Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease


Abstract
1. General Introduction
2. Autophagy: Types and Regulation
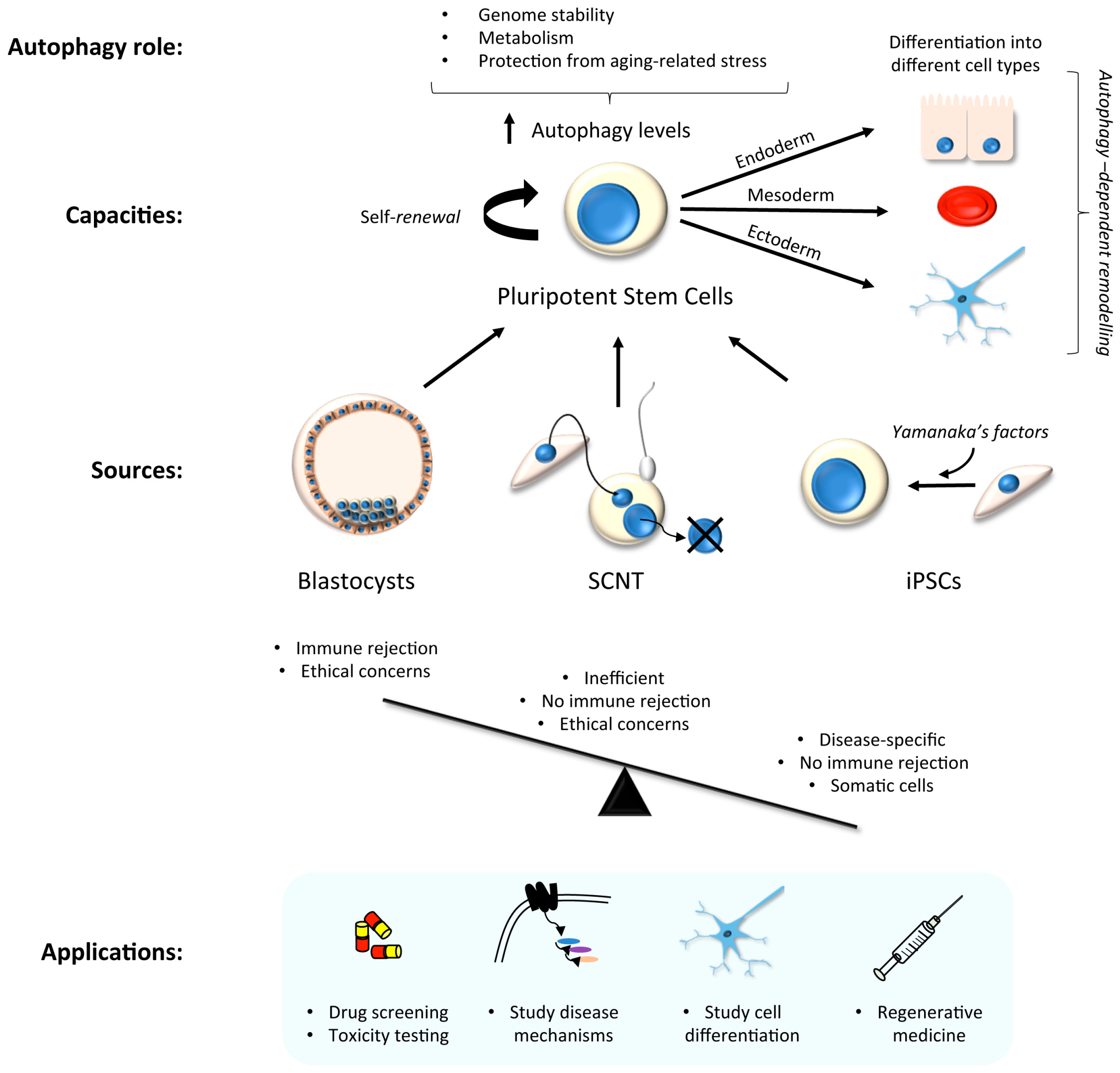
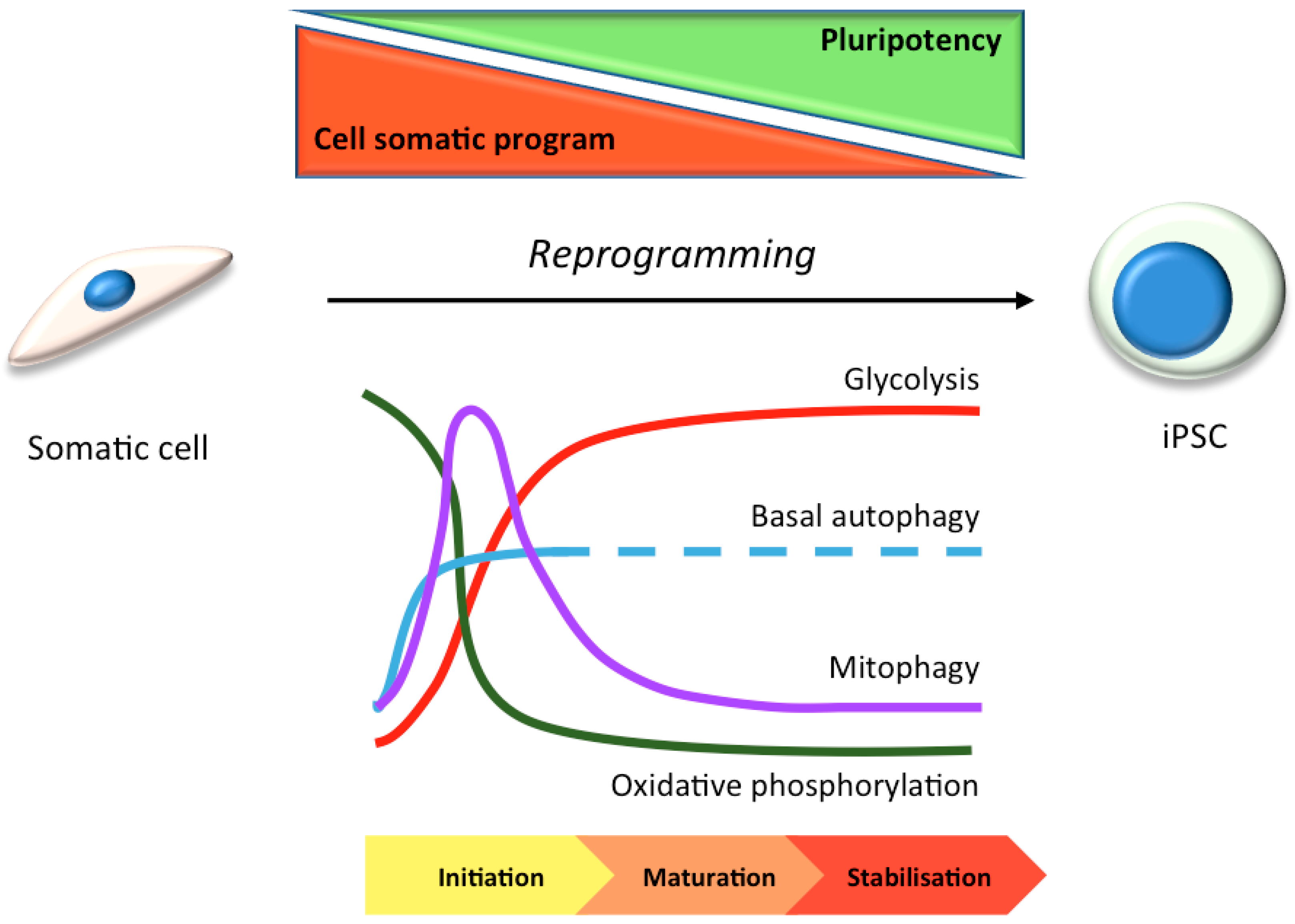
3. Stem Cells in Laboratory Research: Focus on Autophagy
3.1. hiPSC Neuronal Models and Autophagy
4. Studying Autophagy in hiPSC-Derived Neurons
4.1. Activating Autophagy in hiPSC-Derived Neurons
4.2. Taking Account of Autophagic Flux
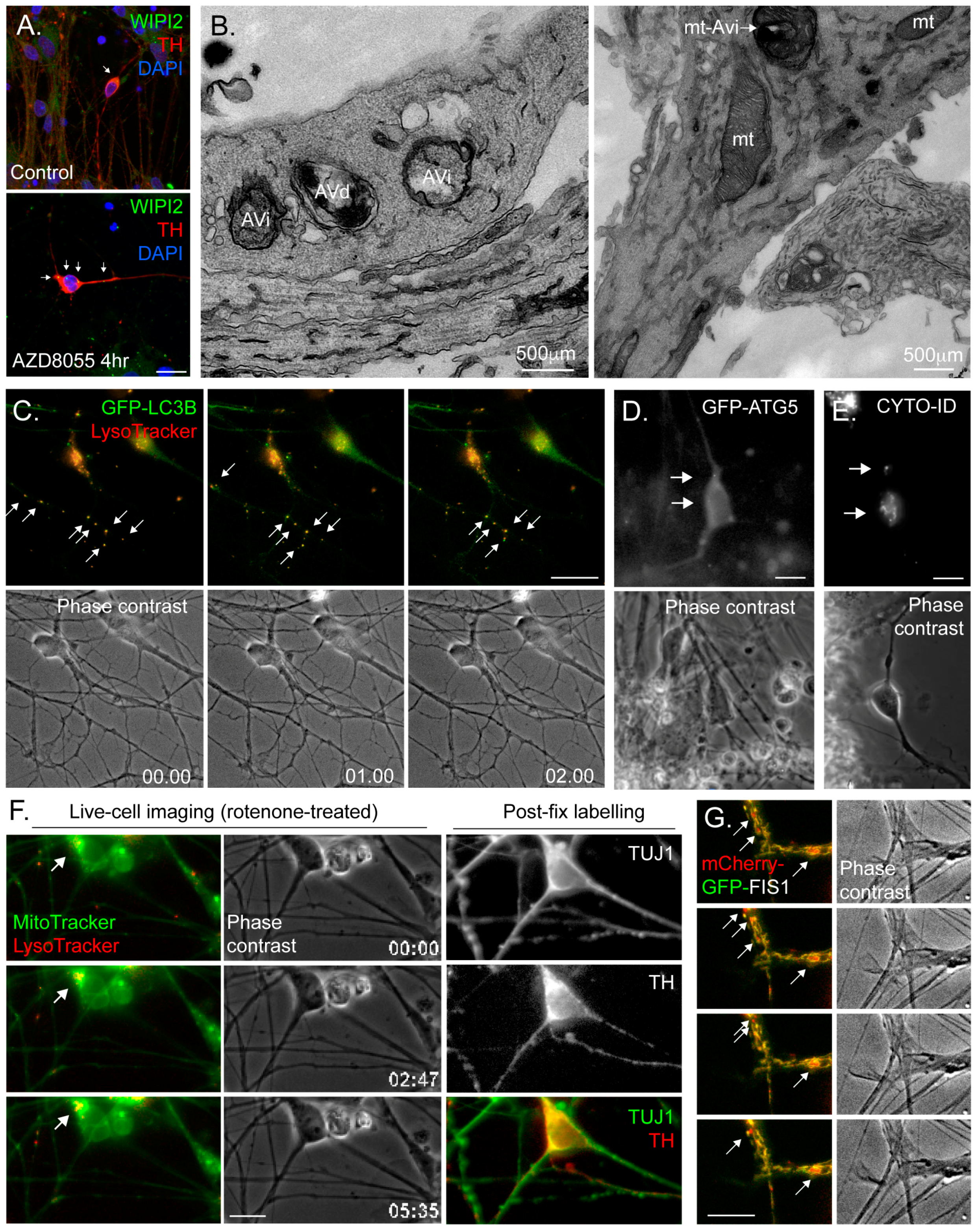
4.3. Common Autophagy Assessment Tools
4.3.1. Immunoblotting
4.3.2. Immunofluorescence Microscopy
4.3.3. Electron Microscopy (EM)
4.3.4. Fluorescence Live-Cell Imaging and Flow Cytometry
4.3.5. Monitoring Mitophagy
4.3.6. Gene Expression
5. Autophagy Studies of Human Neurodegenerative Diseases Using hiPSC-Derived Neuronal Cultures
5.1. Autophagy in hiPSC Models of Alzheimer’s Disease
5.2. Autophagy in iPSC Models of TAUopathy
5.3. Autophagy in hiPSC Models of Parkinson’s Disease
5.4. Autophagy in hiPSC Models of FTD
5.5. Autophagy in hiPSC Models of Other Neurodegenerative Diseases
5.6. Autophagy in hiPSC Models of Neuronal Lysosomal Storage Diseases
5.7. Autophagy in hiPSC Models of Ocular Diseases
6. Future Perspectives and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. Tfeb-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Weick, J.P. Functional properties of human stem cell-derived neurons in health and disease. Stem Cells Int. 2016, 2016, 4190438. [Google Scholar] [CrossRef] [PubMed]
- Jungverdorben, J.; Till, A.; Brustle, O. Induced pluripotent stem cell-based modeling of neurodegenerative diseases: A focus on autophagy. J. Mol. Med. (Berl) 2017, 95, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Maday, S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrian, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z. Neuronal aggregates: Formation, clearance, and spreading. Dev. Cell. 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Muqit, M.M. Ubiquitin and parkinson's disease through the looking glass of genetics. Biochem J. 2017, 474, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Dhaliwal, J.S.; Ceizar, M.; Vaculik, M.; Kumar, K.L.; Lagace, D.C. Knockout of atg5 delays the maturation and reduces the survival of adult-generated neurons in the hippocampus. Cell. Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed]
- Yazdankhah, M.; Farioli-Vecchioli, S.; Tonchev, A.B.; Stoykova, A.; Cecconi, F. The autophagy regulators ambra1 and beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell. Death Dis. 2014, 5, e1403. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Ktistakis, N.T.; Tooze, S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Wilson, M.I.; Tooze, S.A. Wipi2b links ptdins3p to lc3 lipidation through binding atg16l1. Autophagy 2015, 11, 190–191. [Google Scholar] [PubMed]
- Doss, M.X.; Koehler, C.I.; Gissel, C.; Hescheler, J.; Sachinidis, A. Embryonic stem cells: A promising tool for cell replacement therapy. J. Cell. Mol. Med. 2004, 8, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, T.; El Rouby, N.; Fredette, N.C.; Santostefano, K.E.; Terada, N. Concise review: Induced pluripotent stem cell research in the era of precision medicine. Stem Cells 2017, 35, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Izpisua Belmonte, J.C. Looking to the future following 10 years of induced pluripotent stem cell technologies. Nat. Protoc. 2016, 11, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurosci. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Plath, K. The roles of the reprogramming factors oct4, sox2 and klf4 in resetting the somatic cell epigenome during induced pluripotent stem cell generation. Genome Biol. 2012, 13, 251. [Google Scholar] [CrossRef] [PubMed]
- Buganim, Y.; Faddah, D.A.; Jaenisch, R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013, 14, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem Cells 2014, 6, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells--opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Nadri, S.; Ghollasi, M.; Khajeh, K.; Soleimani, M. Comparison of different protocols for neural differentiation of human induced pluripotent stem cells. Mol. Biol. Rep. 2014, 41, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. Fip200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 mapk to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Sotthibundhu, A.; McDonagh, K.; von Kriegsheim, A.; Garcia-Munoz, A.; Klawiter, A.; Thompson, K.; Chauhan, K.D.; Krawczyk, J.; McInerney, V.; Dockery, P.; et al. Rapamycin regulates autophagy and cell adhesion in induced pluripotent stem cells. Stem Cell Res. Ther. 2016, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.; Zhang, H.; Huang, Y.; Zhao, P.; Tang, Y.; Qiu, X.; Ying, Y.; Li, W.; Ni, S.; et al. Autophagy and mtorc1 regulate the stochastic phase of somatic cell reprogramming. Nat. Cell. Biol. 2015, 17, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Shen, L.; Yu, J.; Wan, H.; Guo, A.; Chen, J.; Long, Y.; Zhao, J.; Pei, G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011, 10, 908–911. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kang, L.; Wu, T.; Zhang, J.; Wang, H.; Gao, H.; Zhang, Y.; Huang, B.; Liu, W.; Kou, Z.; et al. An elaborate regulation of mammalian target of rapamycin activity is required for somatic cell reprogramming induced by defined transcription factors. Stem Cells Dev. 2012, 21, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Oliveras-Ferraros, C.; Cufi, S.; Vazquez-Martin, A. Mtor-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011, 10, 3658–3677. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Ye, B.; Huang, G.; Liu, J.; Fan, Z. Transient activation of autophagy via sox2-mediated suppression of mtor is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013, 13, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A.; Sankai, Y. Long-term culture of rat hippocampal neurons at low density in serum-free medium: Combination of the sandwich culture technique with the three-dimensional nanofibrous hydrogel puramatrix. PLoS ONE 2014, 9, e102703. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus rna sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of sh-sy5y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed]
- Ray, B.; Chopra, N.; Long, J.M.; Lahiri, D.K. Human primary mixed brain cultures: Preparation, differentiation, characterization and application to neuroscience research. Mol. Brain 2014, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, H.A.; Ghanbari, K.; Harris, P.L.; Jones, P.K.; Kubat, Z.; Castellani, R.J.; Wolozin, B.L.; Smith, M.A.; Perry, G. Oxidative damage in cultured human olfactory neurons from alzheimer’s disease patients. Aging Cell 2004, 3, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed]
- Spaethling, J.M.; Na, Y.J.; Lee, J.; Ulyanova, A.V.; Baltuch, G.H.; Bell, T.J.; Brem, S.; Chen, H.I.; Dueck, H.; Fisher, S.A.; et al. Primary cell culture of live neurosurgically resected aged adult human brain cells and single cell transcriptomics. Cell Rep. 2017, 18, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Lee, Q.Y.; Camp, J.G.; Mall, M.; Koh, W.; Shariati, S.A.; Sim, S.; Neff, N.F.; Skotheim, J.M.; Wernig, M.; et al. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 2016, 534, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a parkinson's disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of ipsc-derived neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Okano, H. Cell transplantation therapies for spinal cord injury focusing on induced pluripotent stem cells. Cell Res. 2013, 23, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.Y.; Weick, J.P.; Yu, J.; Ma, L.X.; Zhang, X.Q.; Thomson, J.A.; Zhang, S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.K.; Stice, S.L. Neural differentiation of human embryonic stem cells. J. Cell. Biochem. 2008, 105, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Velasco, I.; Salazar, P.; Giorgetti, A.; Ramos-Mejia, V.; Castano, J.; Romero-Moya, D.; Menendez, P. Concise review: Generation of neurons from somatic cells of healthy individuals and neurological patients through induced pluripotency or direct conversion. Stem Cells 2014, 32, 2811–2817. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human es cells efficiently engraft in animal models of parkinson's disease. Nature 2011, 480, 547–551. [Google Scholar] [PubMed]
- D’Aiuto, L.; Zhi, Y.; Kumar Das, D.; Wilcox, M.R.; Johnson, J.W.; McClain, L.; MacDonald, M.L.; Di Maio, R.; Schurdak, M.E.; Piazza, P.; et al. Large-scale generation of human ipsc-derived neural stem cells/early neural progenitor cells and their neuronal differentiation. Organogenesis 2014, 10, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Nistor, P.A.; May, P.W.; Tamagnini, F.; Randall, A.D.; Caldwell, M.A. Long-term culture of pluripotent stem-cell-derived human neurons on diamond--a substrate for neurodegeneration research and therapy. Biomaterials 2015, 61, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.; Zhang, Y.; Chandrasekaran, A.; Phanthong, P.; Schmid, B.; Nielsen, T.T.; Freude, K.K. Modeling neurodegenerative diseases with patient-derived induced pluripotent cells: Possibilities and challenges. New Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.; Denham, M.; Villaescusa, J.C. How to make a midbrain dopaminergic neuron. Development 2015, 142, 1918–1936. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Randall, A.D. Are stem cell-derived neural cells physiologically credible? J. Physiol. 2016, 594, 6569–6572. [Google Scholar] [CrossRef] [PubMed]
- Wakeman, D.R.; Hiller, B.M.; Marmion, D.J.; McMahon, C.W.; Corbett, G.T.; Mangan, K.P.; Ma, J.; Little, L.E.; Xie, Z.; Perez-Rosello, T.; et al. Cryopreservation maintains functionality of human ipsc dopamine neurons and rescues parkinsonian phenotypes in vivo. Stem Cell Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative p53 mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human ipsc-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Gurwitz, D. Human ipsc-derived neurons and lymphoblastoid cells for personalized medicine research in neuropsychiatric disorders. Dialogues Clin. Neurosci. 2016, 18, 267–276. [Google Scholar] [PubMed]
- Yang, J.; Li, S.; He, X.B.; Cheng, C.; Le, W. Induced pluripotent stem cells in alzheimer's disease: Applications for disease modeling and cell-replacement therapy. Mol. Neurodegener. 2016, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.t.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling als with ipscs reveals that mutant sod1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from niemann-pick type c patient-specific ips cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell. Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged lc3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol.Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An autophagic flux probe that releases an internal control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Khayati, K.; Antikainen, H.; Bonder, E.M.; Weber, G.F.; Kruger, W.D.; Jakubowski, H.; Dobrowolski, R. The amino acid metabolite homocysteine activates mtorc1 to inhibit autophagy and form abnormal proteins in human neurons and mice. FASEB J. 2017, 31, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret lc3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Orhon, I.; Reggiori, F. Assays to monitor autophagy progression in cell cultures. Cells 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Nihira, T.; Uchino, A.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Takahashi, K.; Hayakawa, H.; Nagai, M.; Ohyama, M.; et al. I2020t mutant lrrk2 ipsc-derived neurons in the sagamihara family exhibit increased tau phosphorylation through the akt/gsk-3beta signaling pathway. Hum. Mol. Genet. 2015, 24, 4879–4900. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human ips-based models of genetic and sporadic parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for atg4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. Er stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in gba-n370s parkinson's ipsc-derived dopamine neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in ipsc-derived neurons from patients of ftdp-17 with 10+16 mapt mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson’s disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell. 2014, 30, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires pink1 and parkin. J. Cell. Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.F.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers pink1/parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. Mito-qc illuminates mitophagy and mitochondrial architecture in vivo. J. Cell. Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [PubMed]
- Cortes, C.J.; Miranda, H.C.; Frankowski, H.; Batlevi, Y.; Young, J.E.; Le, A.; Ivanov, N.; Sopher, B.L.; Carromeu, C.; Muotri, A.R.; et al. Polyglutamine-expanded androgen receptor interferes with tfeb to elicit autophagy defects in sbma. Nat. Neurosci. 2014, 17, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of nutrient sensing and clearance in presenilin deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Lu, J.; Byrne, S.; Hoffmann, G.F.; Jungbluth, H.; Sahin, M. Congenital disorders of autophagy: An emerging novel class of inborn errors of neuro-metabolism. Brain J. Neuro. 2016, 139, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.H.; Jia, J.P. Dysfunctional autophagy in alzheimer’s disease: Pathogenic roles and therapeutic implications. Neurosci. Bull. 2014, 30, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in alzheimer's disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Matsumoto, A.; Shimazaki, T.; Enoki, R.; Koizumi, A.; Ishii, S.; Itoyama, Y.; Sobue, G.; Okano, H. Spatiotemporal recapitulation of central nervous system development by murine embryonic stem cell-derived neural stem/progenitor cells. Stem Cells 2008, 26, 3086–3098. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human es and ips cells by dual inhibition of smad signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, A.; Diels, A.; Dijkmans, J.; Oyelami, T.; Meneghello, G.; Mertens, L.; Versweyveld, S.; Borgers, M.; Buist, A.; Peeters, P.; et al. Using human ipsc-derived neurons to model tau aggregation. PLoS ONE 2015, 10, e0146127. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Cheng, C.; Mair, W.; Almeida, S.; Fong, H.; Biswas, M.H.; Zhang, Z.; Huang, Y.; Temple, S.; Coppola, G.; et al. Human ipsc-derived neuronal model of tau-a152t frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep. 2016, 7, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Consiglio, A.; Richaud, Y.; Rodriguez-Piza, I.; Dehay, B.; Edel, M.; Bove, J.; Memo, M.; Vila, M.; Raya, A.; et al. Efficient generation of a9 midbrain dopaminergic neurons by lentiviral delivery of lmx1a in human embryonic stem cells and induced pluripotent stem cells. Human Gene Ther. 2012, 23, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Plowey, E.D.; Dagda, R.K.; Hickey, R.W.; Cherra, S.J., 3rd; Clark, R.S. Autophagy in neurite injury and neurodegeneration: In vitro and in vivo models. Methods Enzymol. 2009, 453, 217–249. [Google Scholar] [PubMed]
- Orenstein, S.J.; Kuo, S.H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of lrrk2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. Ipsc-derived neurons from gba1-associated parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial parkin recruitment is impaired in neurons derived from mutant pink1 induced pluripotent stem cells. J. Nurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and pink1 patient ipsc-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in park2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Hargus, G.; Deleidi, M.; Blak, A.; Osborn, T.; Marlow, E.; Lee, K.; Levy, A.; Perez-Torres, E.; Yow, A.; et al. Differentiation of human es and parkinson's disease ips cells into ventral midbrain dopaminergic neurons requires a high activity form of shh, fgf8a and specific regionalization by retinoic acid. Mol. Cell. Neurosci. 2010, 45, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The sac1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef] [PubMed]
- Bott, N.T.; Radke, A.; Stephens, M.L.; Kramer, J.H. Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener. Dis. Manag. 2014, 4, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling key pathological features of frontotemporal dementia with c9orf72 repeat expansion in ipsc-derived human neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Taylor, G.; McEachin, Z.T.; Deng, Q.; Watkins, W.J.; Hudson, K.; Easley, C.A.; Hu, W.T.; Hales, C.M.; Rossoll, W.; et al. Trehalose upregulates progranulin expression in human and mouse models of grn haploinsufficiency: A novel therapeutic lead to treat frontotemporal dementia. Mol. Neurodegener. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances tdp43 turnover and survival in neuronal als models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of tdp-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for huntington's disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, J.C.; Isfort, M.C.; Roggenbuck, J.; Arnold, W.D. The genetics of charcot-marie-tooth disease: Current trends and future implications for diagnosis and management. Appl. Clin. Genet. 2015, 8, 235–243. [Google Scholar] [PubMed]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human charcot-marie-tooth 2a motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Luo, M.; Niu, X.; Chen, Y.; Xie, Y.; He, W.; Song, B.; Xian, Y.; Fan, D.; OuYang, S.; et al. Autophagy promoted the degradation of mutant atxn3 in neurally differentiated spinocerebellar ataxia-3 human induced pluripotent stem cells. Biomed. Res. Int. 2016, 2016, 6701793. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Di Nardo, A.; Turner, D.; Lewis, T.L., Jr.; Conrad, C.; Rothberg, J.M.; Lipton, J.O.; Kolker, S.; et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016, 17, 1053–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Futerman, A.H. Neuronal forms of gaucher disease. Handb Exp. Pharmacol. 2013, 405–419. [Google Scholar]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered tfeb-mediated lysosomal biogenesis in gaucher disease ipsc-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, M.P.; Roberts, E.A.; Kidwell, C.U.; Yuan, S.H.; Plaisted, W.C.; Goldstein, L.S. Disruption and therapeutic rescue of autophagy in a human neuronal model of niemann pick type c1. Hum. Mol. Genet. 2012, 21, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.K.; Park, M.H.; Hong, Y.R.; Marti, H.H.; Kim, H.; Okada, Y.; Otsu, M.; Seo, E.J.; Park, J.H.; et al. Pathological roles of the vegf/sphk pathway in niemann-pick type c neurons. Nat. Commun. 2014, 5, 5514. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Ishitsuka, Y.; Hamasaki, M.; Yoneda, K.; Furuya, H.; Matsuo, M.; Ihn, H.; Fusaki, N.; Nakamura, K.; Nakagata, N.; et al. Hpgcd outperforms hpbcd as a potential treatment for niemann-pick disease type c during disease modeling with ips cells. Stem Cells 2015, 33, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Dulcey, A.E.; Hu, X.; Wassif, C.A.; Porter, F.D.; Austin, C.P.; Ory, D.S.; Marugan, J.; Zheng, W. Methyl-beta-cyclodextrin restores impaired autophagy flux in niemann-pick c1-deficient cells through activation of AMPK. Autophagy 2017. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Solivan-Timpe, F.; Roos, B.R.; Anfinson, K.R.; Robin, A.L.; Wiley, L.A.; Mullins, R.F.; Fingert, J.H. Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J. Stem Cell Res. Ther. 2014, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.P.; Wiley, L.A.; Whitmore, S.S.; Anfinson, K.R.; Cranston, C.M.; Oppedal, D.J.; Daggett, H.T.; Mullins, R.F.; Tucker, B.A.; Stone, E.M. Patient-specific induced pluripotent stem cells to evaluate the pathophysiology of trnt1-associated retinitis pigmentosa. Stem Cell Res. 2017, 21, 58–70. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Postmortem studies | Stable cell lines | Primary human cultures | Biopsies | iNeurons | iPSC-derived neurons |
|---|---|---|---|---|---|
| Brain connectivity | Soma neuronal characteristics | Non-tumour derived | Non-tumour derived | Direct reprogramming | Somatic cells |
| Disease-specific | Unlimited supply | Recapitulation in vivo neurons | Disease-specific | One-step process | Recapitulation in vivo neurons |
| Sample limitation | Physiological differences | Ethical concerns | Sample limitation | Disease-specific | Large supply |
| Static view | No disease-specific | Sample limitation | Surgical procedures | Regenerative medicine | Disease-specific |
| Age-specific characteristics | Regenerative medicine | ||||
| Inability to expand | Study neurogenesis | ||||
| Low efficiency | Immature characteristics | ||||
| Variable efficiency (higher than iNeurons) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Moreno, N.; Stathakos, P.; Caldwell, M.A.; Lane, J.D. Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease. Cells 2017, 6, 24. https://doi.org/10.3390/cells6030024
Jiménez-Moreno N, Stathakos P, Caldwell MA, Lane JD. Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease. Cells. 2017; 6(3):24. https://doi.org/10.3390/cells6030024
Chicago/Turabian StyleJiménez-Moreno, Natalia, Petros Stathakos, Maeve A. Caldwell, and Jon D. Lane. 2017. "Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease" Cells 6, no. 3: 24. https://doi.org/10.3390/cells6030024
APA StyleJiménez-Moreno, N., Stathakos, P., Caldwell, M. A., & Lane, J. D. (2017). Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease. Cells, 6(3), 24. https://doi.org/10.3390/cells6030024