Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. PBMC Donors
2.2. Antigens
2.3. Human Cytokine ELISPOT Assays
2.4. Statistical Analysis
3. Results
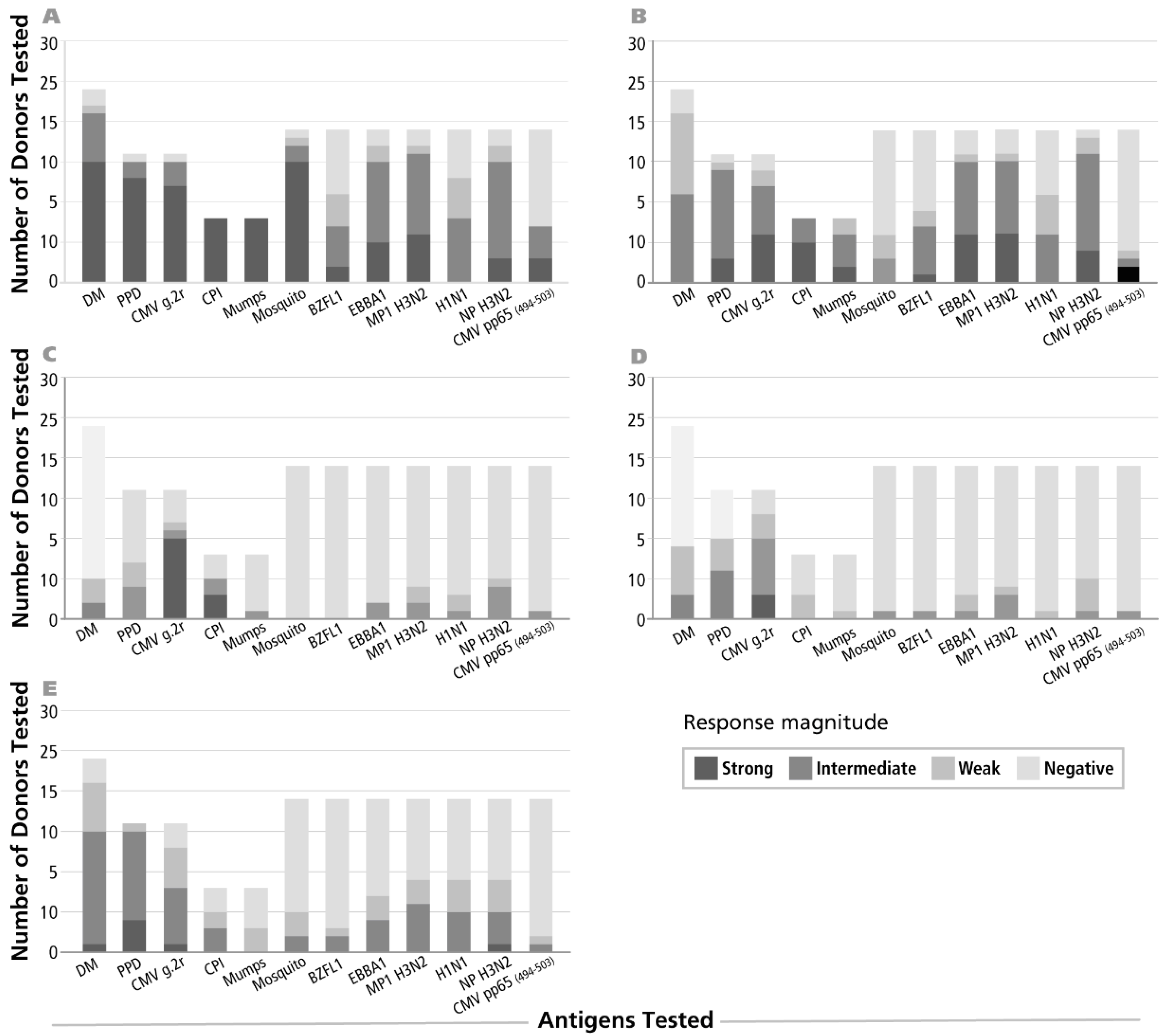
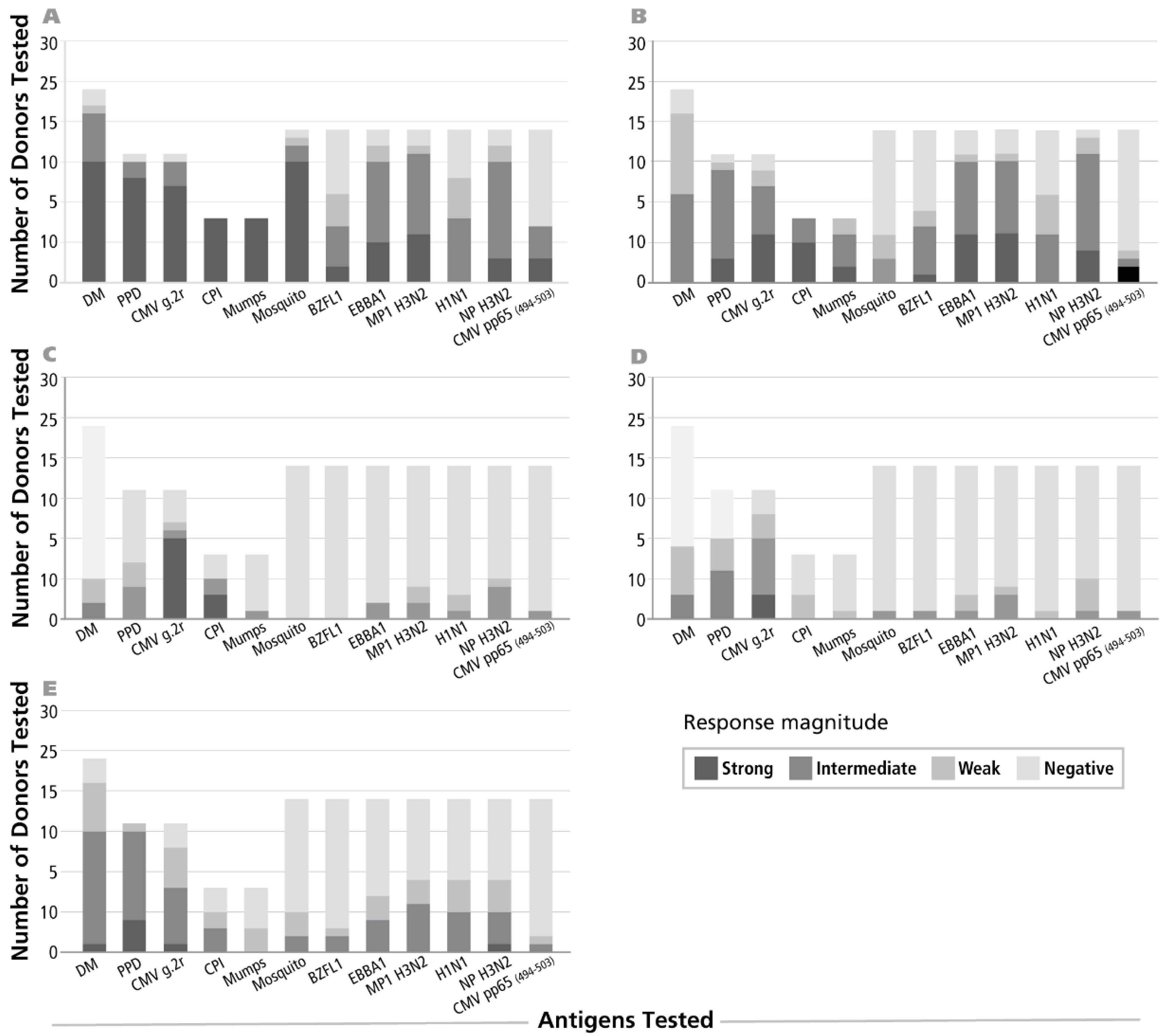
3.1. Identifying Donors Exhibiting IFN-γ, IL-2, IL-4, IL-5 and IL-17 Recall Responses to Select Antigens
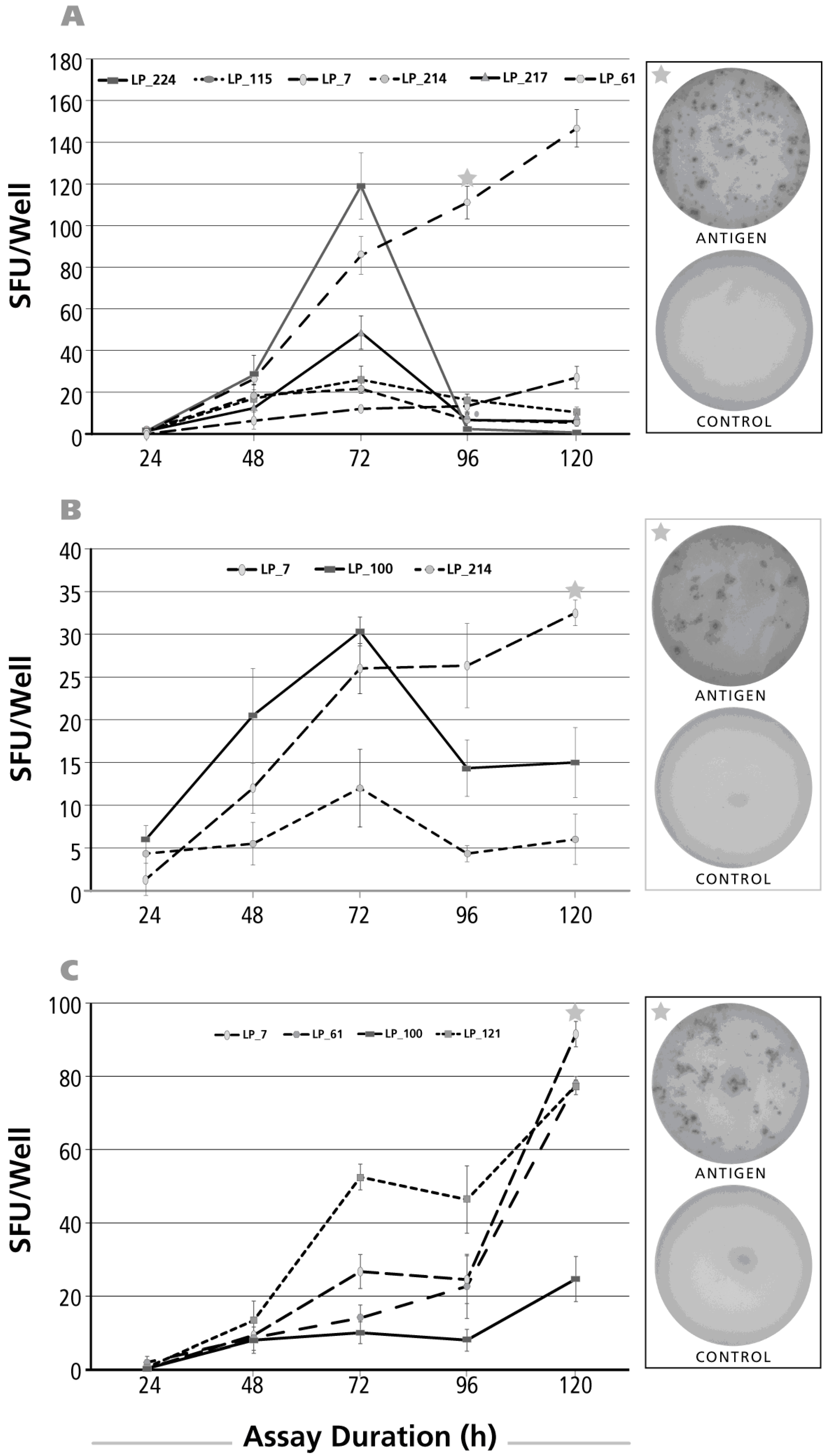
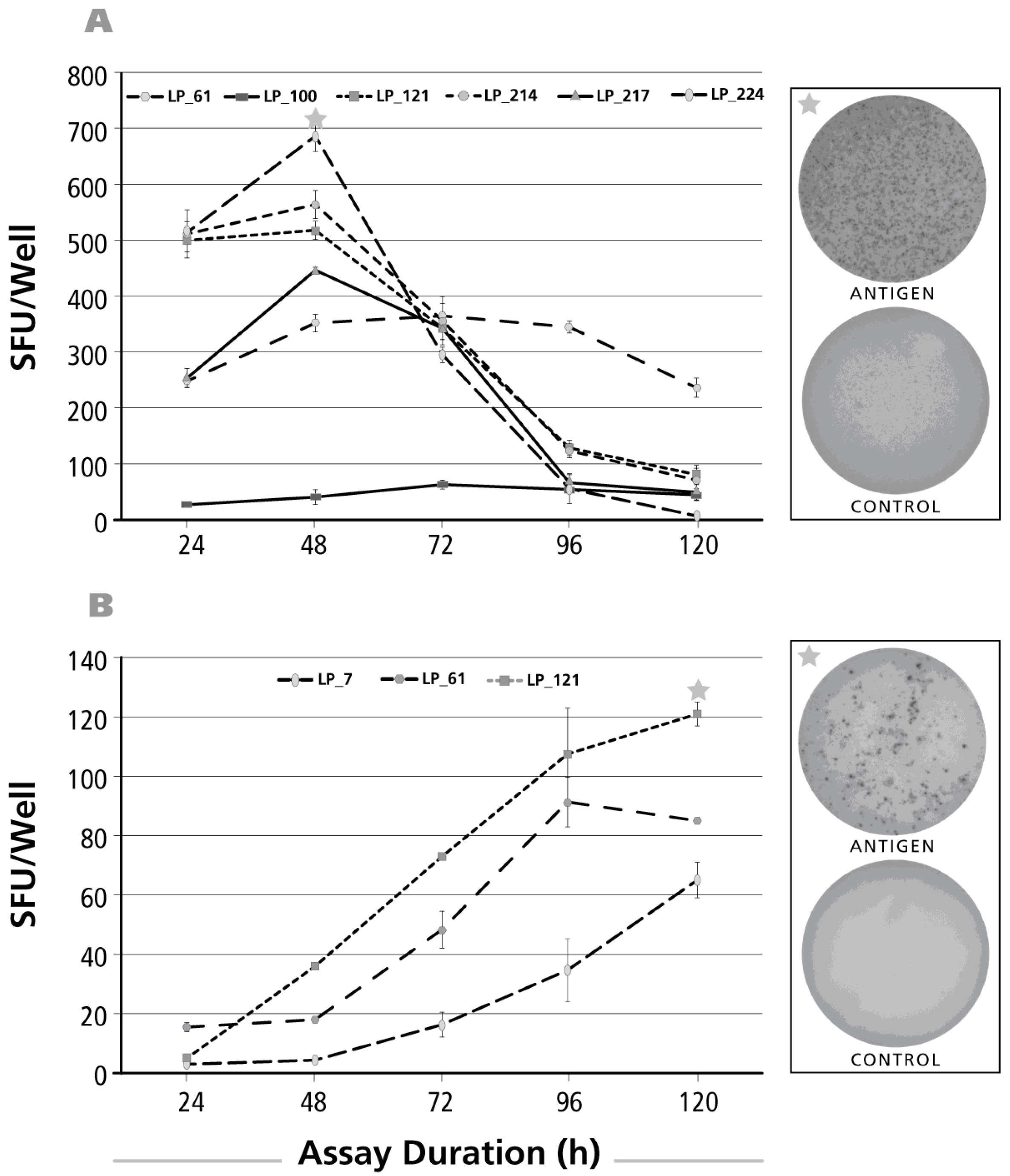
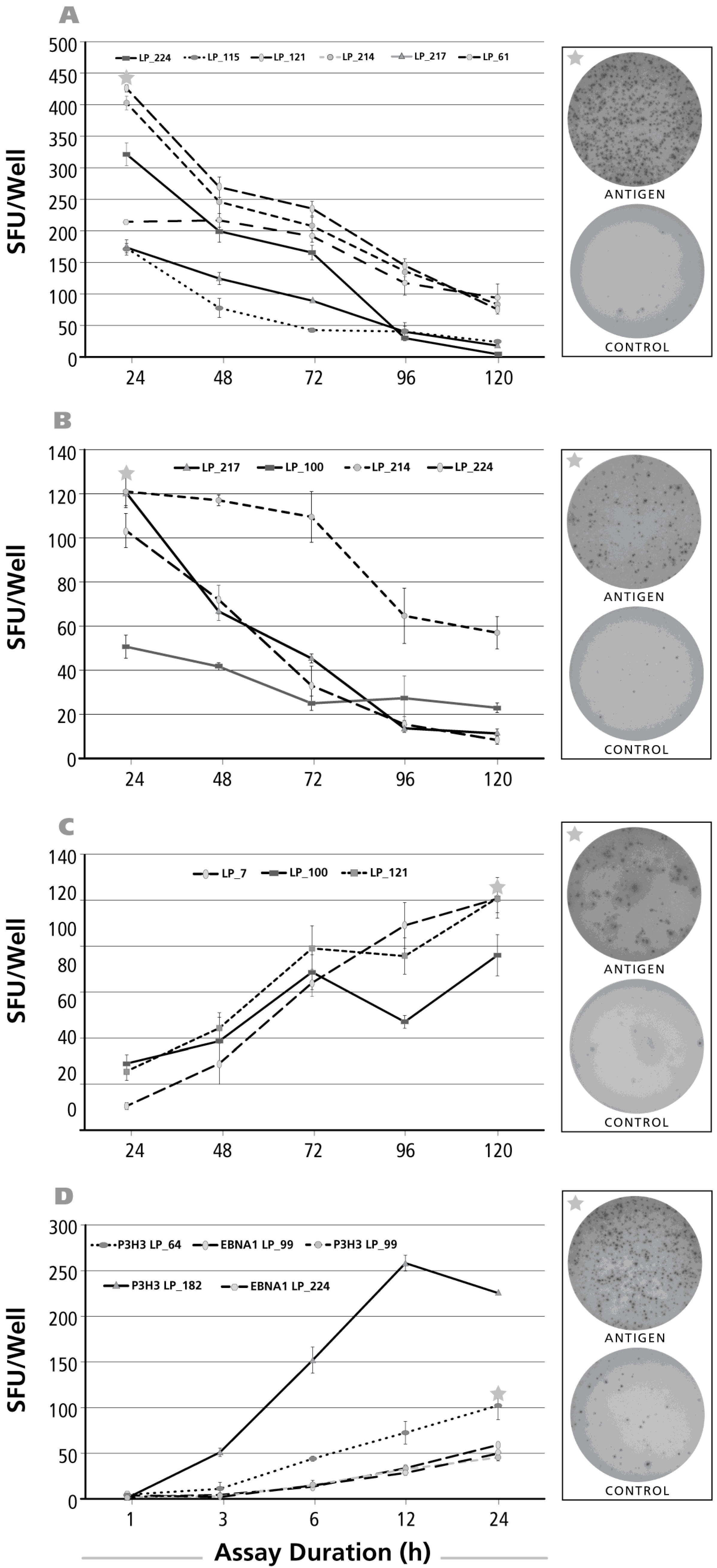
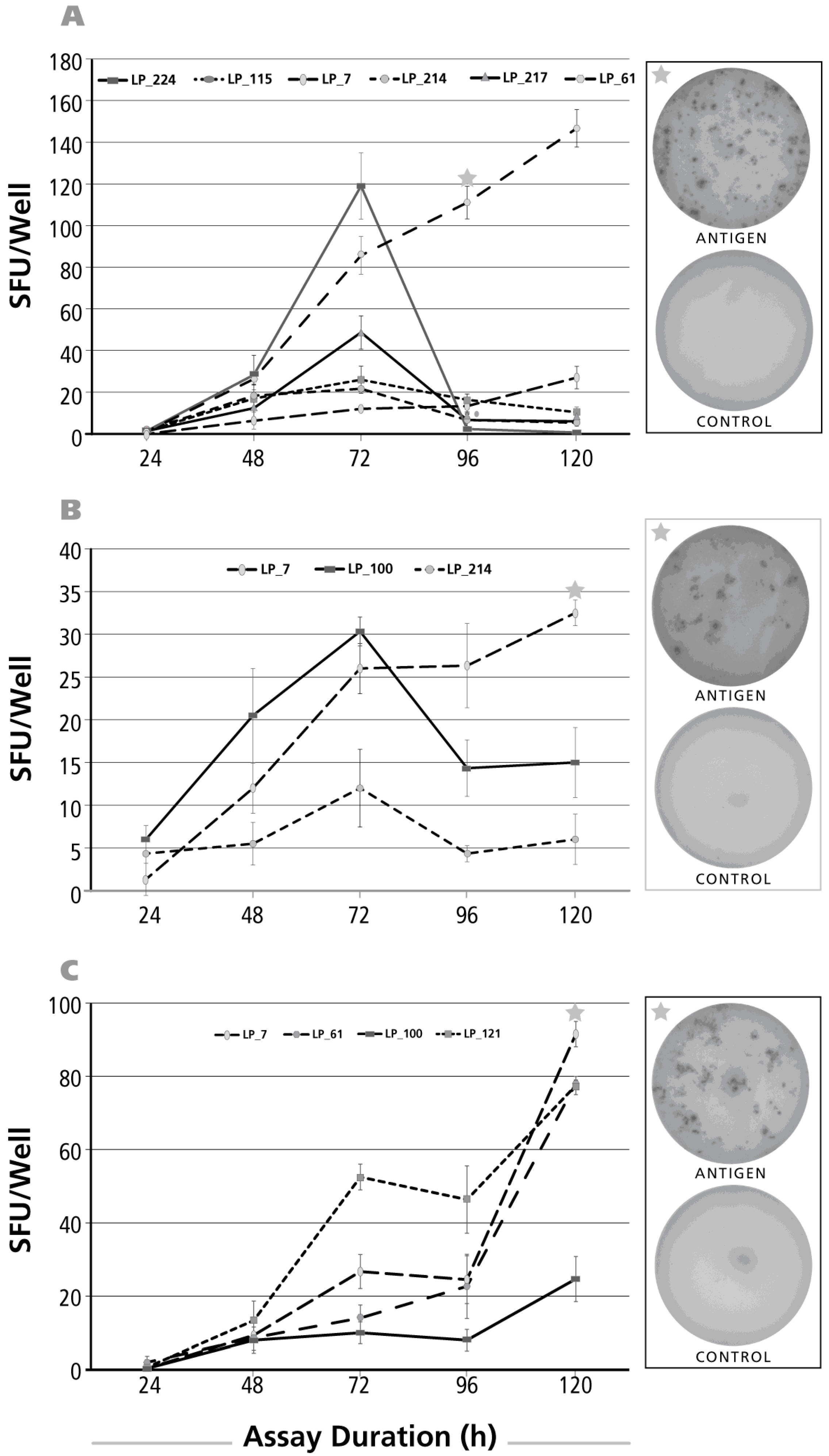
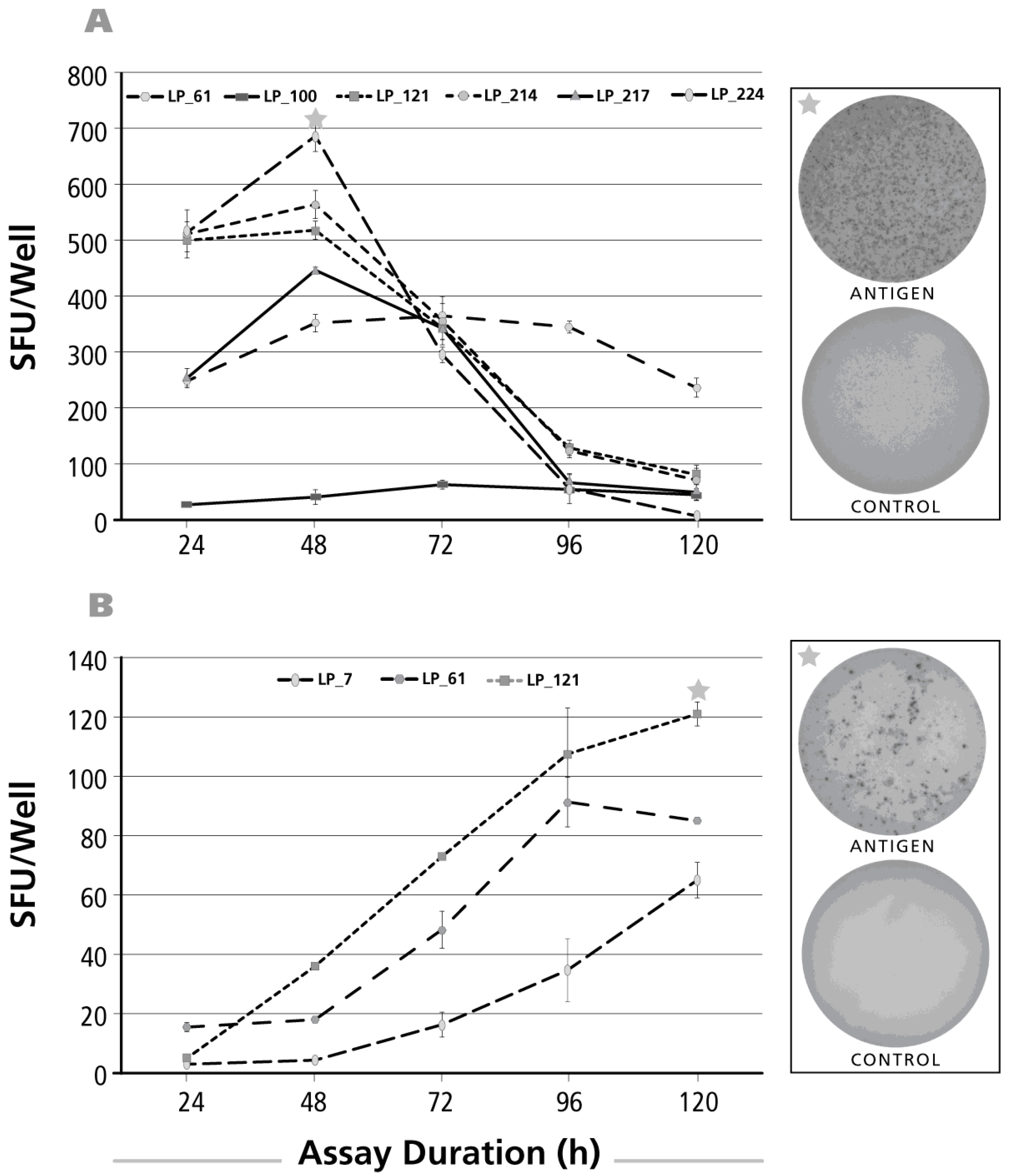
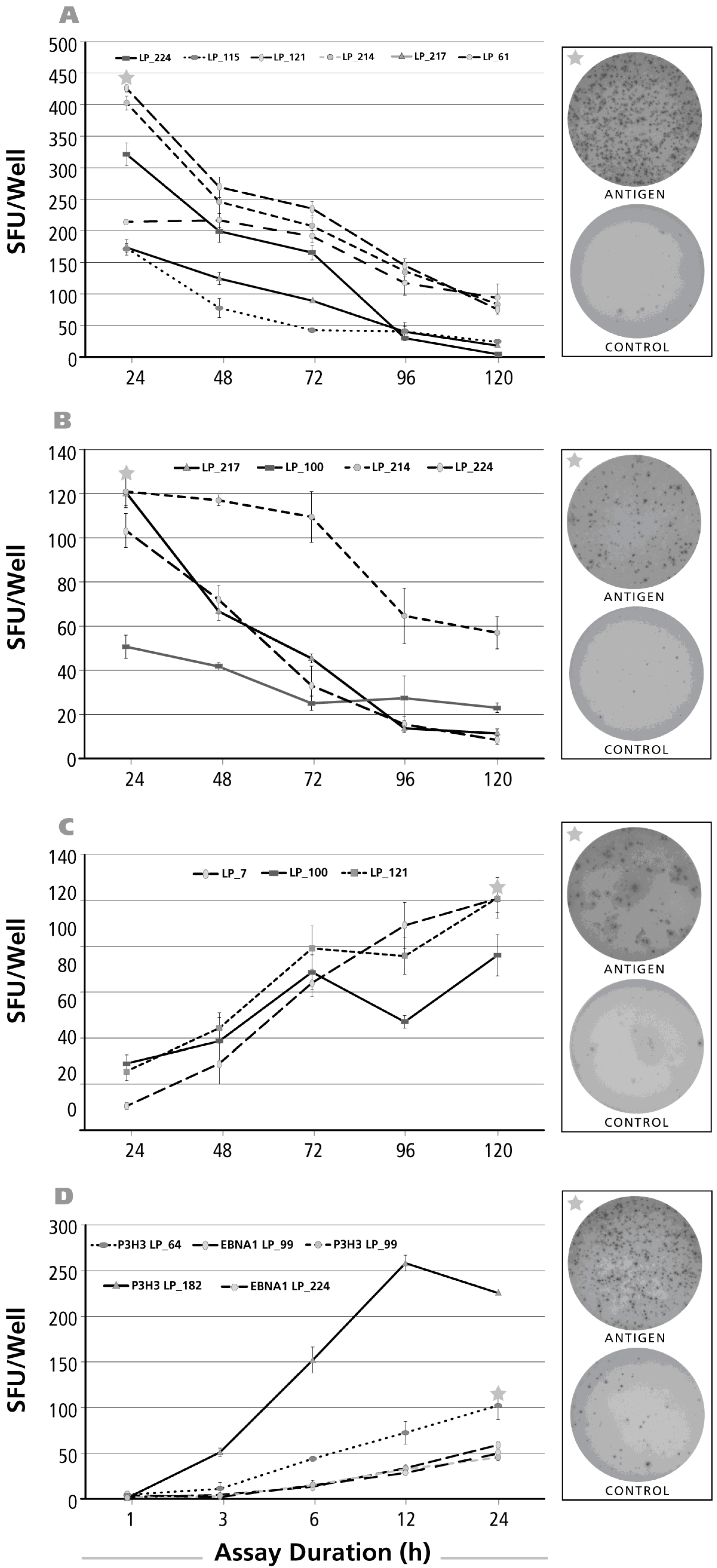
3.2. Kinetics of the IL-17 Recall Response
3.3. Kinetics of the IL-5 Recall Response
3.4. Kinetics of the IL-4 Recall Response
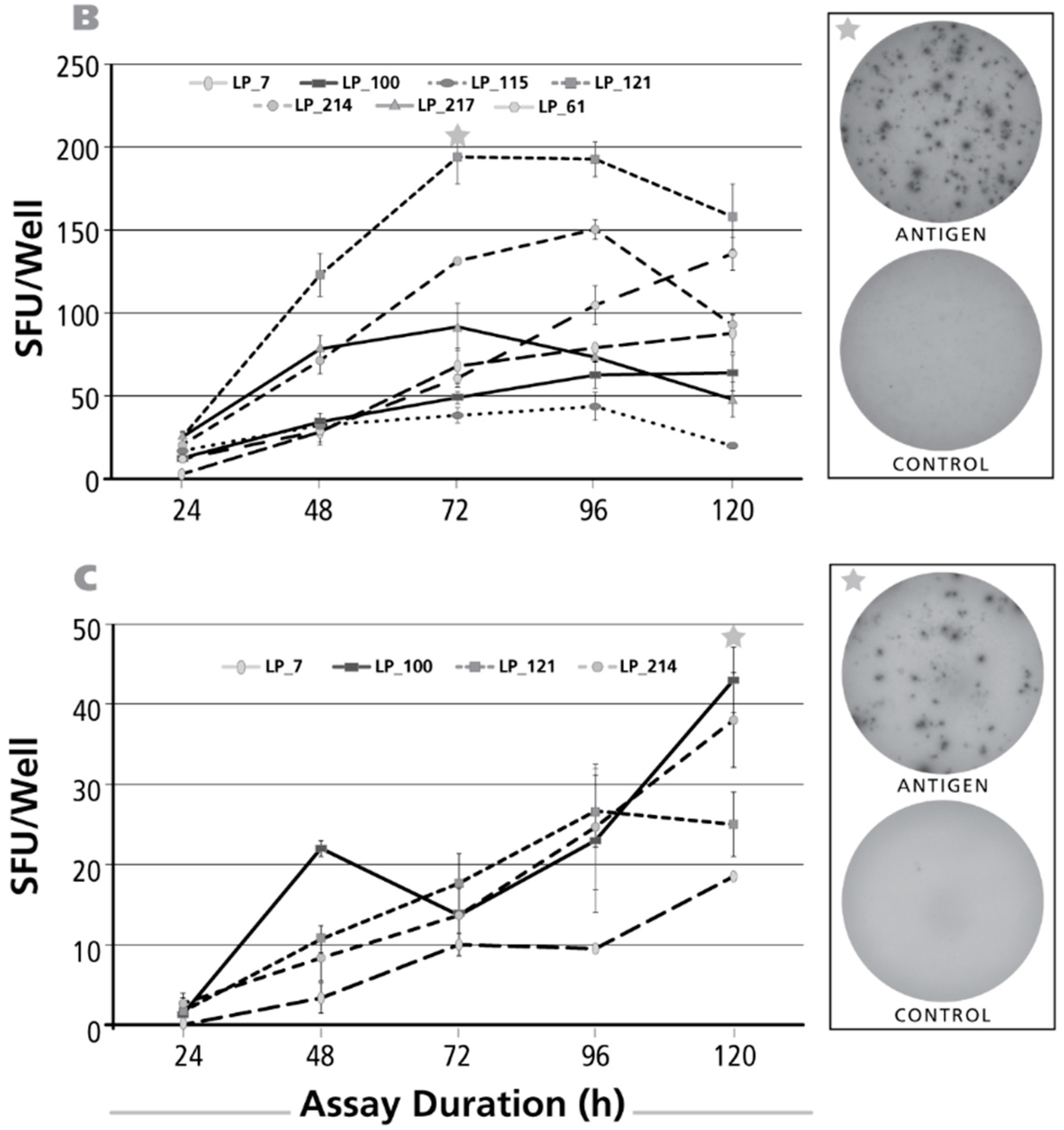
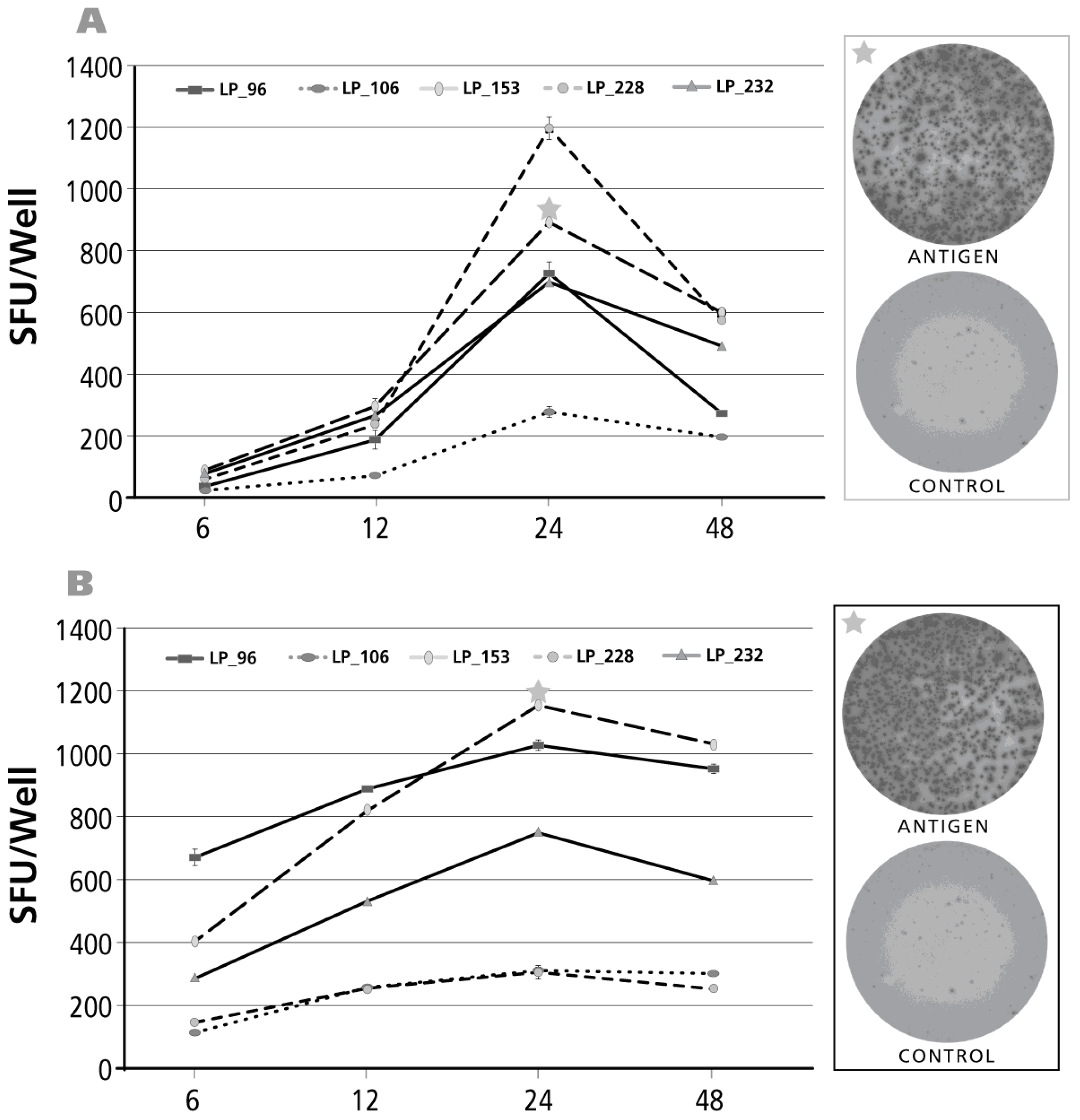
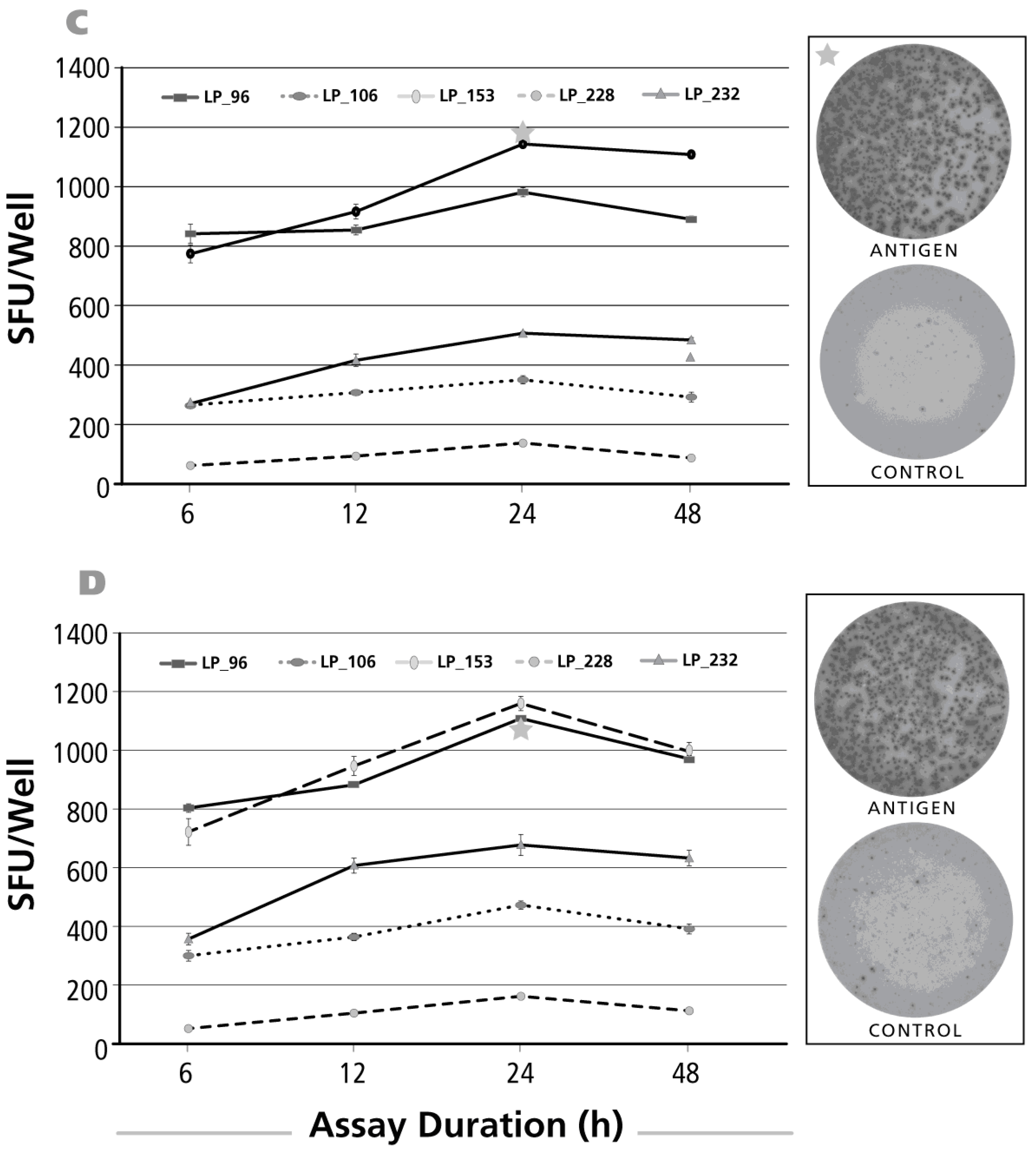
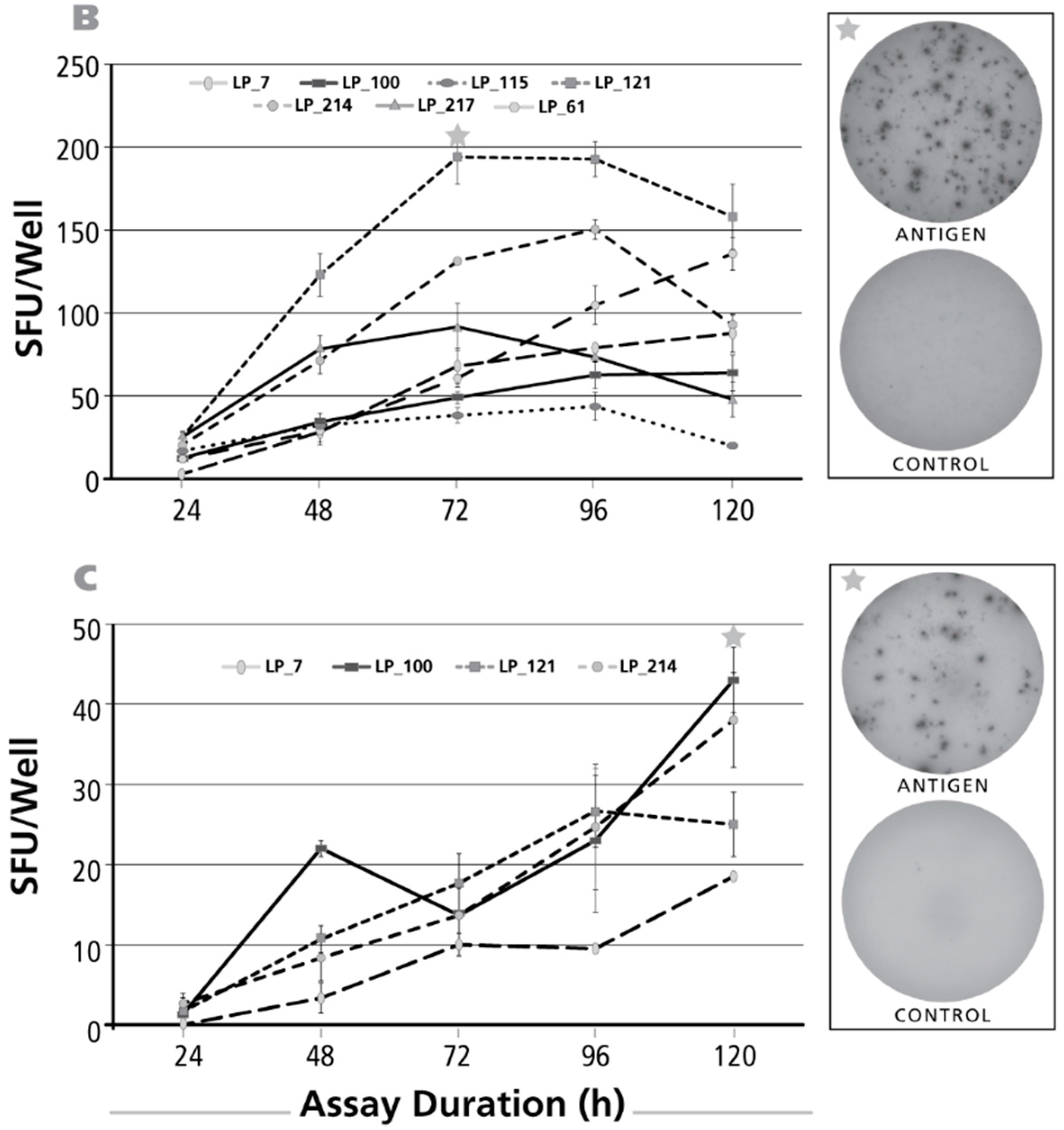
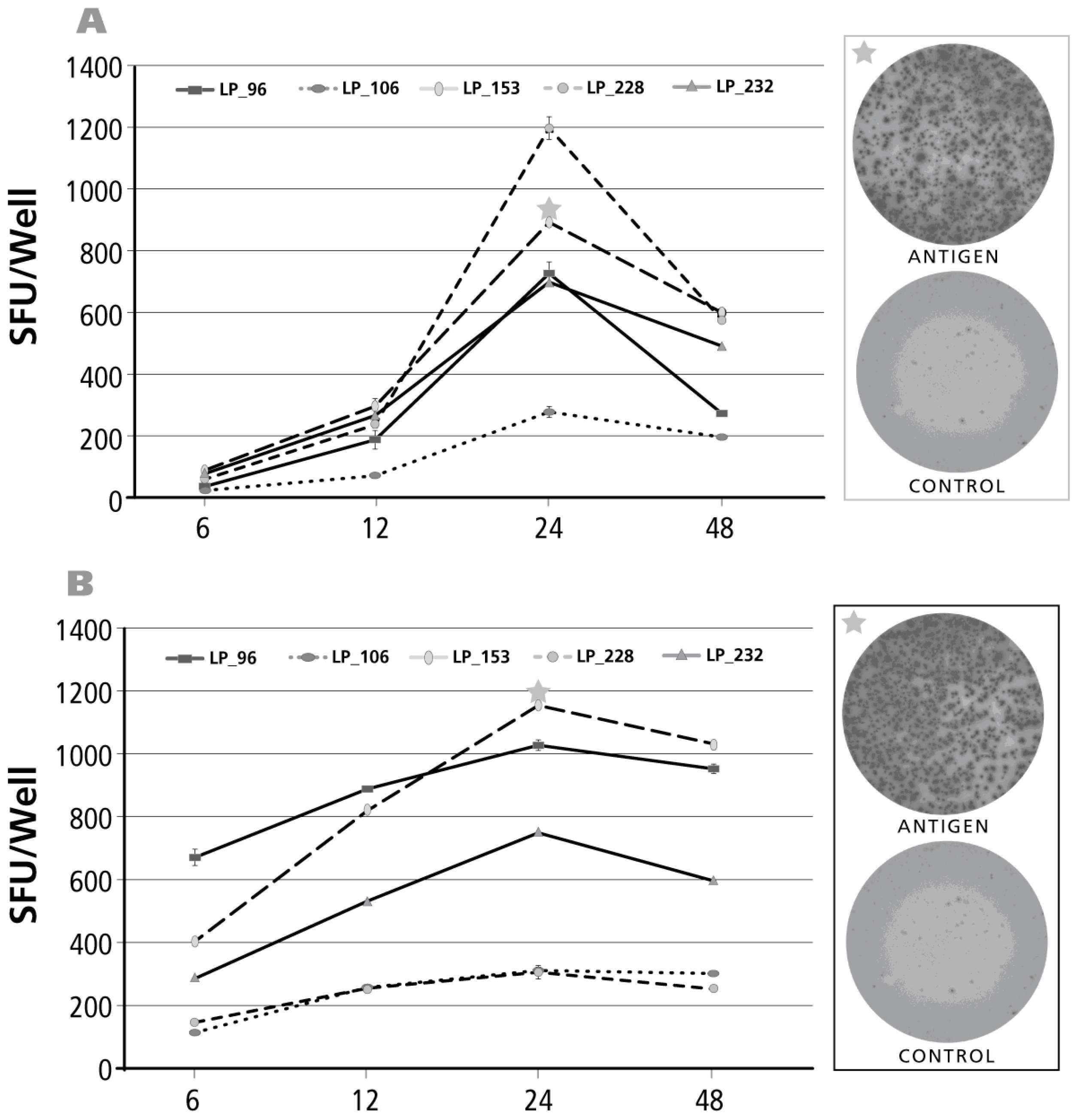
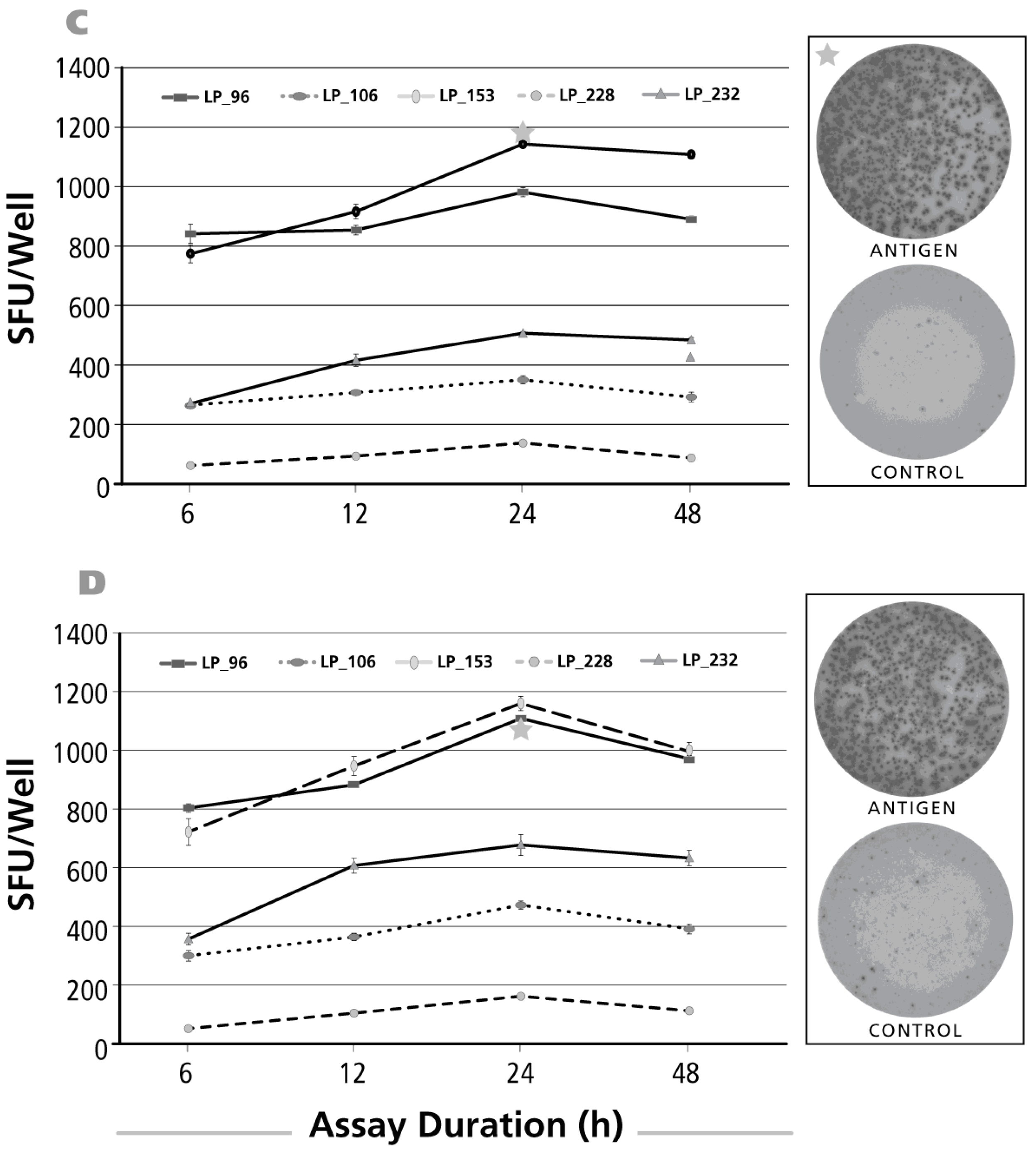
3.5. Kinetics of the IL-2 Recall Response
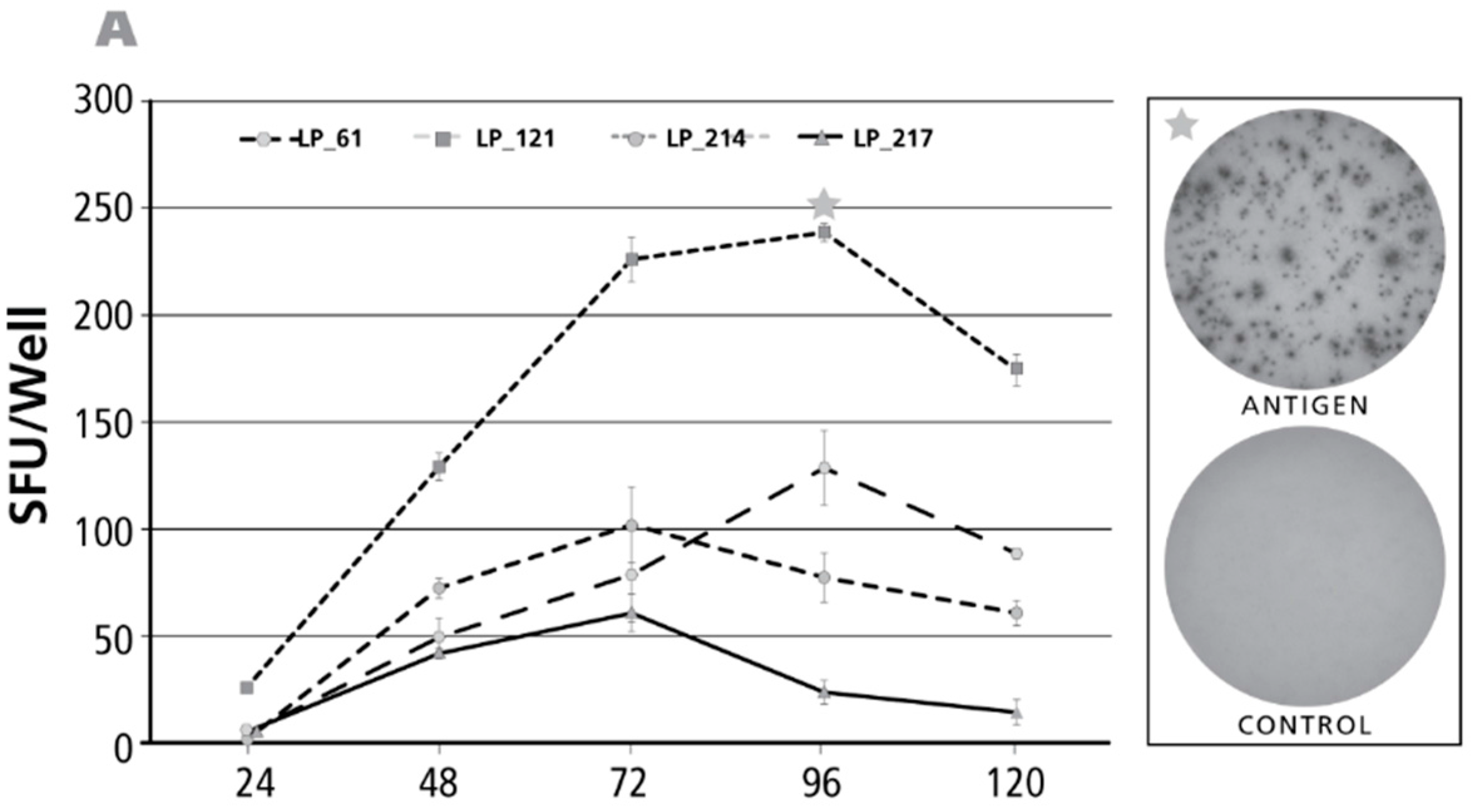
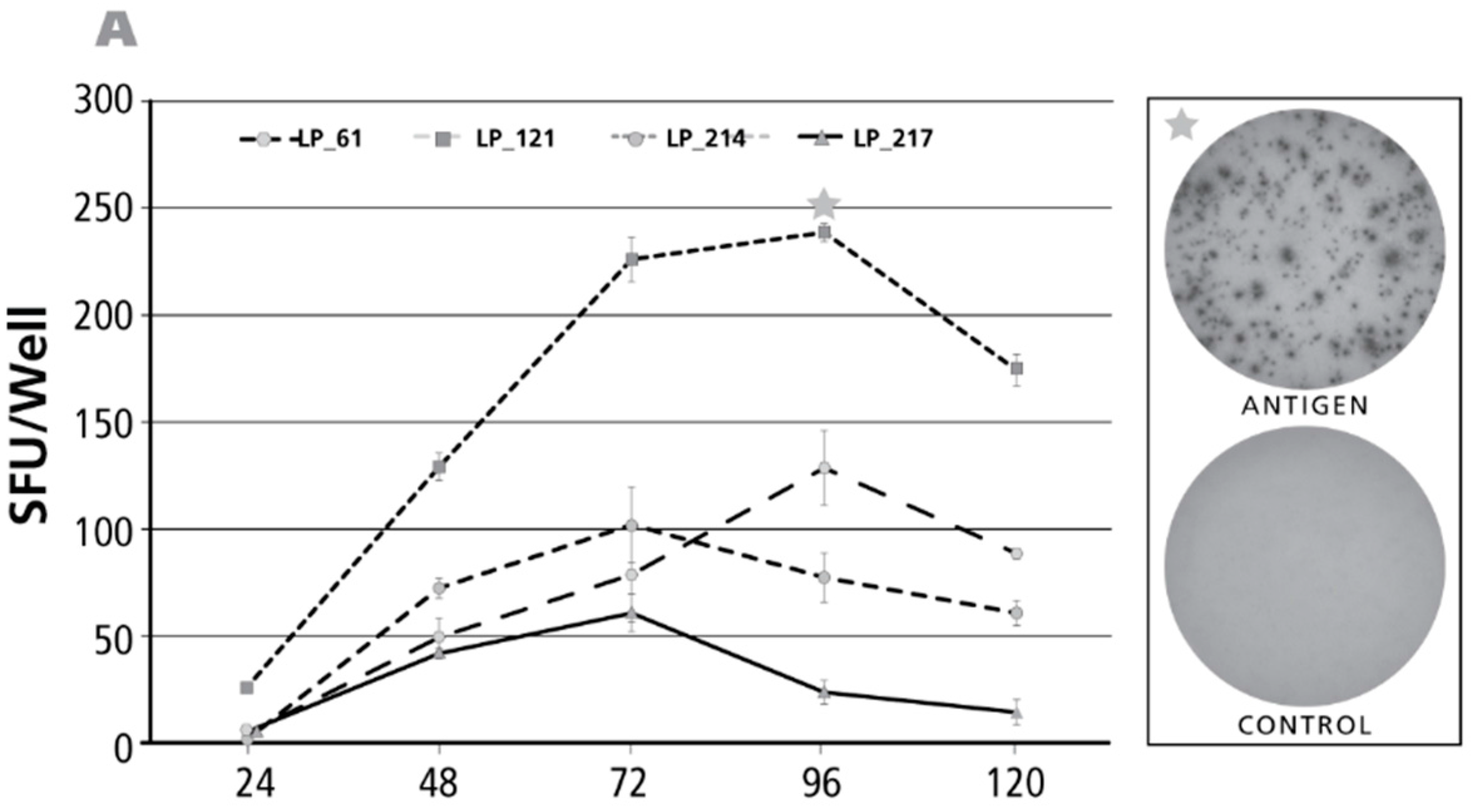
3.6. Kinetics of the IFN-γ Recall Response
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steinman, L. A brief history of t(h)17, the first major revision in the t(h)1/t(h)2 hypothesis of t cell-mediated tissue damage. Nat. Med. 2007, 13, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Lehmann, P.V.; Tary-Lehmann, M. Dissociated induction of cytotoxicity and dth by cfa and cpg. J. Immunother. 2009, 32, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Goldrath, A.W.; Bogatzki, L.Y.; Bevan, M.J. Naive t cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J. Exp. Med. 2000, 192, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of th1 cd4+ t cells through il-12 produced by listeria-induced macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Hosken, N.A.; Litton, M.; Vieira, P.; Hsieh, C.S.; Culpepper, J.A.; Wysocka, M.; Trinchieri, G.; Murphy, K.M.; O’Garra, A. Dendritic cells produce il-12 and direct the development of th1 cells from naive cd4+ t cells. J. Immunol. 1995, 154, 5071–5079. [Google Scholar] [PubMed]
- Seki, N.; Miyazaki, M.; Suzuki, W.; Hayashi, K.; Arima, K.; Myburgh, E.; Izuhara, K.; Brombacher, F.; Kubo, M. Il-4-induced gata-3 expression is a time-restricted instruction switch for th2 cell differentiation. J. Immunol. 2004, 172, 6158–6166. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector th17 and regulatory t cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.C.; Karulin, A.Y.; Tary-Lehmann, M.; Hesse, M.D.; Radeke, H.; Heeger, P.S.; Trezza, R.P.; Heinzel, F.P.; Forsthuber, T.; Lehmann, P.V. Adjuvant-guided type-1 and type-2 immunity: Infectious/noninfectious dichotomy defines the class of response. J. Immunol. 1999, 162, 3942–3949. [Google Scholar] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, t-bet, directs th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Karulin, A.Y.; Hesse, M.D.; Tary-Lehmann, M.; Lehmann, P.V. Single-cytokine-producing cd4 memory cells predominate in type 1 and type 2 immunity. J. Immunol. 2000, 164, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Rottlaender, A.; Rodi, M.; Velasco, V.B., Jr.; Schroeter, M.; Kaiser, C.; Addicks, K.; Tary-Lehmann, M.; Lehmann, P.V. The clinical course of eae is reflected by the dynamics of the neuroantigen-specific t cell compartment in the blood. Clin. Immunol. 2010, 137, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Coffman, R.L. Th1 and th2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Forsthuber, T.; Yip, H.C.; Lehmann, P.V. Induction of th1 and th2 immunity in neonatal mice. Science 1996, 271, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, E.; Della Bella, C.; Bonsignori, F.; Sollai, S.; Amedei, A.; Galli, L.; Niccolai, E.; Del Prete, G.; Singh, M.; D’Elios, M.M.; et al. Potential role of m. Tuberculosis specific ifn-gamma and il-2 elispot assays in discriminating children with active or latent tuberculosis. PLoS ONE 2012, 7, e46041. [Google Scholar] [CrossRef] [PubMed]
- Mackay, C.R.; Marston, W.L.; Dudler, L. Naive and memory t cells show distinct pathways of lymphocyte recirculation. J. Exp. Med. 1990, 171, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Helms, T.; Boehm, B.O.; Asaad, R.J.; Trezza, R.P.; Lehmann, P.V.; Tary-Lehmann, M. Direct visualization of cytokine-producing recall antigen-specific cd4 memory t cells in healthy individuals and hiv patients. J. Immunol. 2000, 164, 3723–3732. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, H.H.; Toyka, K.V.; Tary-Lehmann, M.; Lehmann, P.V. Kinetics and organ distribution of il-17-producing cd4 cells in proteolipid protein 139–151 peptide-induced experimental autoimmune encephalomyelitis of sjl mice. J. Immunol. 2007, 178, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, M.; Zhang, W.; Hanson, J.; Caspell, R.; Karulin, A.Y.; Recks, M.S.; Kuerten, S.; Sundararaman, S.; Lehmann, P.V. Characterization of the hcmv-specific cd4 t cell responses that are associated with protective immunity. Viruses 2015, 7, 4414–4437. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.D.; Karulin, A.Y.; Boehm, B.O.; Lehmann, P.V.; Tary-Lehmann, M. A t cell clone’s avidity is a function of its activation state. J. Immunol. 2001, 167, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Prussin, C.; Metcalfe, D.D. Detection of intracytoplasmic cytokine using flow cytometry and directly conjugated anti-cytokine antibodies. J. Immunol. Methods 1995, 188, 117–128. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Zhang, W. Unique strengths of elispot for t cell diagnostics. In Handbook of Elispot: Methods and Protocols; Kalyuzhny, A.E., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 3–23. [Google Scholar]
- Hanson, J.; Sundararaman, S.; Caspell, R.; Karacsony, E.; Karulin, A.Y.; Lehmann, P.V. Elispot assays in 384-well format: Up to 30 data points with one million cells. Cells 2015, 4, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N. Mhc-dependent antigen processing and peptide presentation: Providing ligands for t lymphocyte activation. Cell 1994, 76, 287–299. [Google Scholar] [CrossRef]
- Rudensky, A.; Preston-Hurlburt, P.; Hong, S.C.; Barlow, A.; Janeway, C.A., Jr. Sequence analysis of peptides bound to mhc class ii molecules. Nature 1991, 353, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, H.; Laux, J.; Moldovan, I.; Caspell, R.; Lehmann, P.V.; Subbramanian, R.A. Optimal thawing of cryopreserved peripheral blood mononuclear cells for use in high-throughput human immune monitoring studies. Cells 2012, 1, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Batoulis, H.; Recks, M.S.; Karacsony, E.; Zhang, W.; Subbramanian, R.A.; Lehmann, P.V. Resting of cryopreserved pbmc does not generally benefit the performance of antigen-specific t cell elispot assays. Cells 2012, 1, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Kutscher, S.; Dembek, C.J.; Deckert, S.; Russo, C.; Korber, N.; Bogner, J.R.; Geisler, F.; Umgelter, A.; Neuenhahn, M.; Albrecht, J.; et al. Overnight resting of pbmc changes functional signatures of antigen specific t- cell responses: Impact for immune monitoring within clinical trials. PLoS ONE 2013, 8, e76215. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Solana, R.; Dela Rosa, O.; Pawelec, G. Human cytomegalovirus infection and t cell immunosenescence: A mini review. Mech. Ageing Dev. 2006, 127, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Currier, J.R.; Kuta, E.G.; Turk, E.; Earhart, L.B.; Loomis-Price, L.; Janetzki, S.; Ferrari, G.; Birx, D.L.; Cox, J.H. A panel of mhc class i restricted viral peptides for use as a quality control for vaccine trial elispot assays. J. Immunol. Methods 2002, 260, 157–172. [Google Scholar] [CrossRef]
- Zhang, W.; Lehmann, P.V. Objective, user-independent elispot data analysis based on scientifically validated principles. In Handbook of Elispot: Methods and Protocols; Kalyuzhny, A.E., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 155–171. [Google Scholar]
- Karulin, A.Y.; Caspell, R.; Dittrich, M.; Lehmann, P.V. Normal distribution of cd8+ t-cell-derived elispot counts within replicates justifies the reliance on parametric statistics for identifying positive responses. Cells 2015, 4, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Karulin, A.Y.; Lehmann, P.V. How elispot morphology reflects on the productivity and kinetics of cells’ secretory activity. In Handbook of Elispot: Methods and Protocols; Kalyuzhny, A.E., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 125–143. [Google Scholar]
- Anthony, D.D.; Lehmann, P.V. T-cell epitope mapping using the elispot approach. Methods 2003, 29, 260–269. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duechting, A.; Przybyla, A.; Kuerten, S.; Lehmann, P.V. Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells. Cells 2017, 6, 29. https://doi.org/10.3390/cells6030029
Duechting A, Przybyla A, Kuerten S, Lehmann PV. Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells. Cells. 2017; 6(3):29. https://doi.org/10.3390/cells6030029
Chicago/Turabian StyleDuechting, Andrea, Anna Przybyla, Stefanie Kuerten, and Paul V. Lehmann. 2017. "Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells" Cells 6, no. 3: 29. https://doi.org/10.3390/cells6030029
APA StyleDuechting, A., Przybyla, A., Kuerten, S., & Lehmann, P. V. (2017). Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells. Cells, 6(3), 29. https://doi.org/10.3390/cells6030029