
Advances in Autophagy Regulatory Mechanisms

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
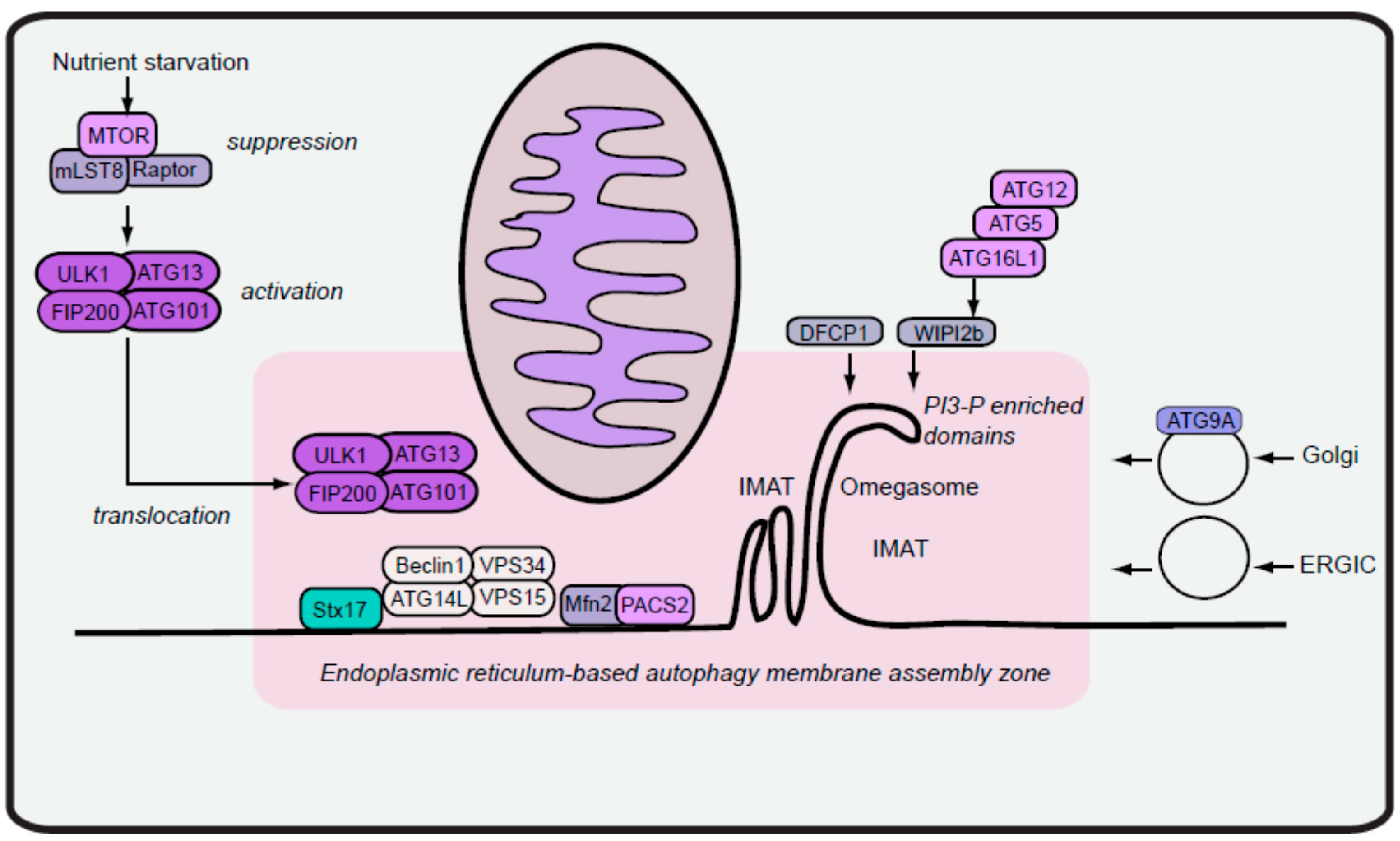
2. Formation of Autophagy Isolation Membranes
2.1. Early Isolation Membranes from ER-Mitochondria Contacts
2.2. Maturation of Isolation Membranes via Vesicles from ER Exit Sites
2.3. Outlook on Autophagy Membrane Initiation
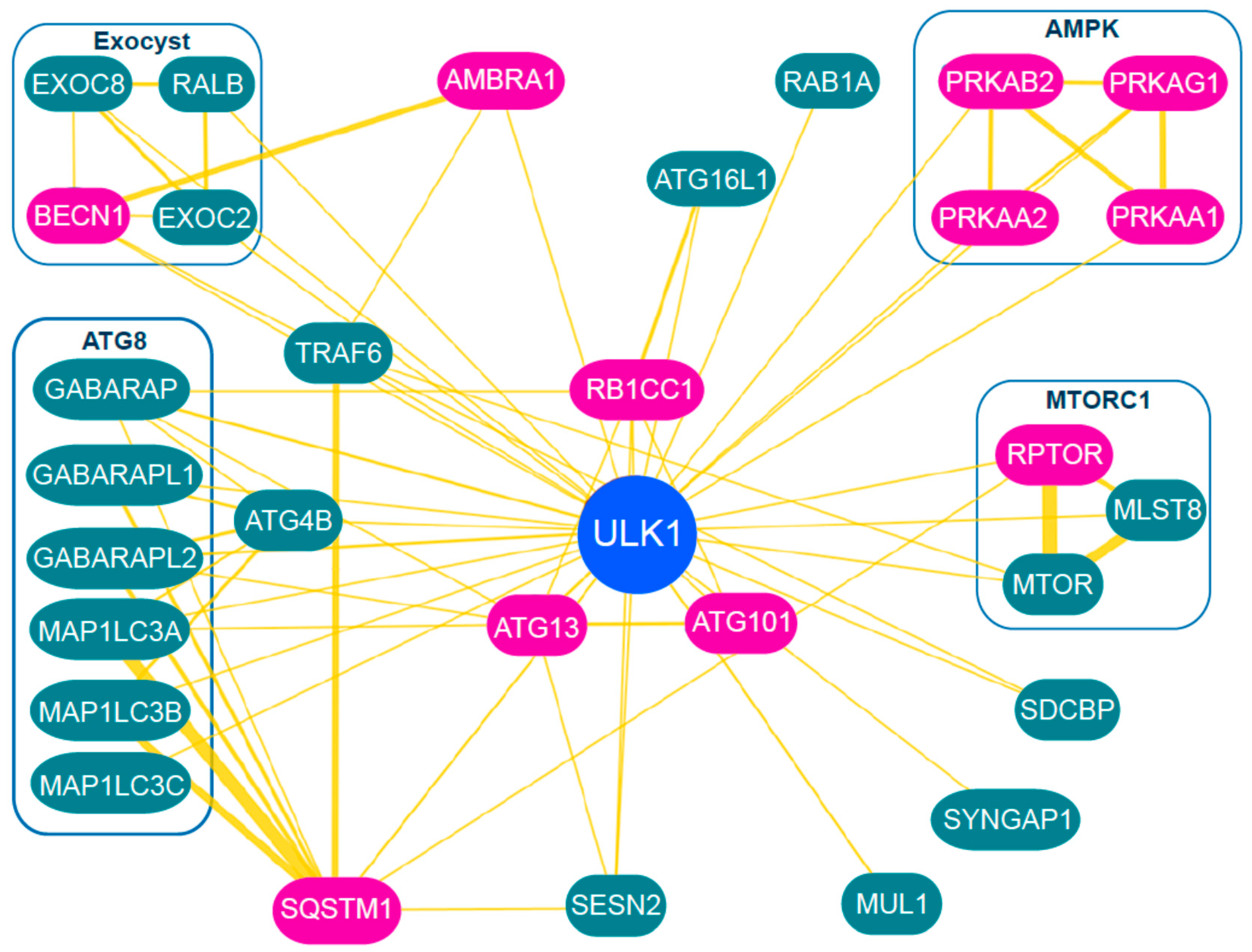
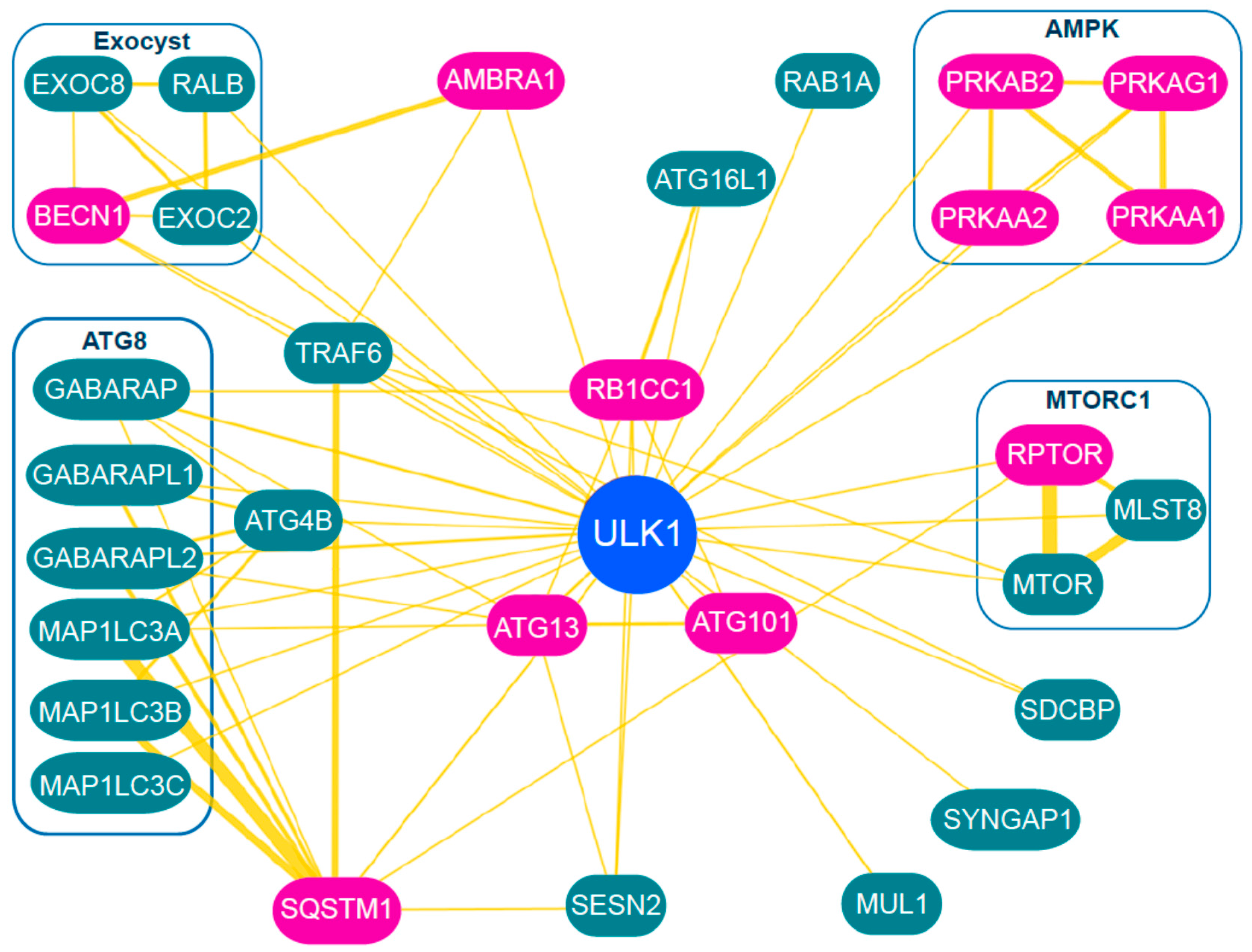
3. Regulation and Role of the ULK1 Complex during Autophagy Initiation
3.1. Phosphorylation of ULK1
3.2. Regulation of ULK1 by MTORC1
3.3. Regulation of ULK1 by AMPK
3.4. Outlook on ULK1 Regulation
3.5. Downstream Targets of ULK1
3.6. ULK1 Phosphorylation of the Beclin 1–ATG14L–VPS34 Complex
3.7. Outlook on Signalling Downstream of ULK1
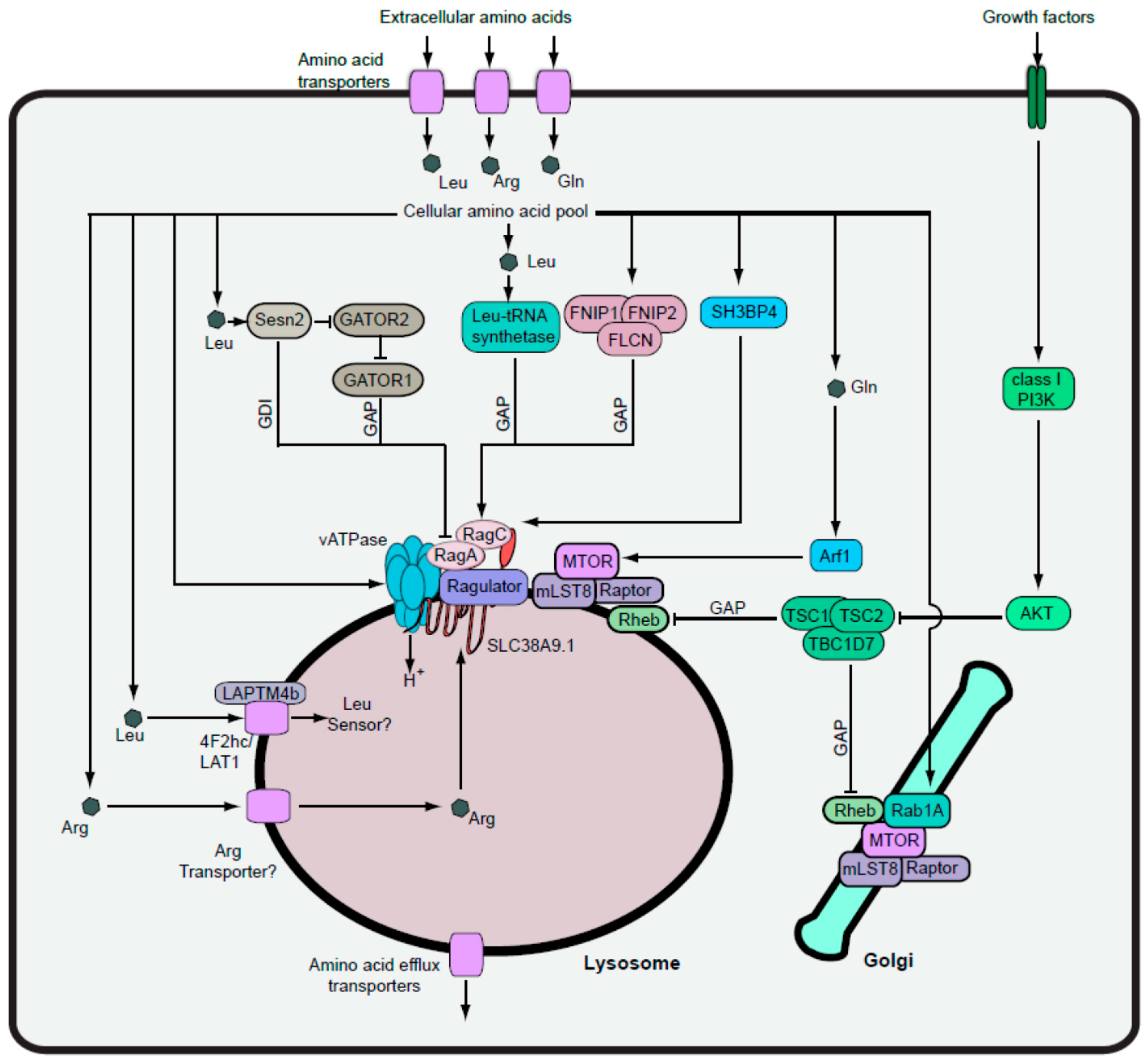
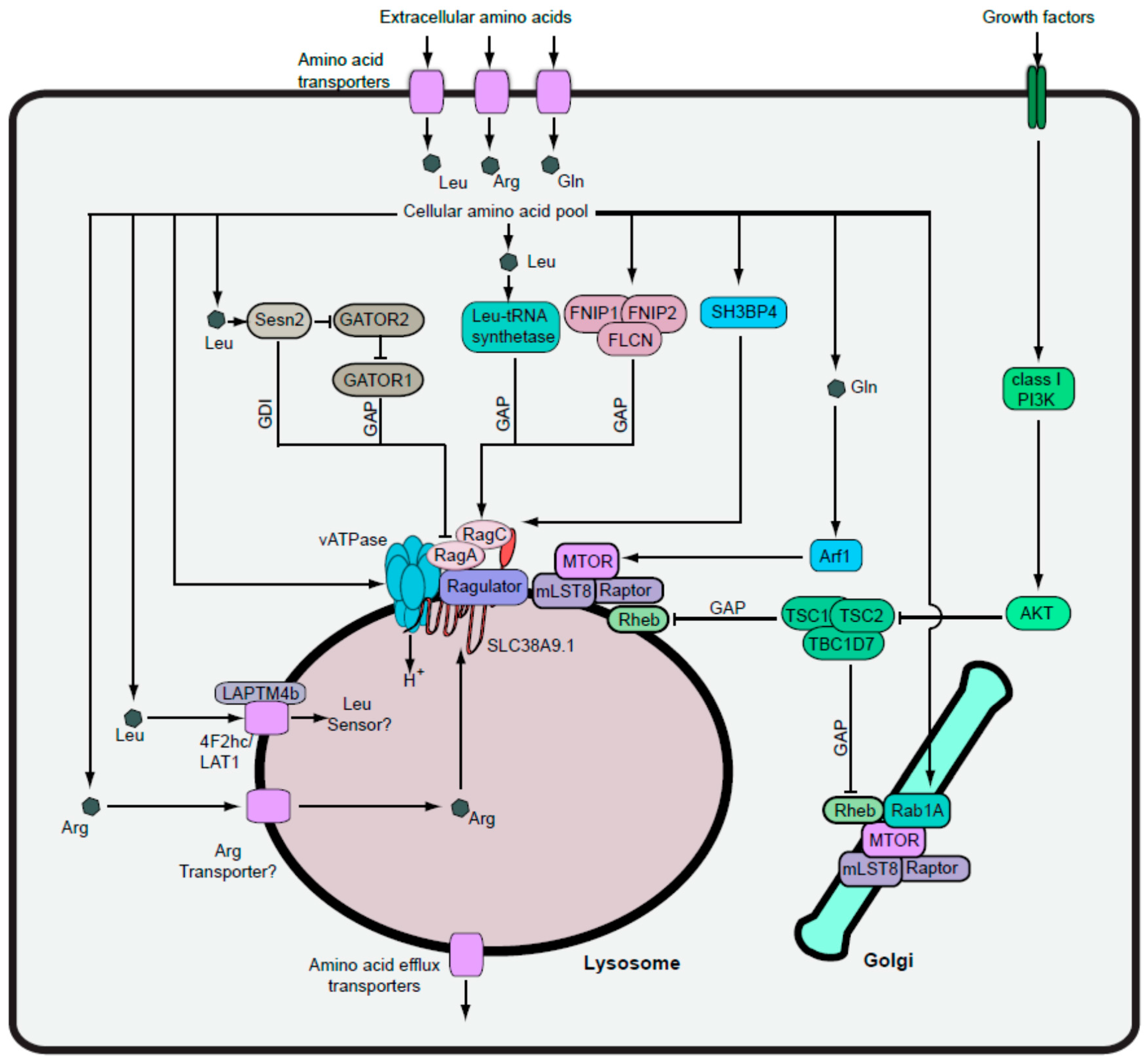
4. Amino Acid Signalling and MTORC1-Dependent Autophagy
4.1. Uptake of Regulatory Amino Acids into Cells
4.2. MTORC1 Activation via the Lysosomal Inside-Out Mechanism
4.3. Transport of Amino Acids into the Lysosome
4.4. Rag-Independent MTORC1 Pathways
4.5. Coordination of MTORC1-Activation Pathways
4.6. MTORC1 Shutdown via the GATOR Complex
4.7. MTORC1 Shutdown via the TSC Complex
5. Summary of MTORC1 Pathways and Autophagy
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Kimura, T.; Timmins, G.; Moseley, P.; Chauhan, S.; Mandell, M. Immunologic manifestations of autophagy. J. Clin. Investig. 2015, 125, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Abada, A.; Elazar, Z. Getting ready for building: Signaling and autophagosome biogenesis. EMBO Rep. 2014, 15, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.A., Jr. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J. Cell Biol. 1990, 110, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagic processes in yeast: Mechanism, machinery and regulation. Genetics 2013, 194, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An ATG4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yamamoto, M.; Kametaka, A.; Sou, Y.S.; Yabashi, A.; Yamada, A.; Annoh, H.; Kametaka, S.; Komatsu, M.; Waguri, S. A cluster of thin tubular structures mediates transformation of the endoplasmic reticulum to autophagic isolation membrane. Mol. Cell. Biol. 2014, 34, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of ATG proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by ATG14l and rubicon associated with beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two beclin 1-binding proteins, ATG14l and rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via ATG14l. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by barkor/ATG14(l). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (mam). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Koyama-Honda, I.; Itakura, E.; Fujiwara, T.K.; Mizushima, N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013, 9, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored snare syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Takacs, Z.; Donnes, P.; Kohlbacher, O. Wipi proteins: Essential Ptdins3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting ATG12-5-16l1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The ATG16l complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Sakoh-Nakatogawa, M.; Matoba, K.; Asai, E.; Kirisako, H.; Ishii, J.; Noda, N.N.; Inagaki, F.; Nakatogawa, H.; Ohsumi, Y. ATG12-ATG5 conjugate enhances E2 activity of ATG3 by rearranging its catalytic site. Nat. Struct. Mol. Biol. 2013, 20, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. Tbc1d14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Joachim, J.; Jefferies, H.B.; Razi, M.; Frith, D.; Snijders, A.P.; Chakravarty, P.; Judith, D.; Tooze, S.A. Activation of ULK kinase and autophagy by GABARAP trafficking from the centrosome is regulated by WAC and GM130. Mol. Cell 2015, 60, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Zoppino, F.C.; Militello, R.D.; Slavin, I.; Alvarez, C.; Colombo, M.I. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 2010, 11, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Wandelmer, J.; Ktistakis, N.T.; Reggiori, F. ERES: Sites for autophagosome biogenesis and maturation? J. Cell Sci. 2015, 128, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Brandizzi, F.; Barlowe, C. Organization of the ER-golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 2013, 14, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013, 2, e00947. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, M.; Schekman, R. Phosphatidylinositol 3-kinase and copii generate LC3 lipidation vesicles from the ER-golgi intermediate compartment. Elife 2014, 3, e04135. [Google Scholar] [CrossRef] [PubMed]
- Biazik, J.; Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of ATG9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Nuhlen, S.; Judith, D.; Frith, D.; Snijders, A.P.; Behrends, C.; Tooze, S.A. TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian ATG proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Dancourt, J.; Shteyn, V.; Puente, G.; Fong, W.M.; Nag, S.; Bewersdorf, J.; Yamamoto, A.; Antonny, B.; Melia, T.J. Lipidation of the LC3/gabarap family of autophagy proteins relies on a membrane-curvature-sensing domain in ATG3. Nat. Cell Biol. 2014, 16, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Mi, N.; Chen, Y.; Wang, S.; Chen, M.; Zhao, M.; Yang, G.; Ma, M.; Su, Q.; Luo, S.; Shi, J.; et al. Capz regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat. Cell Biol. 2015, 17, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective VPS34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Carlson, L.A.; Stjepanovic, G.; Young, L.N.; Kim do, J.; Grob, P.; Stanley, R.E.; Nogales, E.; Hurley, J.H. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.; Williamson, L.E.; Tooze, S.A.; Chan, E.Y. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 2013, 9, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and MTOR regulate autophagy through direct phosphorylation of ULK1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by amp-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y. Regulation and function of uncoordinated-51 like kinase proteins. Antioxid. Redox Signl. 2012, 17, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Driessen, S.; Berleth, N.; Friesen, O.; Loffler, A.S.; Bohler, P.; Hieke, N.; Stuhldreier, F.; Peter, C.; Schink, K.O.; Schultz, S.W.; et al. Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy 2015, 11, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. MTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through ambra1 and traf6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qi, W.; Chen, G.; Feng, D.; Liu, J.; Ma, B.; Zhou, C.; Mu, C.; Zhang, W.; Chen, Q.; et al. Mitochondrial outer-membrane E3 ligase mul1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 2015, 11, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol. Cell 2016, 61, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by ULK1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of MTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 8048. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. MTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of ampk by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [PubMed]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of MTORC1 by the rag gtpases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.G.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls MTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsun, Z.-Y.; Wolfson, R.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. The amino acid transporter SLC38A9 is a key component of a lysosomal membrane complex that signals arginine sufficiency to MTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through Torc1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Saleiro, D.; Mehrotra, S.; Kroczynska, B.; Beauchamp, E.M.; Lisowski, P.; Majchrzak-Kita, B.; Bhagat, T.D.; Stein, B.L.; McMahon, B.; Altman, J.K.; et al. Central role of ULK1 in type I interferon signaling. Cell Rep. 2015, 11, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Gordon, P.B.; Seglen, P.O. Inhibition of hepatocytic autophagy by okadaic acid and other protein phosphatase inhibitors. Eur. J. Biochem. 1993, 215, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Magnaudeix, A.; Wilson, C.M.; Page, G.; Bauvy, C.; Codogno, P.; Leveque, P.; Labrousse, F.; Corre-Delage, M.; Yardin, C.; Terro, F. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol. Aging 2013, 34, 770–790. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.M.; Wu, X.; Laxman, S.; Tu, B.P. Methionine inhibits autophagy and promotes growth by inducing the sam-responsive methylation of PP2A. Cell 2013, 154, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Banreti, A.; Lukacsovich, T.; Csikos, G.; Erdelyi, M.; Sass, M. PP2A regulates autophagy in two alternative ways in drosophila. Autophagy 2012, 8, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Okada, T.; Mitani, S.; Gengyo-Ando, K.; Baillie, D.L.; Kohara, Y.; Goshima, Y. Protein phosphatase 2A cooperates with the autophagy-related kinase unc-51 to regulate axon guidance in caenorhabditis elegans. Development 2010, 137, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; He, C.; Wang, K.; Klionsky, D.J. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy 2009, 5, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Kiely, M.; Kiely, P.A. PP2A: The wolf in sheep’s clothing? Cancers 2015, 7, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Stanevich, V.; Satyshur, K.A.; Kong, M.; Watkins, G.R.; Wadzinski, B.E.; Sengupta, R.; Xing, Y. Structural basis of protein phosphatase 2A stable latency. Nat. Commun. 2013, 4, 1699. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of ampk with ULK1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of sting to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J. AMPK → ULK1 → Autophagy. Mol. Cell. Biol. 2011, 31, 3082–3084. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Li, W.; Chen, Y.; Yan, Z.; Huang, X.; Zhuang, H.; Zhong, W.; Chen, Y.; Wu, W.; Lin, C.; et al. Phosphorylation of ULK1 by ampk regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015, 589, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.H.; Dorsey, F.C.; Joshi, A.; Hennessy-Walters, K.M.; Rose, K.L.; McCastlain, K.; Zhang, J.; Iyengar, R.; Jung, C.H.; Suen, D.F.; et al. Hsp90-Cdc37 chaperone complex regulates ULK1- and ATG13-mediated mitophagy. Mol. Cell 2011, 43, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Mizushima, N. ATG13 is essential for autophagy and cardiac development in mice. Mol. Cell Biol. 2015, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates MTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent MTORC1 association with the ULK1-ATG13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Longatti, A.; McKnight, N.C.; Tooze, S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an ATG13-independent mechanism. Mol. Cell Biol. 2009, 29, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Kir, S.; Tooze, S.A. Sirna screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kaizuka, T.; Mizushima, N.; Noda, N.N. Structure of the ATG101-ATG13 complex reveals essential roles of ATG101 in autophagy initiation. Nat. Struct. Mol. Biol. 2015, 22, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Florey, O.; Overholtzer, M.; Jiang, X. Interaction between FIP200 and ATG16l1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 2013, 20, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaizuka, T.; Cadwell, K.; Sahani, M.H.; Saitoh, T.; Akira, S.; Virgin, H.W.; Mizushima, N. FIP200 regulates targeting of ATG16l1 to the isolation membrane. EMBO Rep. 2013, 14, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the ATG1 kinase complex to regulate autophagy. Mol. Cell. Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-ATG13-FIP200 complexes mediate MTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (MTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.J.; Stanley, R.E.; Hurley, J.H. Architecture of the ATG17 complex as a scaffold for autophagosome biogenesis. Cell 2012, 151, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Kofinger, J.; Ragusa, M.J.; Lee, I.H.; Hummer, G.; Hurley, J.H. Solution structure of the ATG1 complex: Implications for the architecture of the phagophore assembly site. Structure 2015, 23, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.H.; Lu, S.; Liu, X.; Li, F.K.; Yu, A.Y.; Klionsky, D.J.; Dong, M.Q.; Yip, C.K. Molecular interactions of the saccharomyces cerevisiae ATG1 complex provide insights into assembly and regulatory mechanisms. Autophagy 2015, 11, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Kim, D.J.; Stjepanovic, G.; Hurley, J.H. Structure of the human ATG13-ATG101 horma heterodimer: An interaction hub within the ULK1 complex. Structure 2015, 23, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.W.; Yamamoto, H.; Oikawa, Y.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ohsumi, Y. ATG13 horma domain recruits ATG9 vesicles during autophagosome formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3350–3355. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The biogrid interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits MTORC1 signaling, promotes multisite raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of ambra1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Semple, I.A.; Park, H.; Park, H.; Park, H.W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. ULK1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.W.; Wang, Y.B.; Wang, S.L.; Wu, M.H.; Lin, S.Y.; Chen, G.C. ATG1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011, 30, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Seifan, S.; Claessens, T.; Behrends, C.; Kamps, M.A.; Rozycka, E.; Kemp, A.J.; Nookala, R.K.; Blenis, J.; Coull, B.J.; et al. Flcn, a novel autophagy component, interacts with gabarap and is regulated by ULK1 phosphorylation. Autophagy 2014, 10, 1749–1760. [Google Scholar] [CrossRef]
- Papinski, D.; Schuschnig, M.; Reiter, W.; Wilhelm, L.; Barnes, C.A.; Maiolica, A.; Hansmann, I.; Pfaffenwimmer, T.; Kijanska, M.; Stoffel, I.; et al. Early steps in autophagy depend on direct phosphorylation of ATG9 by the ATG1 kinase. Mol. Cell 2014, 53, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Garriga, G. The conserved kinase unc-51 acts with VAB-8 and unc-14 to regulate axon outgrowth in C. elegans. Development 2004, 131, 5991–6000. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Mochizuki, H.; Flores, R., 3rd; Josowitz, R.; Krasieva, T.B.; Lamorte, V.J.; Suzuki, E.; Gindhart, J.G.; Furukubo-Tokunaga, K.; Tomoda, T. Unc-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008, 22, 3292–3307. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.C.; Ragusa, M.J.; Stanley, R.E.; Hurley, J.H. A horma domain in ATG13 mediates PI 3-kinase recruitment in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 5486–5491. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct VPS34 complexes by ampk in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Fogel, A.I.; Dlouhy, B.J.; Wang, C.; Ryu, S.W.; Neutzner, A.; Hasson, S.A.; Sideris, D.P.; Abeliovich, H.; Youle, R.J. Role of membrane association and ATG14-dependent phosphorylation in beclin-1-mediated autophagy. Mol. Cell Biol. 2013, 33, 3675–3688. [Google Scholar] [CrossRef]
- Gurkar, A.U.; Chu, K.; Raj, L.; Bouley, R.; Lee, S.H.; Kim, Y.B.; Dunn, S.E.; Mandinova, A.; Lee, S.W. Identification of ROCK1 kinase as a critical regulator of beclin1-mediated autophagy during metabolic stress. Nat. Commun. 2013, 4, 2189. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through beclin 1 phosphorylation. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Babu, J.R.; da Silva, S.; Shu, Q.; Graef, I.A.; Oliver, T.; Tomoda, T.; Tani, T.; Wooten, M.W.; Wang, F. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc. Natl. Acad. Sci. USA 2007, 104, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of MTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Venerando, R.; Marin, O.; Siliprandi, N.; Mortimore, G.E. Inhibition of macroautophagy and proteolysis in the isolated rat hepatocyte by a nontransportable derivative of the multiple antigen peptide Leu8-Lys4-Lys2-Lys-beta Ala. J. Biol. Chem. 1994, 269, 25348–25353. [Google Scholar] [PubMed]
- Wang, Q.; Holst, J. L-type amino acid transport and cancer: Targeting the MTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar] [PubMed]
- Closs, E.I.; Boissel, J.P.; Habermeier, A.; Rotmann, A. Structure and function of cationic amino acid transporters (CATs). J. Membr. Biol. 2006, 213, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Wagner, C.A.; Lang, F.; Broer, S. The heterodimeric amino acid transporter 4F2hc/y+LAT2 mediates arginine efflux in exchange with glutamine. Biochem. J. 2000, 349 Pt 3, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates MTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. MTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human MTOR complex i and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human MTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to MTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases bind raptor and mediate amino acid signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of torc1 by rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets MTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a gef for the rag GTPases that signal amino acid levels to MTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Genau, H.M.; Behrends, C. Amino acid-dependent MTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol. Cell. Biol. 2015, 35, 2479–2494. [Google Scholar] [CrossRef] [PubMed]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4B recruits the LAT1–4F2hc Leu transporter to lysosomes and promotes MTORC1 activation. Nat. Commun. 2015, 6, 7250. [Google Scholar] [CrossRef] [PubMed]
- Sagne, C.; Gasnier, B. Molecular physiology and pathophysiology of lysosomal membrane transporters. J. Inherit. Metab. Dis. 2008, 31, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, H.; Rutkowski, R.; Gartner, A.; Wang, X. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science 2012, 337, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the MTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to MTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.S.; Roczniak-Ferguson, A.; Ferguson, S.M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of rag GTPases. J. Cell Biol. 2013, 202, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Stone, M.; Hwang, T.H.; Kim, Y.-G.; Dunlevy, J.R.; Griffin, T.J.; Kim, D.-H. SH3BP4 is a negative regulator of amino acid-rag GTPase-MTORC1 signaling. Mol. Cell 2012, 46, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F.S. Rab1A is an MTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A. Review series: Rab GTPases and membrane identity: Causal or inconsequential? J. Cell Biol. 2013, 202, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Mitin, N.; Wilder, R.S.; Henske, E.P.; Tamanoi, F.; Cox, A.D.; Der, C.J. Differential requirement of CAAX-mediated posttranslational processing for RHEB localization and signaling. Oncogene 2009, 29, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the tsc complex integrates insulin and nutrient regulation of MTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Menon, S.; Yamasaki, A.; Chou, H.T.; Walz, T.; Jiang, Y.; Ferro-Novick, S. YPT1 recruits the ATG1 kinase to the preautophagosomal structure. Proc. Natl. Acad. Sci. USA 2013, 110, 9800–9805. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A tumor suppressor complex with GAP activity for the rag GTPases that signal amino acid sufficiency to MTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.X.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit MTORC1 kinase activation through the Gator complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of MTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 inhibits MTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the sestrin2-MTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the MTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Bae, S.H.; Park, S.; Rhee, S.G. Sestrin 2 is not a reductase for cysteine sulfinic acid of peroxiredoxins. Antioxid. Redox signal. 2009, 11, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and MTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for rag GTPases to control MTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of MTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.-L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. Tsc2 is phosphorylated and inhibited by AKT and suppresses MTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of torc1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Parkhitko, A.; Myachina, F.; Morrison, T.A.; Hindi, K.M.; Auricchio, N.; Karbowniczek, M.; Wu, J.J.; Finkel, T.; Kwiatkowski, D.J.; Yu, J.J.; et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc. Natl. Acad. Sci. USA 2011, 108, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Wu, Y.T.; Chen, B.; Zhou, J.; Shen, H.M. Impaired autophagy due to constitutive MTOR activation sensitizes TSC2-null cells to cell death under stress. Autophagy 2011, 7, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via MTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor tfeb links MTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by MTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Moruno-Manchon, J.F.; Perez-Jimenez, E.; Knecht, E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem. J. 2013, 449, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.J.; Martinez-Marquez, J.Y.; Prosser, D.C.; Ganser, L.R.; Buelto, D.; Wendland, B.; Duncan, M.C. Glucose starvation inhibits autophagy via vacuolar hydrolysis and induces plasma membrane internalization by down-regulating recycling. J. Biol. Chem. 2014, 289, 16736–16747. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Peinado, S.; Leon-Annicchiarico, C.L.; Galindo-Moreno, J.; Iurlaro, R.; Caro-Maldonado, A.; Prehn, J.H.; Ryan, K.M.; Munoz-Pinedo, C. Glucose-starved cells do not engage in prosurvival autophagy. J. Biol. Chem. 2013, 288, 30387–30398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal V-ATPase-ragulator complex is a common activator for ampk and MTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallagher, L.E.; Williamson, L.E.; Chan, E.Y.W. Advances in Autophagy Regulatory Mechanisms. Cells 2016, 5, 24. https://doi.org/10.3390/cells5020024
Gallagher LE, Williamson LE, Chan EYW. Advances in Autophagy Regulatory Mechanisms. Cells. 2016; 5(2):24. https://doi.org/10.3390/cells5020024
Chicago/Turabian StyleGallagher, Laura E., Leon E. Williamson, and Edmond Y. W. Chan. 2016. "Advances in Autophagy Regulatory Mechanisms" Cells 5, no. 2: 24. https://doi.org/10.3390/cells5020024
APA StyleGallagher, L. E., Williamson, L. E., & Chan, E. Y. W. (2016). Advances in Autophagy Regulatory Mechanisms. Cells, 5(2), 24. https://doi.org/10.3390/cells5020024