
Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin A Responsible for Congenital Muscular Dystrophy in a Patient

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Production
2.2. Intramuscular Delivery of AAV Vectors
2.3. Removal and Fixation of TA Muscles
2.4. Analysis of TA Muscle Protein Extracts
2.5. Histology of TA Muscle Sections
2.6. Primary Antibodies
2.7. Immunofluorescence of TA Muscle Sections
- Single immunostaining for FLAG-lamin A: Rabbit anti-FLAG (1:350) for 1.5 h at room temperature, followed by donkey anti-rabbit IgG coupled to Cy5 (1:150) for 1 h at room temperature.
- Double immunostaining for desmin/FLAG, which required two successive incubations: (1) mix of mouse anti-desmin (1:50) and rabbit anti-FLAG (1:350) for 1.5 h at room temperature; and (2) mix of goat anti-mouse IgG coupled to fluorescein (488) (1:400) with donkey anti-rabbit IgG coupled to Cy5 (1:150) for 1 h at room temperature.
- Double immunostaining for MHC I/FLAG, which required four successive incubations: (1) mouse anti-MHC 1 (1:20) overnight at 4 °C; (2) goat anti-mouse IgG coupled to fluorescein (488) (1:400) for 1 h at room temperature; (3) rabbit anti-FLAG (1:350) for 1.5 h at room temperature; and (4) donkey anti-rabbit IgG coupled to Cy5 (1:150) for 1 h at room temperature.
- Triple immunostaining for MHC IIB or MHC IIX/FLAG/perlecan, which required six successive incubations: (1) rabbit anti-FLAG (1:350) for 1.5 h at room temperature; (2) donkey anti-rabbit IgG coupled to Cy5 (1:150) for 1 h at room temperature; (3) mouse anti-MHC IIX (1:30) or mouse anti-MHC IIB (1:2) overnight at 4 °C; (4) rabbit anti-mouse IgM coupled to Cy3 (1:250) for 1 h at room temperature; (5) rat anti-perlecan (1:300) for 1.5 h at room temperature; and (6) goat anti-rat coupled to fluorescein (488) (1:400) for 1 h at room temperature.
- Triple immunostaining for MHC IIA/FLAG/perlecan, which required four successive incubations: (1) mouse anti-MHC IIA (1:20) overnight at 4 °C; (2) goat anti-mouse IgG1 coupled to Cy3 (1:250) for 1 h at room temperature; (3) mix of rabbit anti-FLAG (1:350) and rat anti-perlecan (1:300) for 1.5 h at room temperature; and (4) mix of donkey anti-rabbit IgG coupled to Cy5 (1:150) and goat anti-rat coupled to fluorescein (488) (1:400) for 1 h at room temperature.
2.8. Analysis of TA Muscle Characteristics
2.9. Quantitative RT-PCR
2.10. Statistical Analysis
3. Results
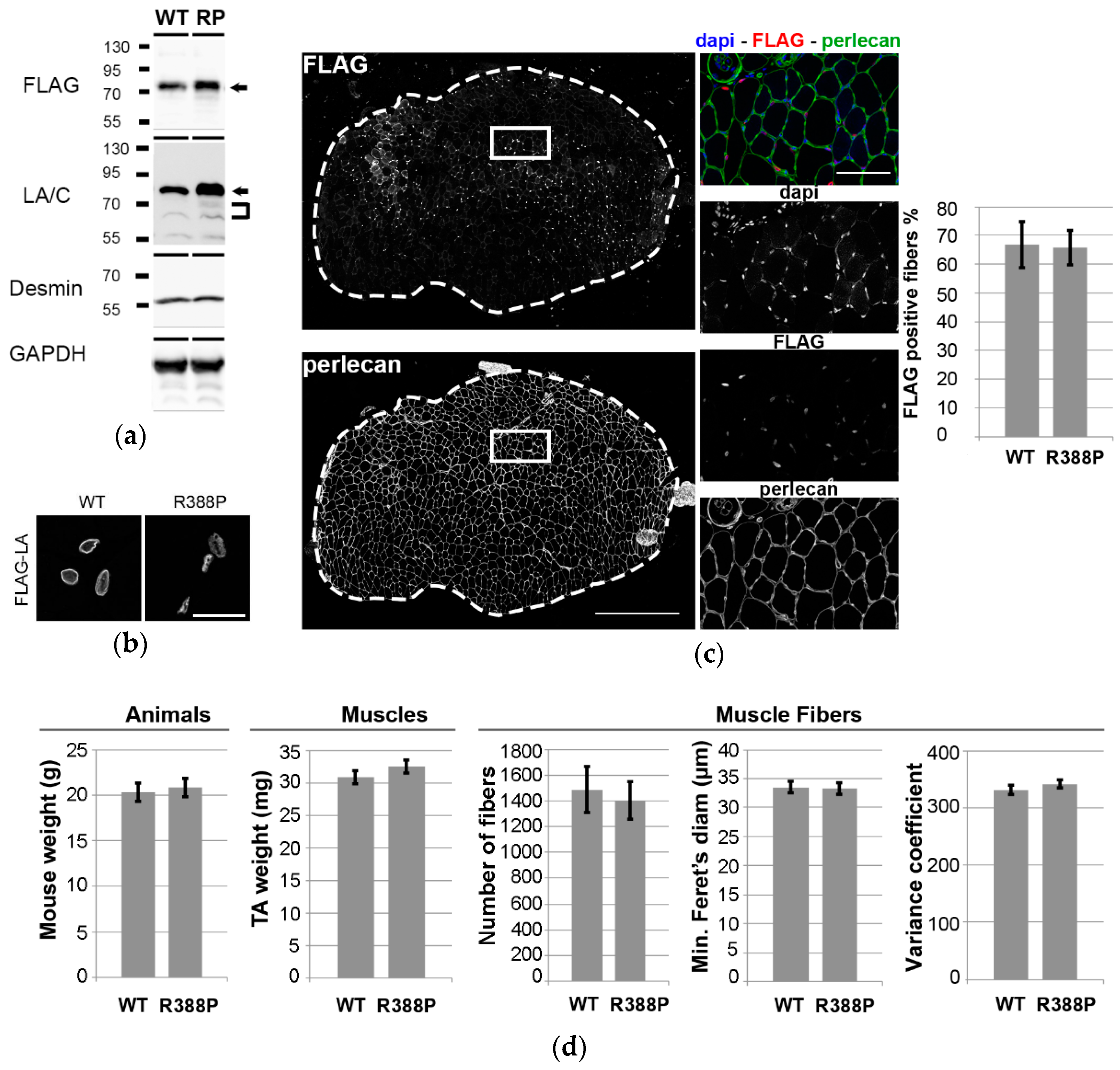
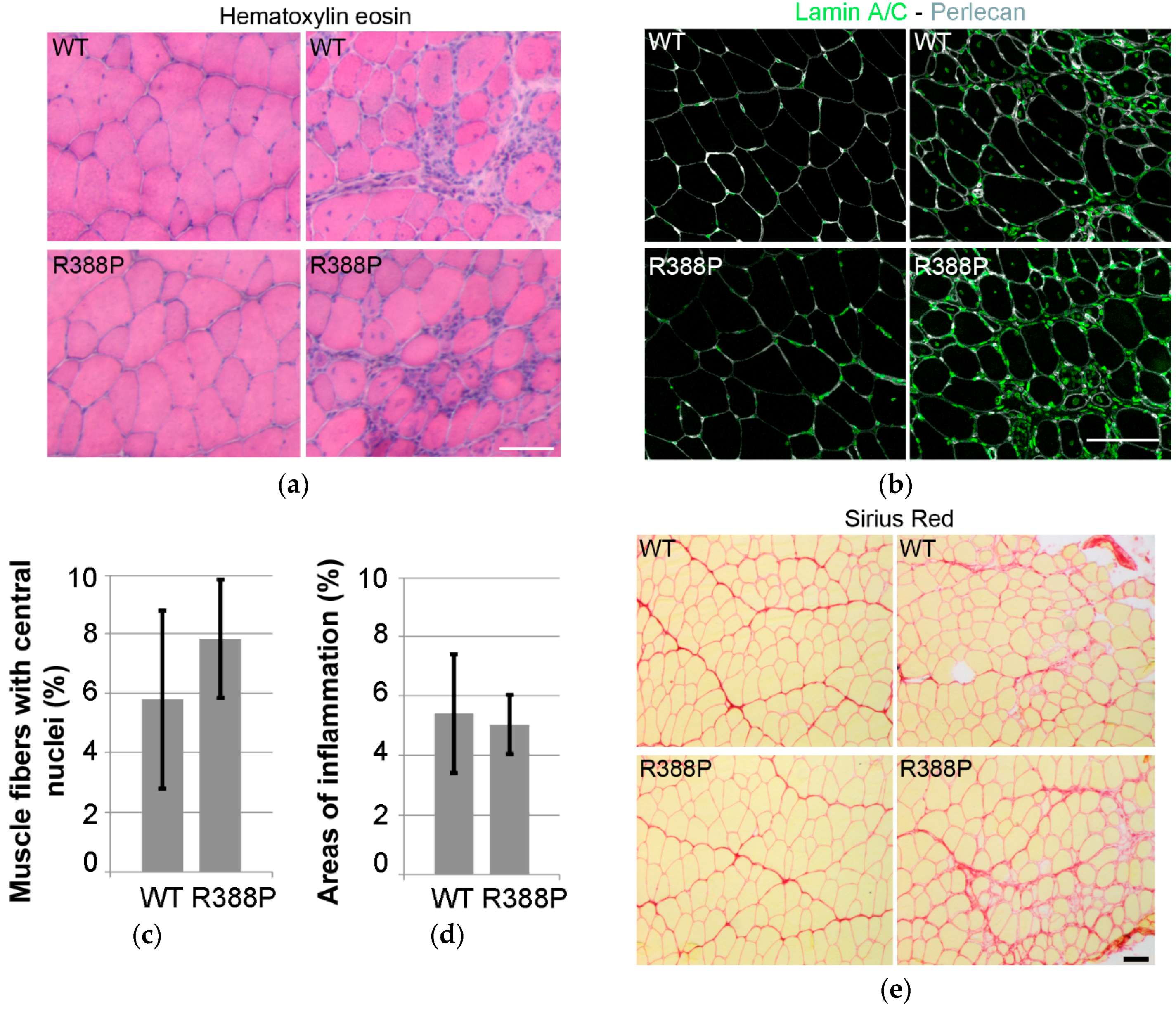
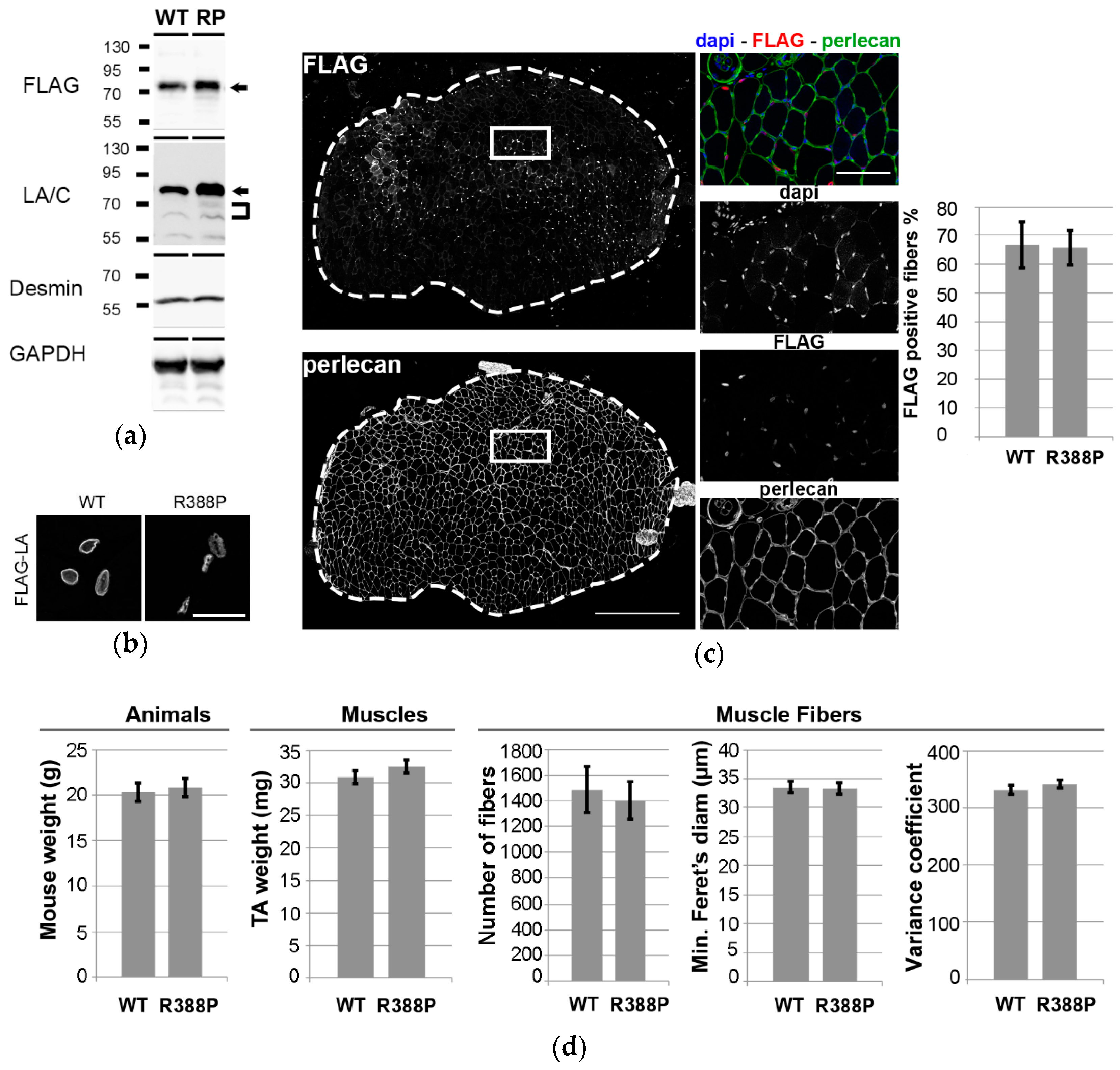
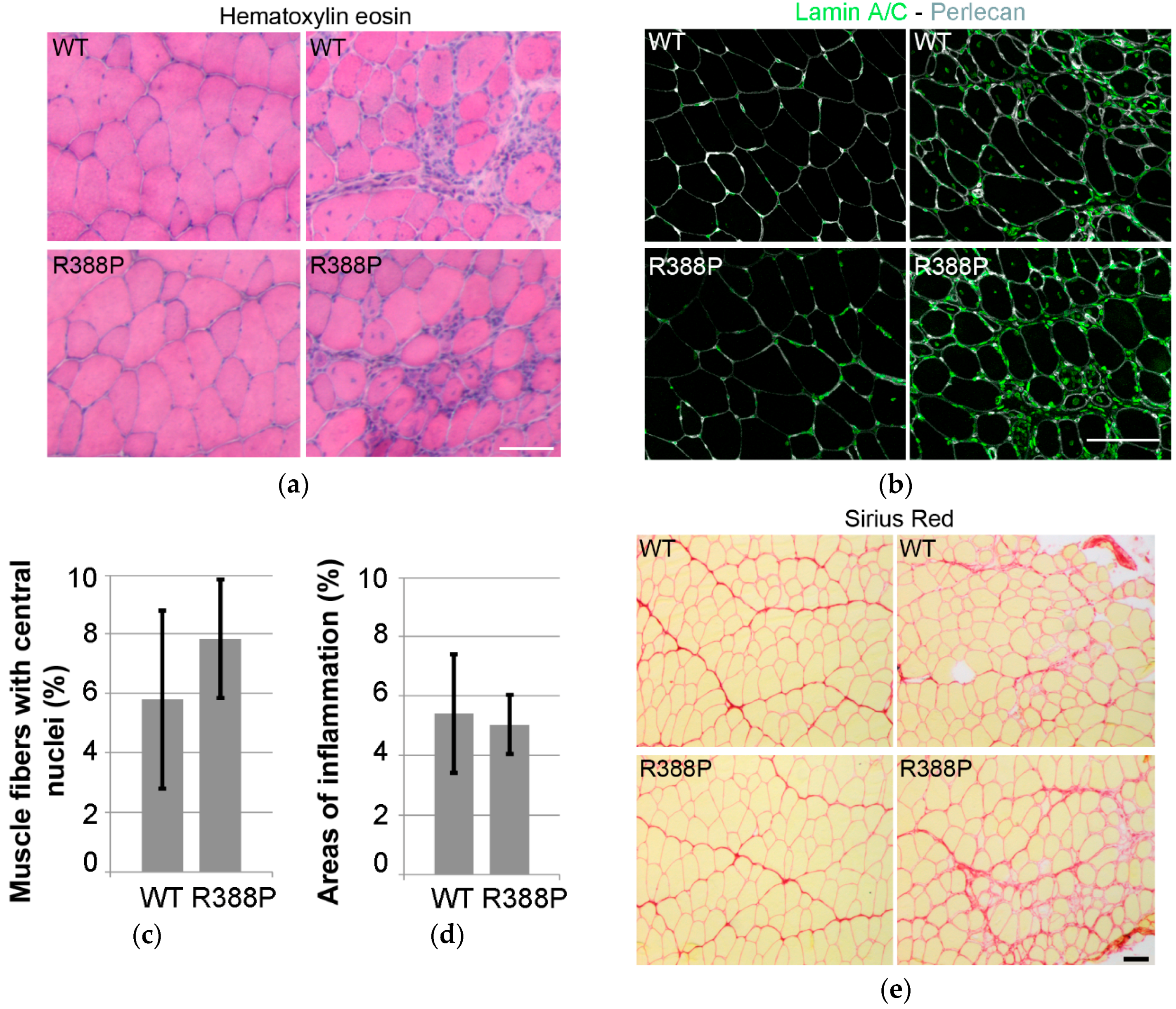
3.1. Similar Morphological and Histological Properties of TA Transduced with AAV-FLAG-LA WT or Mutant R388P
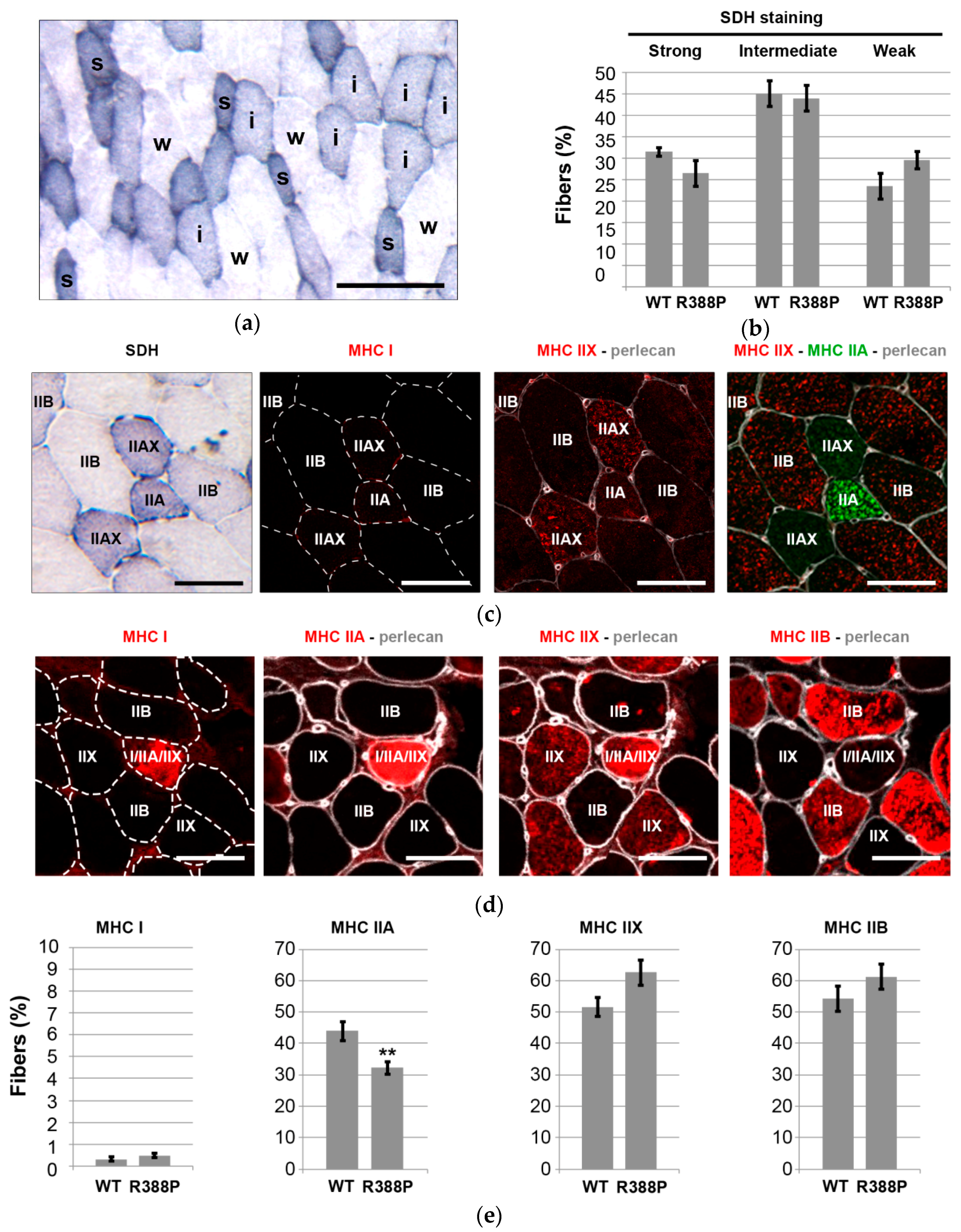
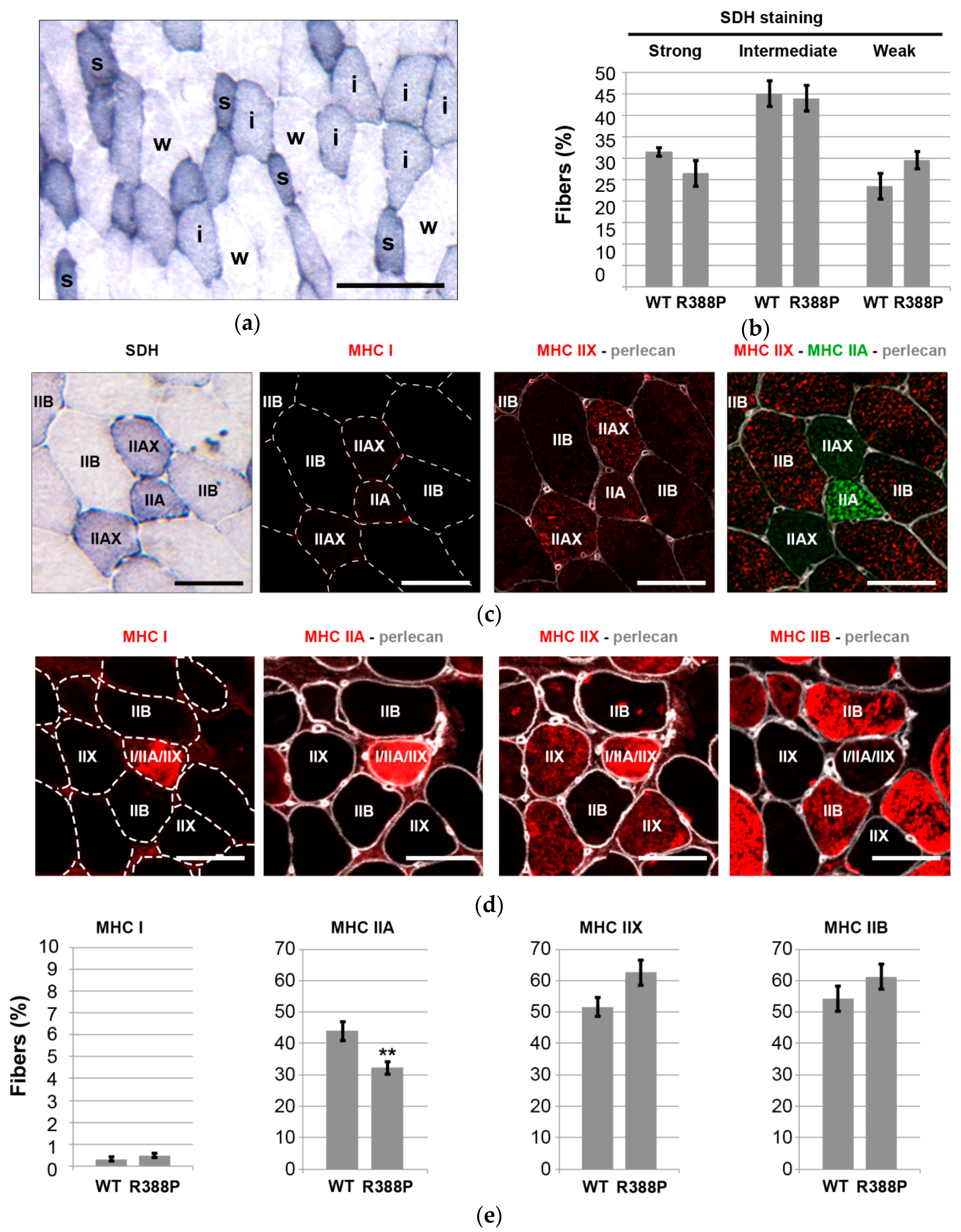
3.2. Expression of Mutant Lamin A Lowers the Frequency of Fast Oxidative Fibers in TA Muscle
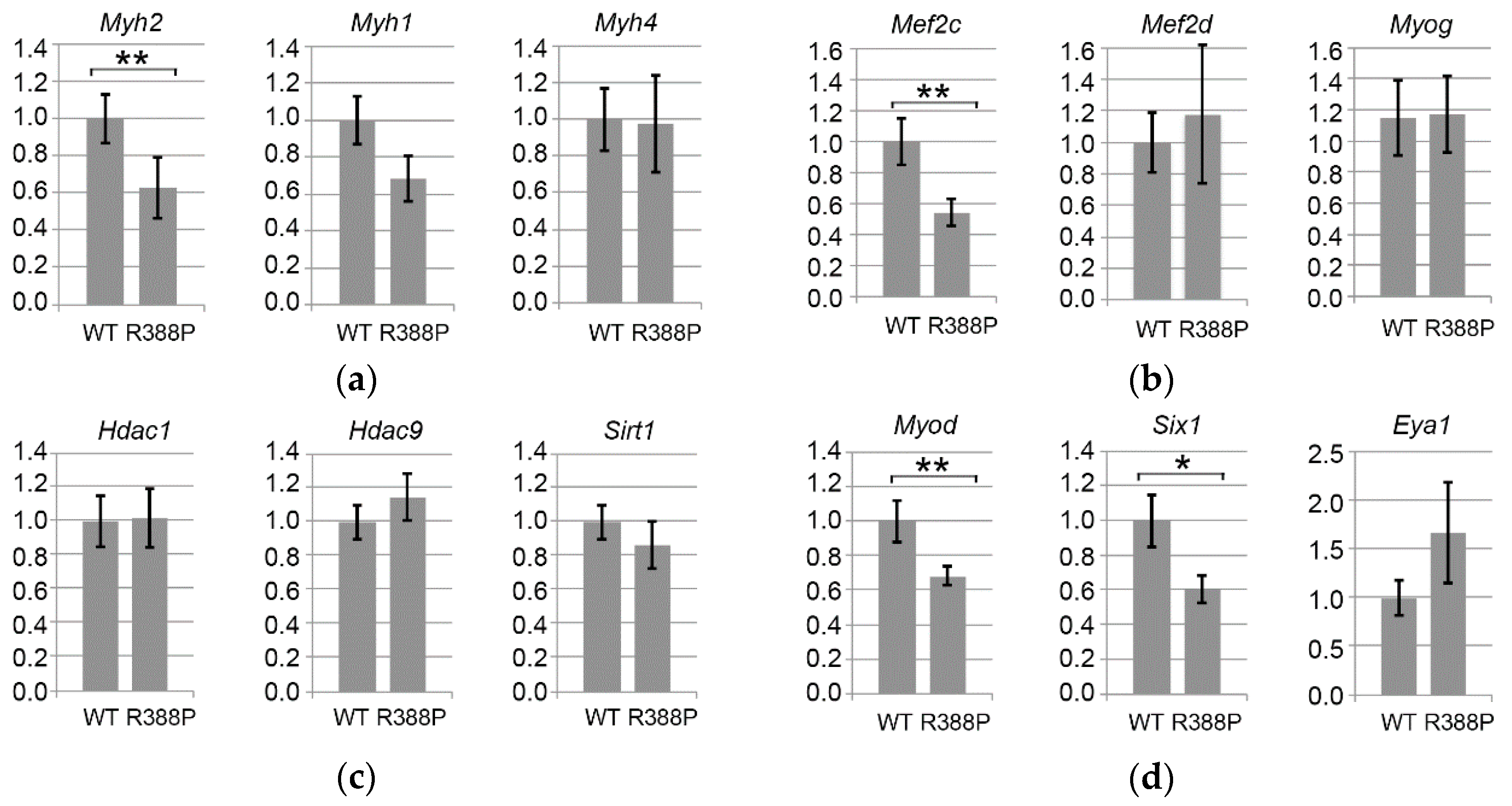
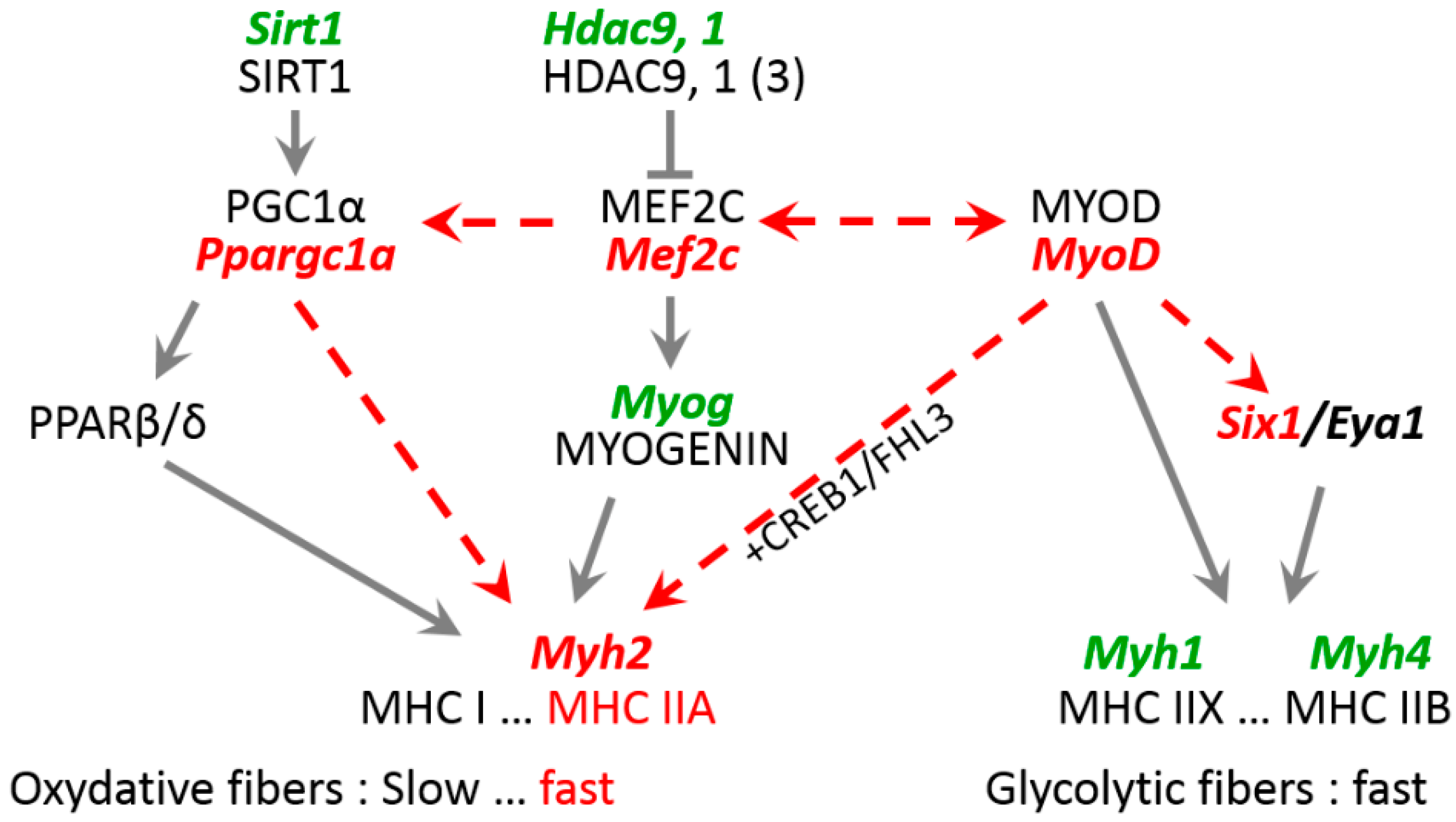
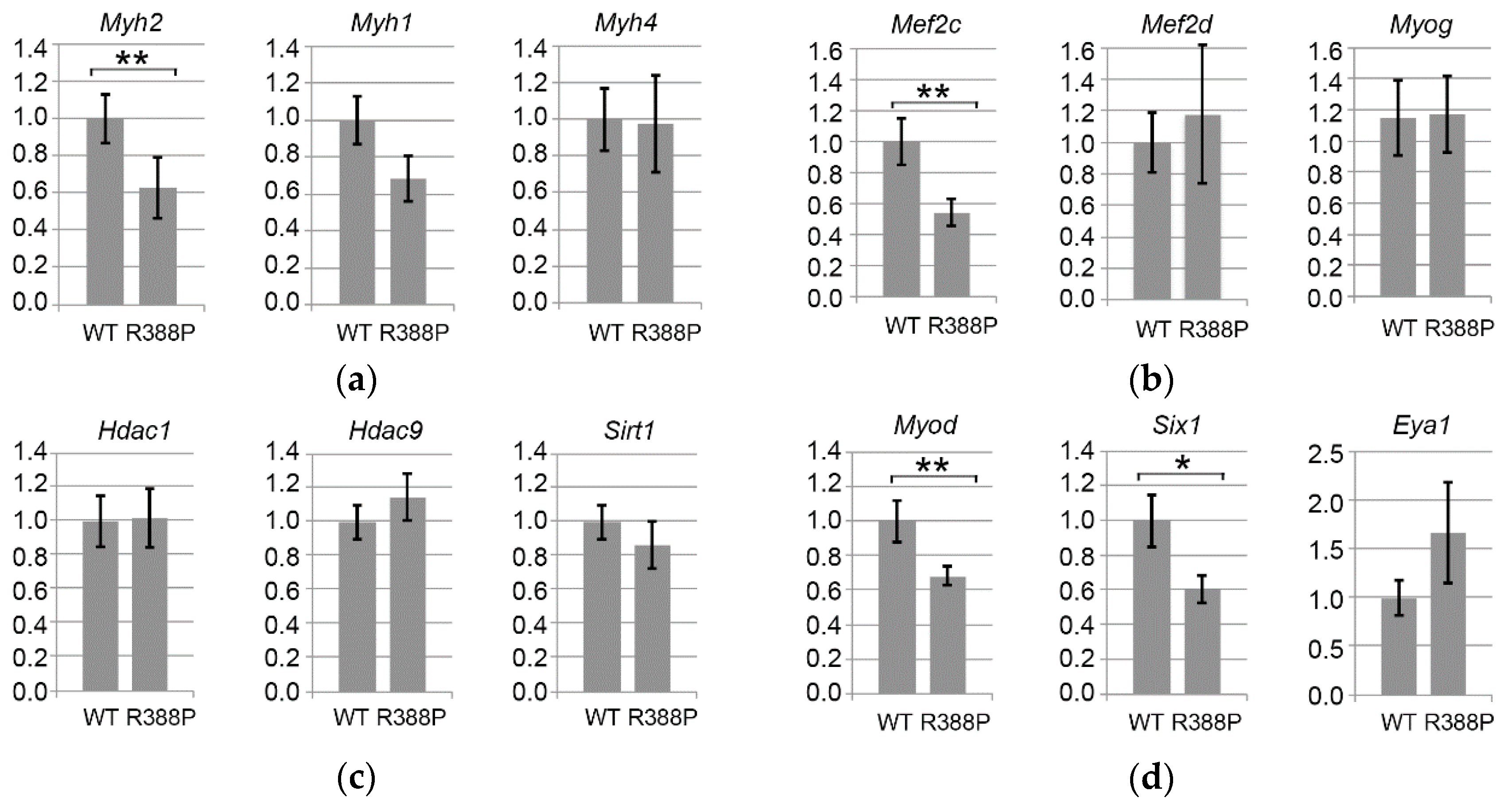
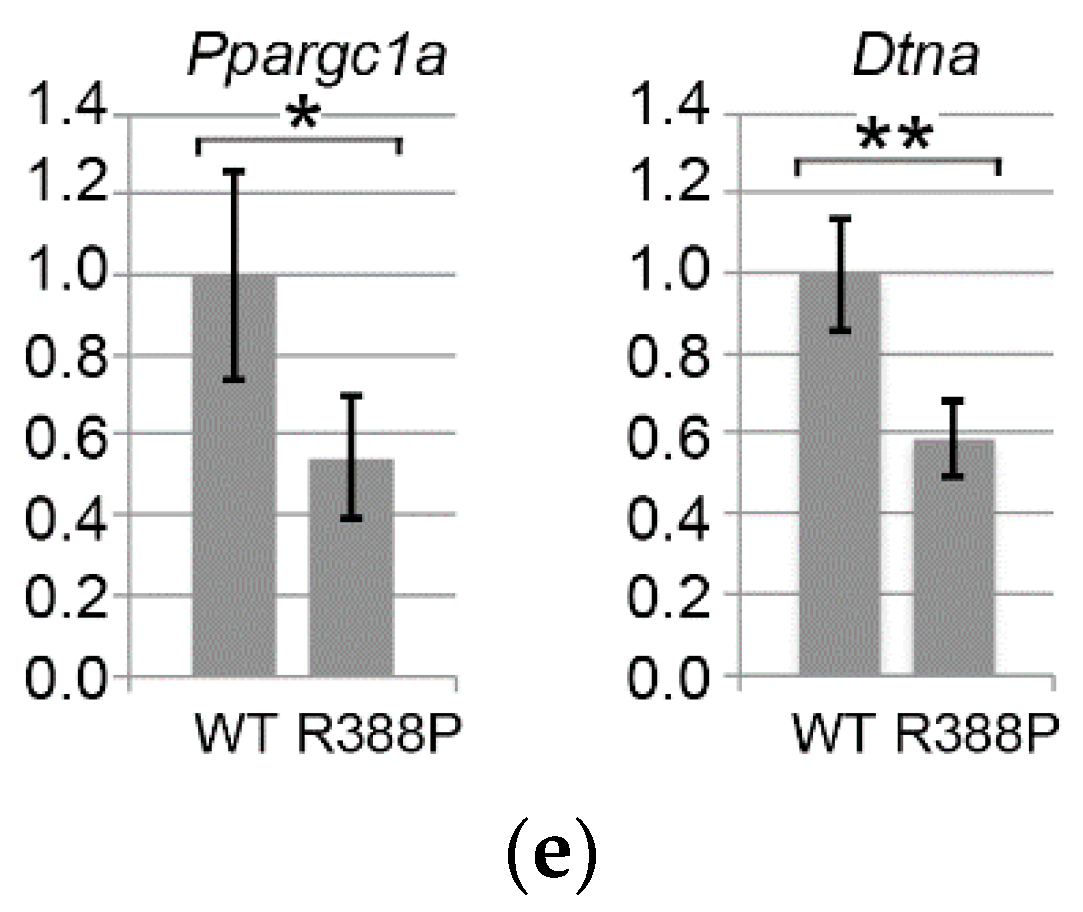
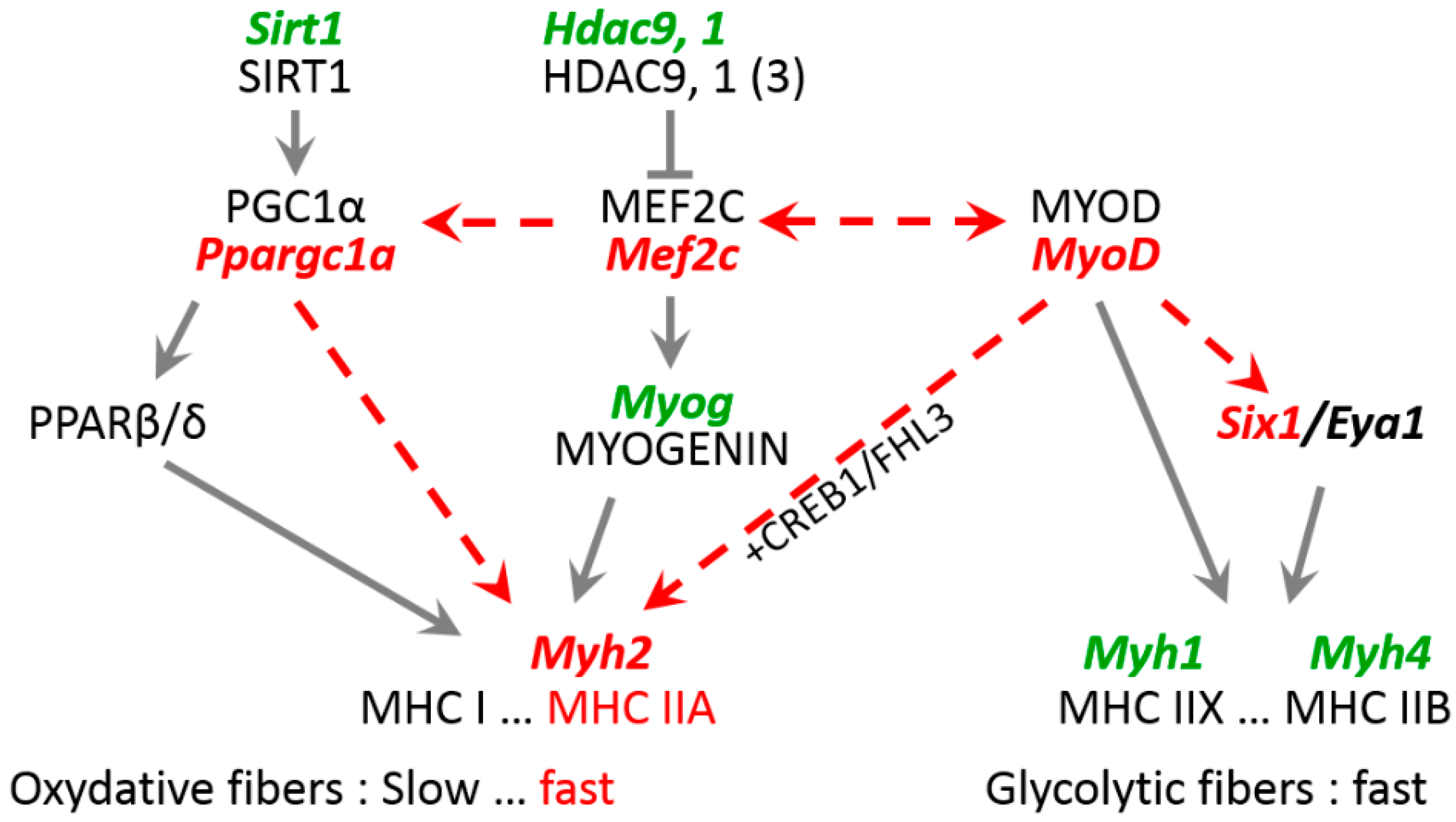
3.3. Expression of Mutant Lamin A Alters Expression of Genes Involved in Fiber Type Determination
3.4. Expression of Dystrobrevin α in TA Muscles Expressing WT or Mutant Lamin A
4. Discussion
4.1. Validation of the AAV Experimental Model to Explore Laminopathies
4.2. Role of Key Transcription Factors in Laminopathy Induced by R388P Mutant Lamin A
4.3. Mechanisms Underlying Deregulation of Key Genes upon Expression of R388P Mutant Lamin A
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kieckhaefer, J.E.; Cao, K. Mouse models of laminopathies. Aging Cell. 2013, 12, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Joanne, P.; Chourbagi, O.; Hourde, C.; Ferry, A.; Butler-Browne, G.; Vicart, P.; Dumonceaux, J.; Agbulut, O. Viral-mediated expression of desmin mutants to create mouse models of myofibrillar myopathy. Skelet. Muscle 2013, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, D.; Quadrilatero, J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Kajino, S.; Ishihara, K.; Goto, K.; Ishigaki, K.; Noguchi, S.; Nonaka, I.; Osawa, M.; Nishino, I.; Hayashi, Y.K. Congenital fiber type disproportion myopathy caused by LMNA mutations. J. Neurol. Sci. 2014, 340, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, L.; Fiorillo, C.; Tessa, A.; Manganelli, F.; Iodice, R.; Dubbioso, R.; Vitale, F.; Storti, E.; Soscia, E.; Santorelli, F.; et al. Muscle fiber type disproportion (FTD) in a family with mutations in the LMNA gene. Muscle Nerve 2015, 51, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [PubMed]
- Barateau, A.; Vadrot, N.; Vicart, P.; Ferreiro, A.; Mayer, M.; Héron, D.; Vigouroux, C.; Buendia, B. A novel lamin a mutant responsible for congenital muscular dystrophy causes distinct abnormalities of the cell nucleus. PLoS ONE 2017, 12, e0169189. [Google Scholar] [CrossRef] [PubMed]
- Favreau, C.; Higuet, D.; Courvalin, J.C.; Buendia, B. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol. Cell. Biol. 2004, 24, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Estrella, N.L.; Desjardins, C.A.; Nocco, S.E.; Clark, A.L.; Maksimenko, Y.; Naya, F.J. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem. 2015, 290, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Macpherson, P.; Marvin, M.; Meadows, E.; Klein, W.H.; Yang, X.J.; Goldman, D. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol. Biol. Cell 2009, 20, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Mejat, A.; Ramond, F.; Bassel-Duby, R.; Khochbin, S.; Olson, E.N.; Schaeffer, L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat. Neurosci. 2005, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sternsdorf, T.; Bolger, T.A.; Evans, R.M.; Yao, T.P. Regulation of MEF2 by histone deacetylase 4-and SIRT1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 2005, 25, 8456–8464. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, K. Excitation-transcription coupling in skeletal muscle: The molecular pathways of exercise. Biol. Rev. Camb. Philos. Soc. 2011, 86, 564–600. [Google Scholar] [CrossRef] [PubMed]
- Grifone, R.; Laclef, C.; Spitz, F.; Lopez, S.; Demignon, J.; Guidotti, J.E.; Kawakami, K.; Xu, P.X.; Kelly, R.; Petrof, B.J.; et al. Six1 and Eya1 expression can reprogram adult muscle from the slow-twitch phenotype into the fast-twitch phenotype. Mol. Cell. Biol. 2004, 24, 6253–6267. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.L.; Lien, C.F.; Gorecki, D.C. Dystrobrevins in muscle and non-muscle tissues. Neuromuscul. Disord. 2007, 17, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.; Sekelja, M.; Oldenburg, A.; Barateau, A.; Briand, N.; Delbarre, E.; Shah, A.; Sorensen, A.; Vigouroux, C.; Buendia, B.; et al. Chrom3D: Three dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 2017, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Requena, T.; Cabrera, S.; Martin-Sierra, C.; Price, S.D.; Lysakowski, A.; Lopez-Escamez, J.A. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum. Mol. Genet. 2015, 24, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Haenggi, T.; Fritschy, J.M. Role ofrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell. Mol. Life Sci. 2006, 63, 1614–1631. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Kumar, A. Therapeutic targeting of signaling pathways in muscular dystrophy. J. Mol. Med. (Berl.) 2010, 88, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Collas, P.; Lund, E.G.; Oldenburg, A.R. Closing the (nuclear) envelope on the genome: How nuclear lamins interact with promoters and modulate gene expression. Bioessays 2013, 36, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Duband-Goulet, I.; Oldenburg, A.; Buendia, B.; Collas, P. Distinct features of lamin A-interacting chromatin domains mapped by ChIP-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus 2015, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma. 2013, 122, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ghosh, S.; Yang, X.; Zheng, H.; Liu, X.; Wang, Z.; Jin, G.; Zheng, B.; Kennedy, B.K.; Suh, Y.; et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell. Metab. 2012, 16, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 2013, 4, 1868. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′… 3′) | Reverse Primer (5′… 3′) |
|---|---|---|
| 18S rRNA | CGGCTACCACATCCAAGGAA | TATACGCTATTGGAGCTGGAA |
| Myh2 | AAAGCTCCAAGGACCCTCTT | AGCTCATGACTGCTGAACTC |
| Myh1 | CCAAGGAGGAGGAACAGCAG | TTTCGTCTAGCTGGCGTGAG |
| Myh4 | GTCCTTCCTCAAACCCTTAAAGT | ATCTCAGCGTCGGAACTCAT |
| Mef2c | AGAGTTTGGACAACAAAGCCC | CACGCTTCACTTCATCTCTCC |
| Mef2d | AGGGAGGCAAAGGGTTAATGC | CCTGGCTGAGTAAACTTGGTG |
| Myog | GTCCCAACCCAGGAGATCAT | CTGTCCACGATGGACGTAAG |
| Myod | CTGCTCTGATGGCATGATGG | TGTAGTAGGCGGTGTCGTAG |
| Six1 | AAGAACGAGAGCGTGCTCAA | CATCCCTTCAAGGCCCCAAT |
| Eya1 | TGGCCCTACCCCTTCCCCAC | TGACAATCCACTTTCCGTCTT |
| Hdac1 | TACTACGACGGGGATGTTGGAAAC | TCCTCAGCATTGGCTTTGTGA |
| Hdac9 | GCGGTCCAGGTTAAAACAGA | GAGCTGAAGCCTCATTTTCG |
| Sirt1 | AGAACCACCAAAGCGGAAA | TCCCACAGGAGACAGAAACC |
| Ppargc1a | CGGAAATCATATCCAACCAG | TGAGGACCGCTAGCAAGTTTG |
| Dtna | GCAGAGATGAGGGCTCAAG | TGACGTTCCAAATGTCCACC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barateau, A.; Vadrot, N.; Agbulut, O.; Vicart, P.; Batonnet-Pichon, S.; Buendia, B. Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin A Responsible for Congenital Muscular Dystrophy in a Patient. Cells 2017, 6, 10. https://doi.org/10.3390/cells6020010
Barateau A, Vadrot N, Agbulut O, Vicart P, Batonnet-Pichon S, Buendia B. Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin A Responsible for Congenital Muscular Dystrophy in a Patient. Cells. 2017; 6(2):10. https://doi.org/10.3390/cells6020010
Chicago/Turabian StyleBarateau, Alice, Nathalie Vadrot, Onnik Agbulut, Patrick Vicart, Sabrina Batonnet-Pichon, and Brigitte Buendia. 2017. "Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin A Responsible for Congenital Muscular Dystrophy in a Patient" Cells 6, no. 2: 10. https://doi.org/10.3390/cells6020010
APA StyleBarateau, A., Vadrot, N., Agbulut, O., Vicart, P., Batonnet-Pichon, S., & Buendia, B. (2017). Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin A Responsible for Congenital Muscular Dystrophy in a Patient. Cells, 6(2), 10. https://doi.org/10.3390/cells6020010