1223–1236. [Google Scholar] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Looi, C.Y.; Arya, A.; Cheah, F.K.; Muharram, B.; Leong, K.H.; Mohamad, K.; Wong, W.F.; Rai, N.; Mustafa, M.R. Induction of apoptosis in human breast cancer cells via caspase pathway by vernodalin isolated from Centratherumanthelminticum (L.) seeds. PLoS ONE 2013, 8, e56643. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–422940. [Google Scholar] [CrossRef] [PubMed]
- Brauns, S.C.; Dealtry, G.; Milne, P.; Naudé, R.; Van de Venter, M. Caspase-3 activation and induction of PARP cleavage by cyclic dipeptide cyclo(Phe-Pro) in HT-29 cells. Anticancer Res. 2005, 25, 4197–4202. [Google Scholar] [PubMed]
- Pink, J.J.; Wuerzberger-Davies, S.; Tagliarino, C.; Planchon, S.M.; Yang, X.; Froelich, C.J.; Boothman, D.A. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during ‚-lapachone-mediated apoptosis. Exp. Cell. Res. 2000, 255, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Holst, S.; Belo, A.L.; Giovannetti, E.; van Die, I.; Wuhrer, M. Profiling of different pancreatic cancer cells used as models for metastatic behaviour shows large variation in their N-glycosylation. Sci. Rep. 2017, 7, 16623. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.S.; Liu, C.C.; Lin, J.H.; Hsu, T.W.; Hsu, J.W.; Su, K.; Hung, S.C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Byrd, J.C.; Mazurek, N.; Liu, K.; Koo, J.S.; Bresalier, R.S. Galectin-3 modulates Muc2 mucin expression in human colon cancer cells at the level of transcription via AP-1 activation. Gastroenterology 2005, 129, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, I.; Ponnusamy, M.P.; Macha, M.A.; Haridas, D.; Majhi, P.D.; Kaur, S.; Jain, M.; Batra, S.K.; Ganti, A.K. Mucins in lung cancer: Diagnostic, prognostic, and therapeutic implications. J. Thorac. Oncol. 2015, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Zhang, M.Y.; Wu, X.; Sun, X.J.; Xu, T.; He, Q.Z.; Di, W. High Muc2 expression in ovarian cancer is inversely associated with the M1/M2 ratio of tumor-associated macrophages and patient survival time. PLoS ONE 2013, 8, e79769. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Nakata, M.; Kaneko, N.; Fushiki, H.; Kita, A.; Nakahara, T.; Koutoku, H.; Sasamata, M. YM155, a selective survivin suppressant, inhibits tumor spread and prolongs survival in a spontaneous metastatic model of human triple negative breast cancer. Int. J. Oncol. 2011, 39, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.D.; Samlowski, W.; Ward, J.; Catlett, J.; Cranmer, L.; Kirkwood, J.; Lawson, D.; Whitman, E.; Gonzalez, R. A multi-center phase II evaluation of the small molecule Survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Investig. New Drugs 2011, 29, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.; Hose, D.; Seckinger, A.; Weiz, L.; Meiβner, T.; Rème, T.; Breitkreutz, I.; Podar, K.; Ho, A.D.; Goldschmidt, H.; et al. Preclinical efficacy of sepantronium bromide (YM155) in multiple myeloma is conferred by down regulation of Mcl-1. Oncotarget 2014, 5, 10237–10250. [Google Scholar] [CrossRef] [PubMed]
- Oberoi-khanuja, T.K.; Murali, A.; Rajalingam, K. IAPs on the move: Role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013, 4, e784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, W.; Wang, Y.F.; Liu, B.; Zhang, W.F.; Zhao, Y.F.; Kulkarni, A.B.; Sun, Z.J. Dual induction of apoptotic and autophagic cell death by targeting survivin in head neck squamous cell carcinoma. Cell Death Dis. 2016, 6, e1771. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gundamaraju, R.; Vemuri, R.; Chong, W.C.; Myers, S.; Norouzi, S.; Shastri, M.D.; Eri, R. Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells. Cells 2018, 7, 171. https://doi.org/10.3390/cells7100171
Gundamaraju R, Vemuri R, Chong WC, Myers S, Norouzi S, Shastri MD, Eri R. Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells. Cells. 2018; 7(10):171. https://doi.org/10.3390/cells7100171
Chicago/Turabian StyleGundamaraju, Rohit, Ravichandra Vemuri, Wai Chin Chong, Stephen Myers, Shaghayegh Norouzi, Madhur D. Shastri, and Rajaraman Eri. 2018. "Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells" Cells 7, no. 10: 171. https://doi.org/10.3390/cells7100171
APA StyleGundamaraju, R., Vemuri, R., Chong, W. C., Myers, S., Norouzi, S., Shastri, M. D., & Eri, R. (2018). Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells. Cells, 7(10), 171. https://doi.org/10.3390/cells7100171