
Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout



Abstract
:1. Introduction
2. Materials and Methods
2.1. CRISPR/Cas9 Plasmids for FLOT1 and AGA
2.2. Cell Culture and Transfections
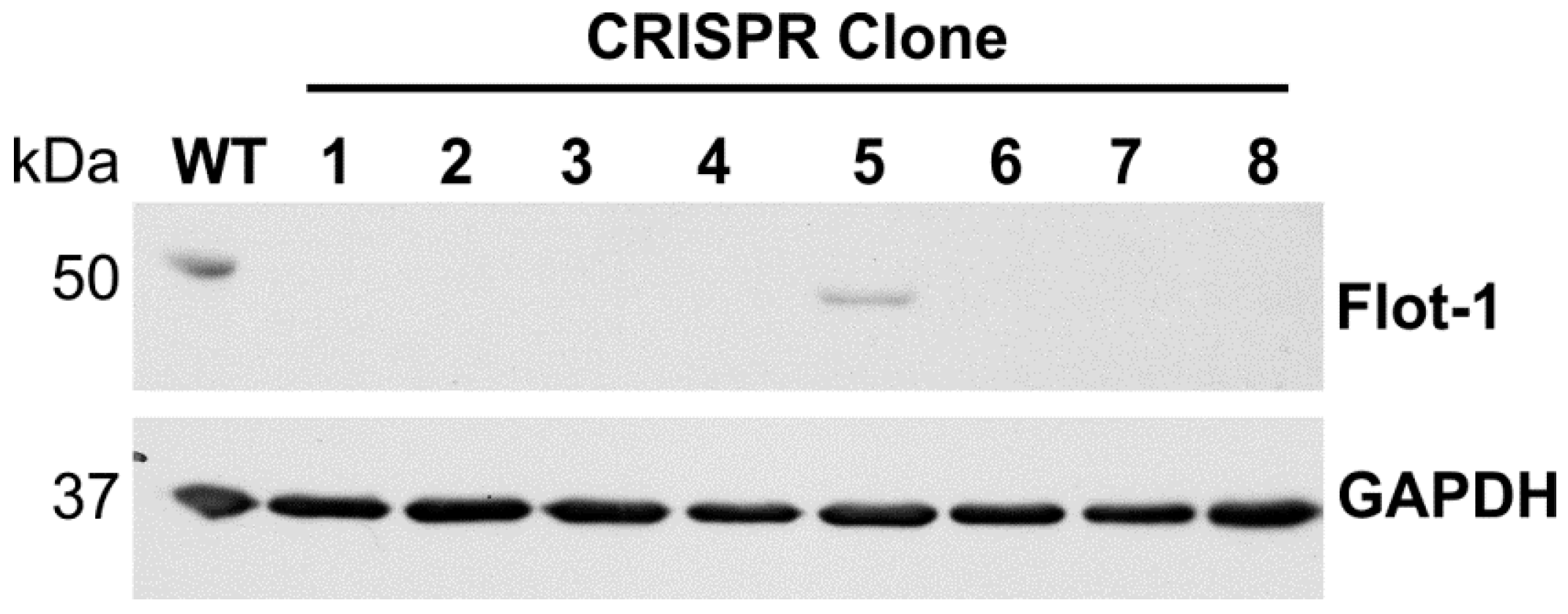
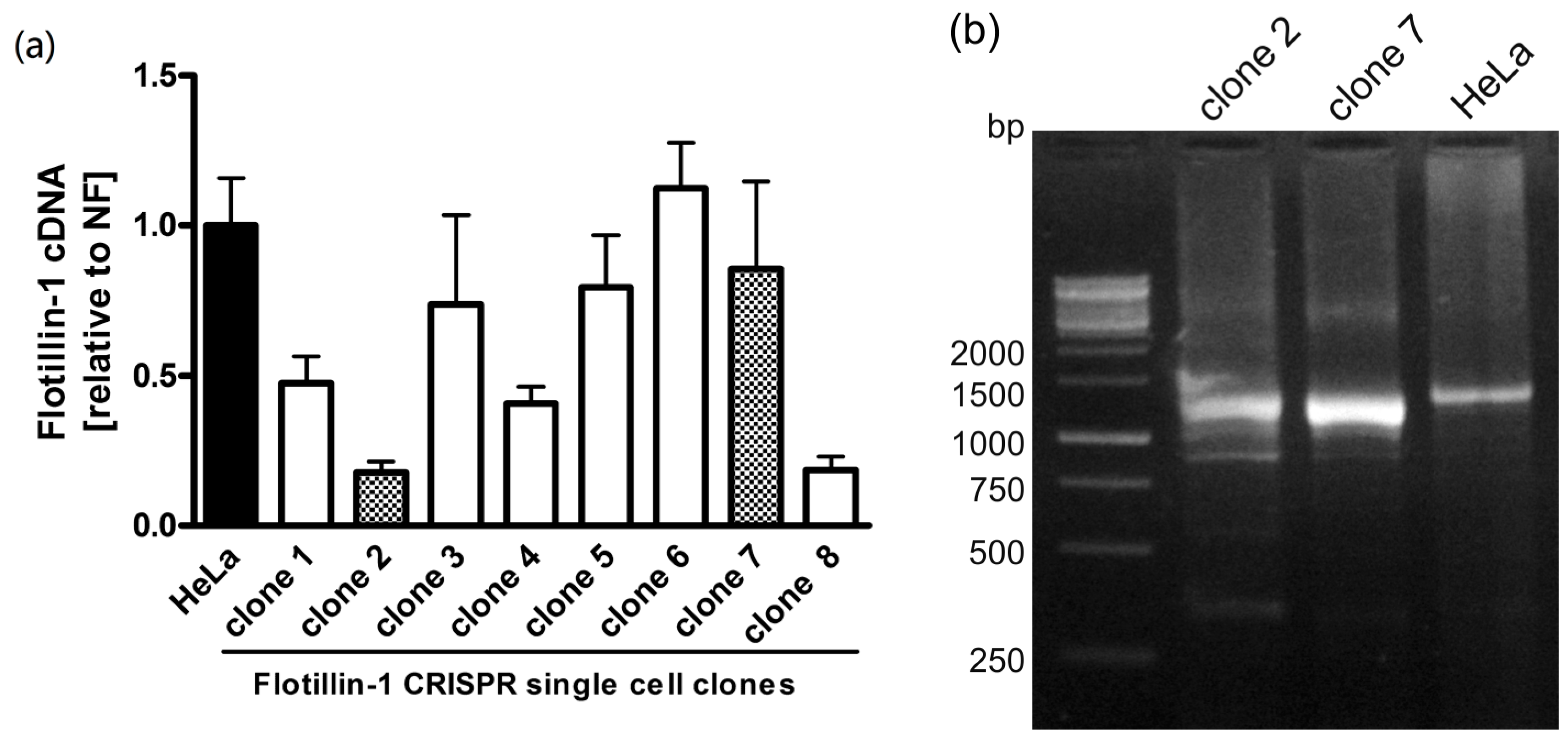
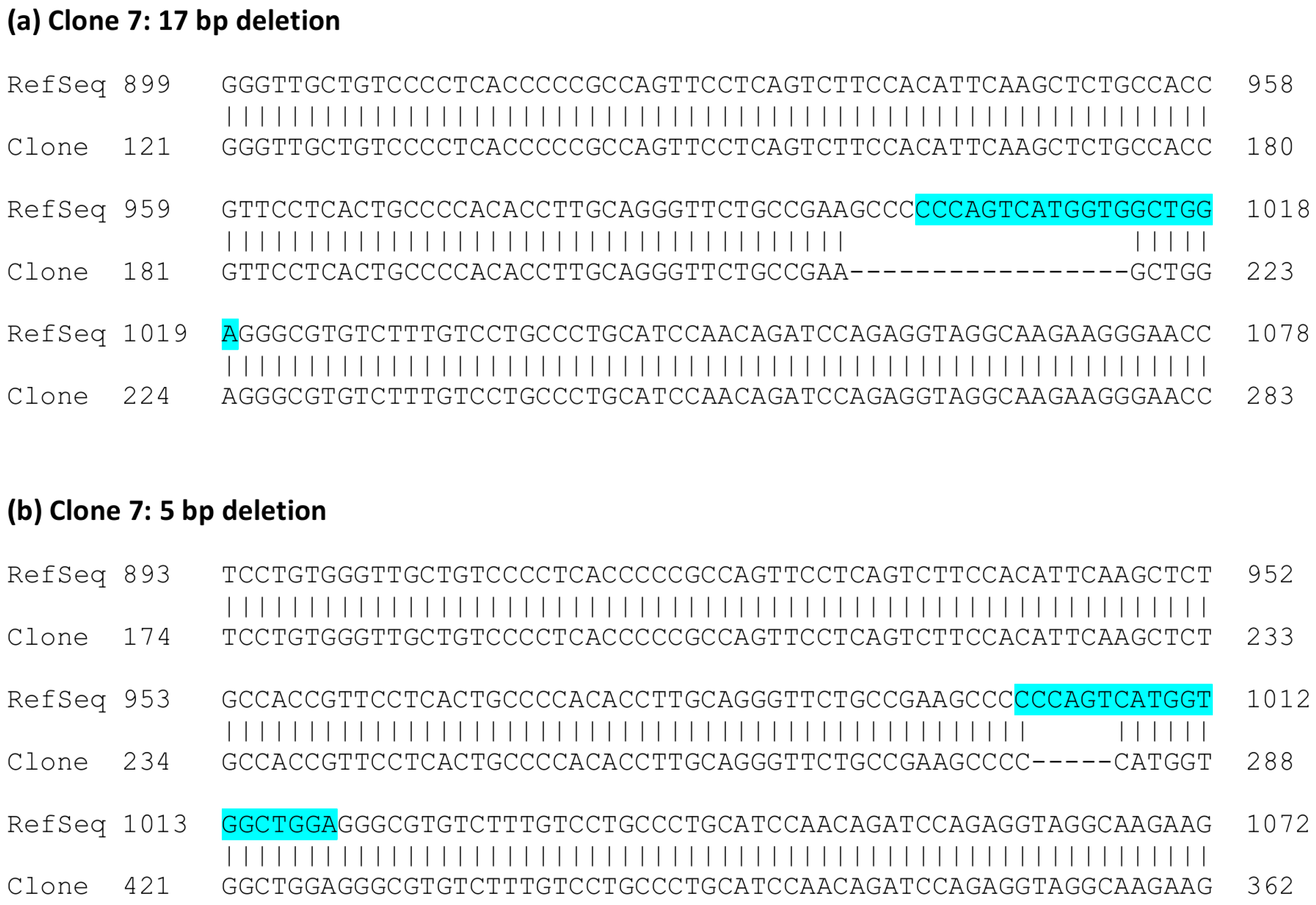
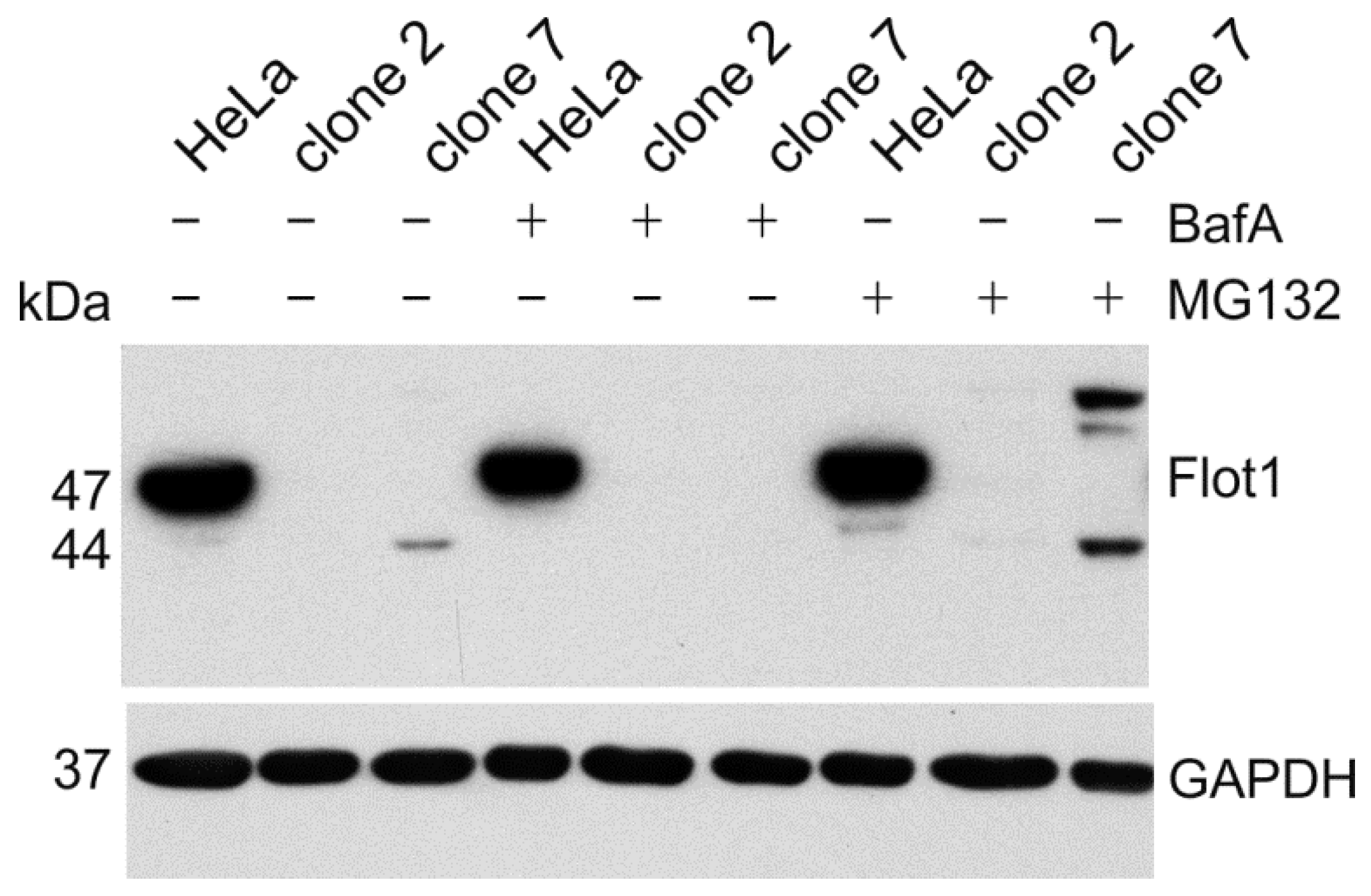
2.3. Analysis of Single-Cell Clones
2.4. Quantitative Real-Time PCR
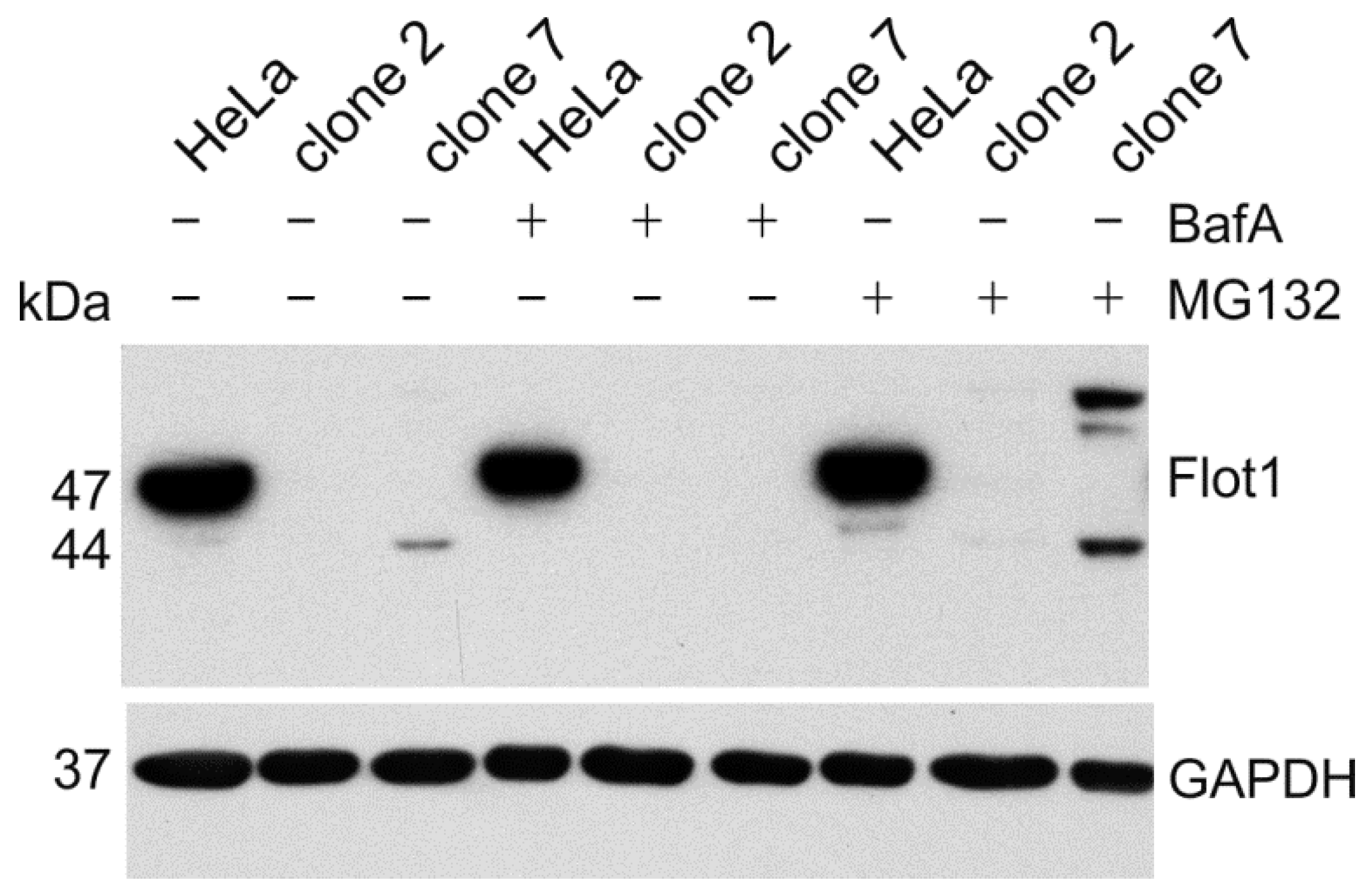
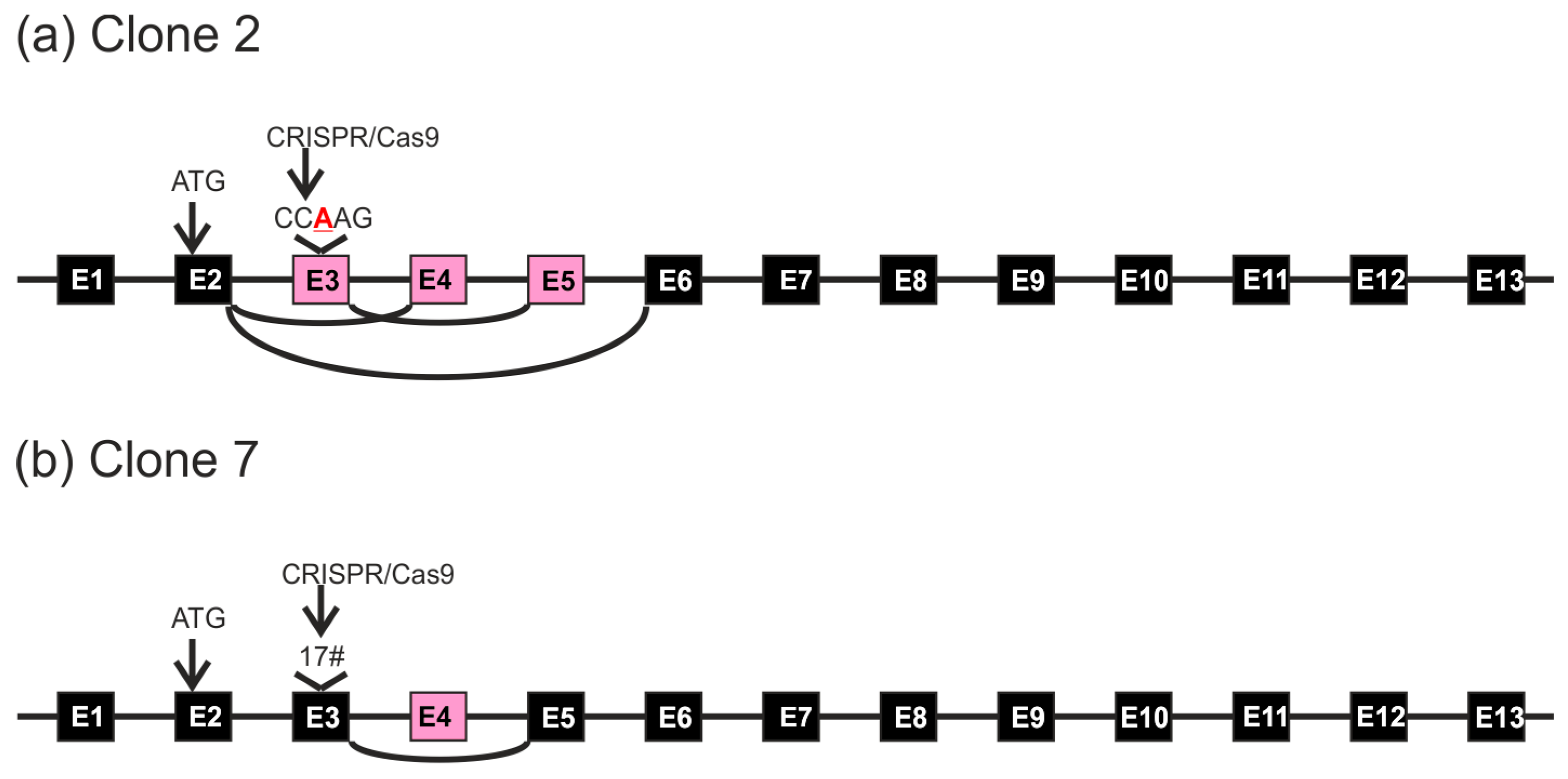
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGA: | Aspartylglucosaminidase |
| Cas9: | CRISPR associated sequence 9 |
| CRISPR: | Clustered regularly interspaced short palindromic repeats |
| DMEM: | Dulbecco’s modified Eagle’s medium |
| EGFR: | Epidermal Growth Factor Receptor |
| ESE: | Exonic splicing enhancers |
| ESS: | Exonic splicing silencers |
| FCS: | Fetal calf serum |
| gRNA: | Guide RNA |
| hnRNA: | Heteronuclear RNA |
| indels: | Insertions/deletions |
| MAPK: | Mitogen-activated protein kinase |
| mRNA: | Messenger RNA |
| NHEJ: | Non-homologous-end-joining |
| shRNA: | Small hairpin RNA |
| siRNA: | Small interfering RNA |
| qPCR: | Quantitative PCR |
| WT: | Wildtype |
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [PubMed]
- Banning, A.; Kurrle, N.; Meister, M.; Tikkanen, R. Flotillins in receptor tyrosine kinase signaling and cancer. Cells 2014, 3, 129–149. [Google Scholar] [PubMed]
- Meister, M.; Tomasovic, A.; Banning, A.; Tikkanen, R. Mitogen-activated protein (MAP) kinase scaffolding proteins: A recount. Int. J. Mol. Sci. 2013, 14, 4854–4884. [Google Scholar] [PubMed]
- Amaddii, M.; Meister, M.; Banning, A.; Tomasovic, A.; Mooz, J.; Rajalingam, K.; Tikkanen, R. Flotillin-1/reggie-2 protein plays dual role in activation of receptor-tyrosine kinase/mitogen-activated protein kinase signaling. J. Biol. Chem. 2012, 287, 7265–7278. [Google Scholar] [PubMed]
- Banning, A.; Ockenga, W.; Finger, F.; Siebrasse, P.; Tikkanen, R. Transcriptional regulation of flotillins by the extracellularly regulated kinases and retinoid X receptor complexes. PLoS ONE 2012, 7, e45514. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, S.; Ockenga, W.; Banning, A.; Tikkanen, R. Cholinergic Transactivation of the EGFR in HaCaT Keratinocytes Stimulates a Flotillin-1 Dependent MAPK-Mediated Transcriptional Response. Int. J. Mol. Sci. 2015, 16, 6447–6463. [Google Scholar] [CrossRef] [PubMed]
- Edgar, A.J.; Polak, J.M. Flotillin-1: Gene structure: cDNA cloning from human lung and the identification of alternative polyadenylation signals. Int. J. Biochem. Cell Biol. 2001, 33, 53–64. [Google Scholar] [CrossRef]
- Kurrle, N.; Vollner, F.; Eming, R.; Hertl, M.; Banning, A.; Tikkanen, R. Flotillins directly interact with gamma-catenin and regulate epithelial cell-cell adhesion. PLoS ONE 2013, 8, e84393. [Google Scholar] [CrossRef] [PubMed]
- Vollner, F.; Ali, J.; Kurrle, N.; Exner, Y.; Eming, R.; Hertl, M.; Banning, A.; Tikkanen, R. Loss of flotillin expression results in weakened desmosomal adhesion and Pemphigus vulgaris-like localisation of desmoglein-3 in human keratinocytes. Sci. Rep. 2016, 6, 28820. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Gulec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of small molecule compounds for pharmacological chaperone therapy of aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; Laine, M.; Oinonen, C.; von Schantz, C.; Jalanko, A.; Rouvinen, J.; Peltonen, L. Molecular pathogenesis of a disease: Structural consequences of aspartylglucosaminuria mutations. Hum. Mol. Genet. 2001, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Charpentier, E. Toward whole-transcriptome editing with CRISPR-Cas9. Mol. Cell 2015, 58, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Bosley, K.S.; Botchan, M.; Bredenoord, A.L.; Carroll, D.; Charo, R.A.; Charpentier, E.; Cohen, R.; Corn, J.; Doudna, J.; Feng, G.; et al. CRISPR germline engineering--the community speaks. Nat. Biotechnol. 2015, 33, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E. CRISPR-Cas9: How research on a bacterial RNA-guided mechanism opened new perspectives in biotechnology and biomedicine. EMBO Mol. Med. 2015, 7, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Disset, A.; Bourgeois, C.F.; Benmalek, N.; Claustres, M.; Stevenin, J.; Tuffery-Giraud, S. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum. Mol. Genet. 2006, 15, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.N.; Pearce, D.A. Nonsense-mediated decay in genetic disease: Friend or foe? Mutat. Res. Rev. Mutat. Res. 2014, 762, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Morrow, I.C.; Rea, S.; Martin, S.; Prior, I.A.; Prohaska, R.; Hancock, J.F.; James, D.E.; Parton, R.G. Flotillin-1/reggie-2 traffics to surface raft domains via a novel golgi-independent pathway. Identification of a novel membrane targeting domain and a role for palmitoylation. J. Biol. Chem. 2002, 277, 48834–48841. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Deyoung, S.M.; Zhang, M.; Dold, L.H.; Saltiel, A.R. The stomatin/prohibitin/flotillin/HflK/C domain of flotillin-1 contains distinct sequences that direct plasma membrane localization and protein interactions in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 16125–16134. [Google Scholar] [CrossRef] [PubMed]




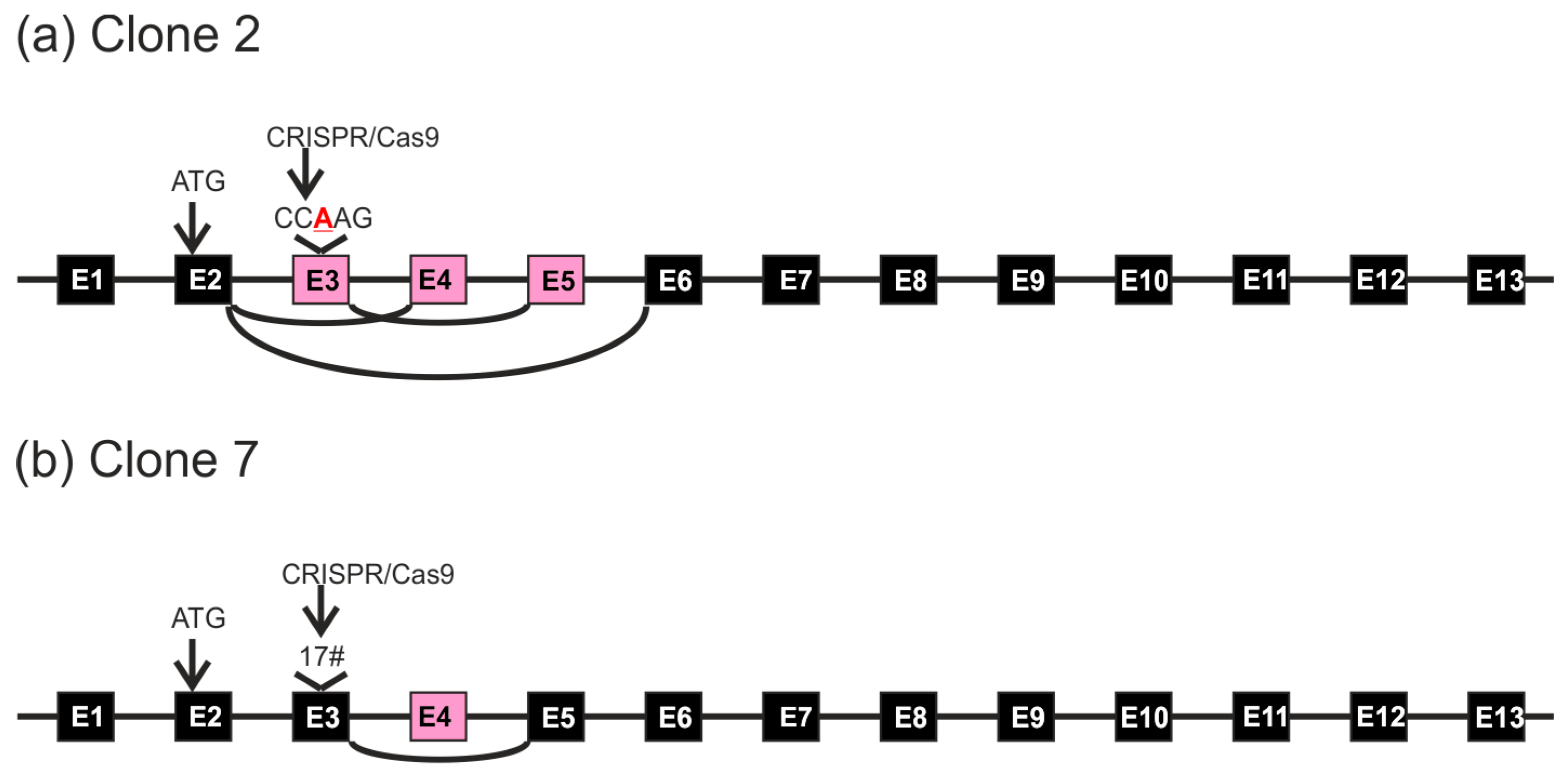

{kind=link}
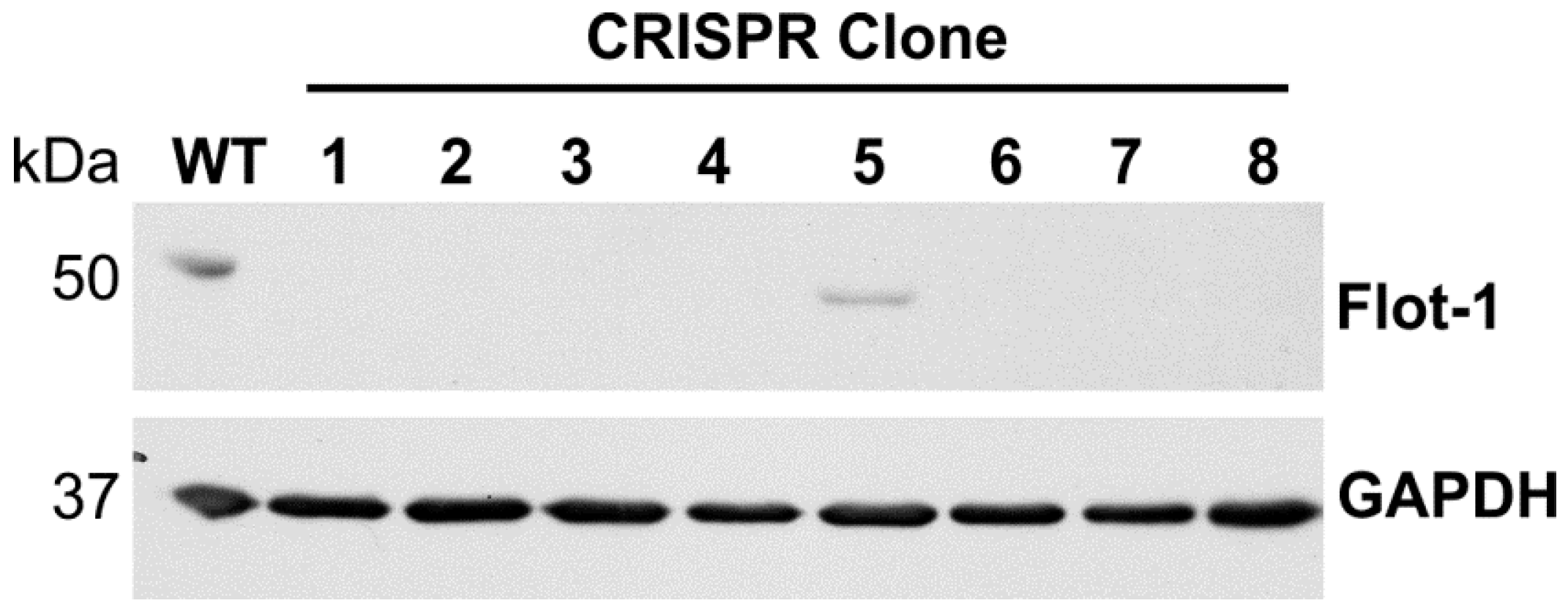
{kind=link}
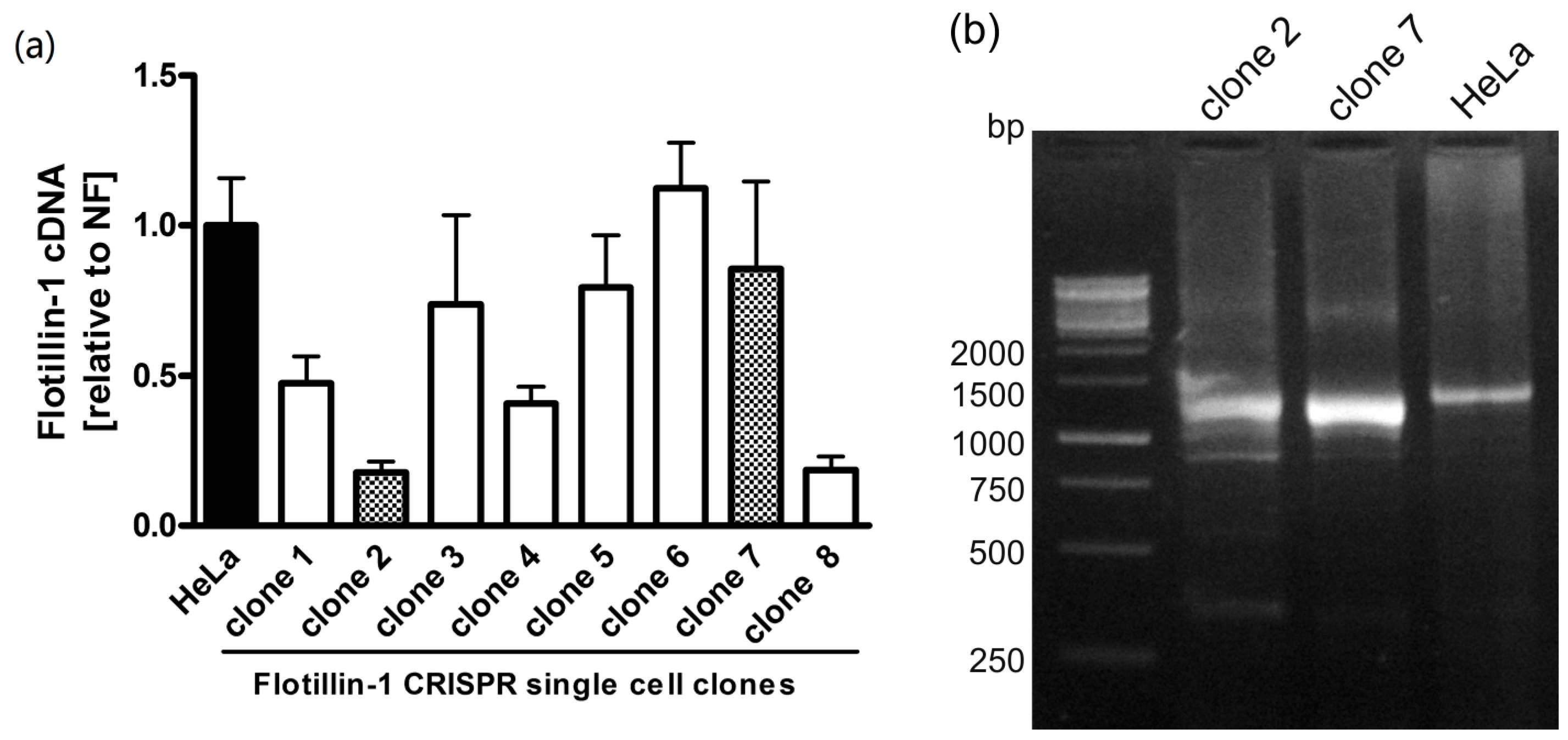
{kind=link}
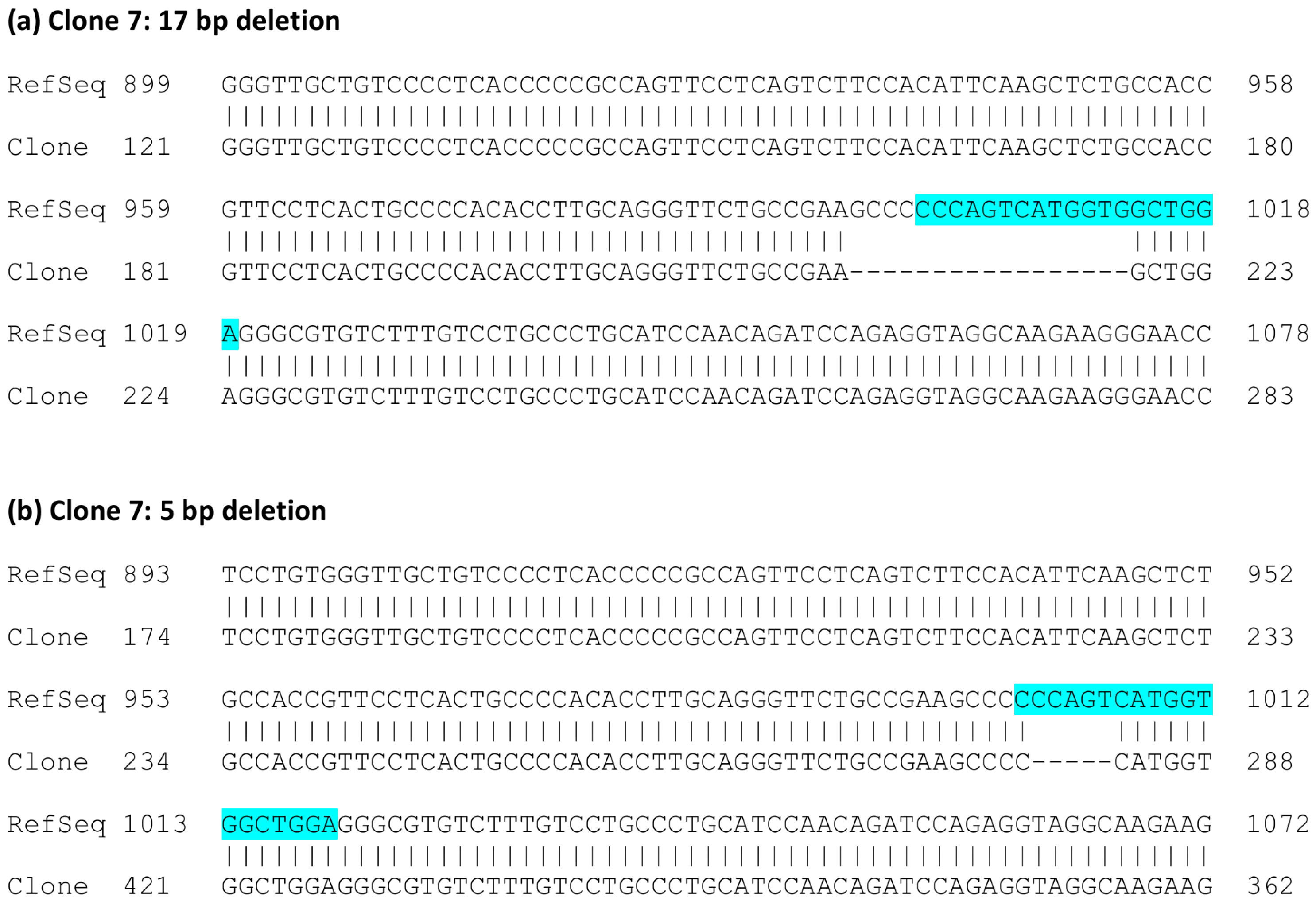{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guide ID | Sequence 5′-3′ | Target Exon |
|---|---|---|
| 165719 | ATGGTTCAGGCTGGAGCTGG | 2 |
| 165720 | TTTCACTTGTGGCCCAAATG | 2 |
| 165721 | GTACTTACCGGAGACCACCA | 2 |
| 165722 | GCAGAACCCTGCAAGGTGTG | 3 |
| 165723 | CTCCAGCCACCATGACTGGG | 3 |
| Primer | Sequence 5′-3′ |
|---|---|
| Flot-1-genomic-fwd | CTATAGGTACCTCCCTCTCCCTACCAACTTCCC |
| Flot-1-genomic-rev | CTATAGAATTCTGCAGGCAAGGGTTGAGAAGAC |
| Flot-1-cDNA-fwd | CTATAGGTACCATGTTTTTCACTTGTGGCCC |
| Flot-1-cDNA-rev | CTATAGAATTCCCGGCTGTTCTCAAAGGCTTG |
| AGA-pcDNA3-fwd | CTATAGGATCCATGGCGCGGAAGTCGAACTTG |
| AGA-pcDNA3-rev | CTATACTCGAGTTAGATGCAGTCCACTTTTTCC |
| Target Gene | Primer Name | Sequence 5′-3′ |
|---|---|---|
| Flot-1 | Flot1-exon10-fwd | TATGCAGGCGGAGGCAGAAG |
| Flot-1 | Flot1-exon12-rev | CAGTGTGATCTTATTGGCTGAA |
| Rpl13a | Rpl13a-fwd | CCTGGAGGAGAAGAGGAAAGAGA |
| Rpl13a | Rpl13a-rev | TTGAGGACCTCTGTGTATTTGTCAA |
| B2M | B2M-fwd | AGATGAGTATGCCTGCCGTGTG |
| B2M | B2M-rev | TGCGGCATCTTCAAACCTCCA |
| Ywhaz | Ywhaz-fwd | AGGTTGCCGCTGGTGATGAC |
| Ywhaz | Ywhaz-rev | GGCCAGACCCAGTCTGATAGGA |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapahnke, M.; Banning, A.; Tikkanen, R. Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout. Cells 2016, 5, 45. https://doi.org/10.3390/cells5040045
Kapahnke M, Banning A, Tikkanen R. Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout. Cells. 2016; 5(4):45. https://doi.org/10.3390/cells5040045
Chicago/Turabian StyleKapahnke, Marcel, Antje Banning, and Ritva Tikkanen. 2016. "Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout" Cells 5, no. 4: 45. https://doi.org/10.3390/cells5040045
APA StyleKapahnke, M., Banning, A., & Tikkanen, R. (2016). Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout. Cells, 5(4), 45. https://doi.org/10.3390/cells5040045