Systems Analysis of Drug-Induced Receptor Tyrosine Kinase Reprogramming Following Targeted Mono- and Combination Anti-Cancer Therapy


 and
and
Abstract
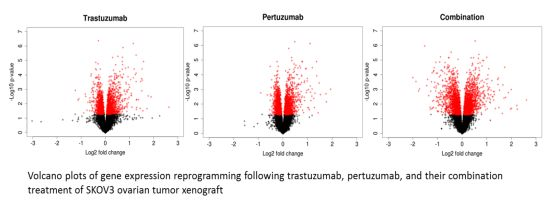
:
1. Introduction
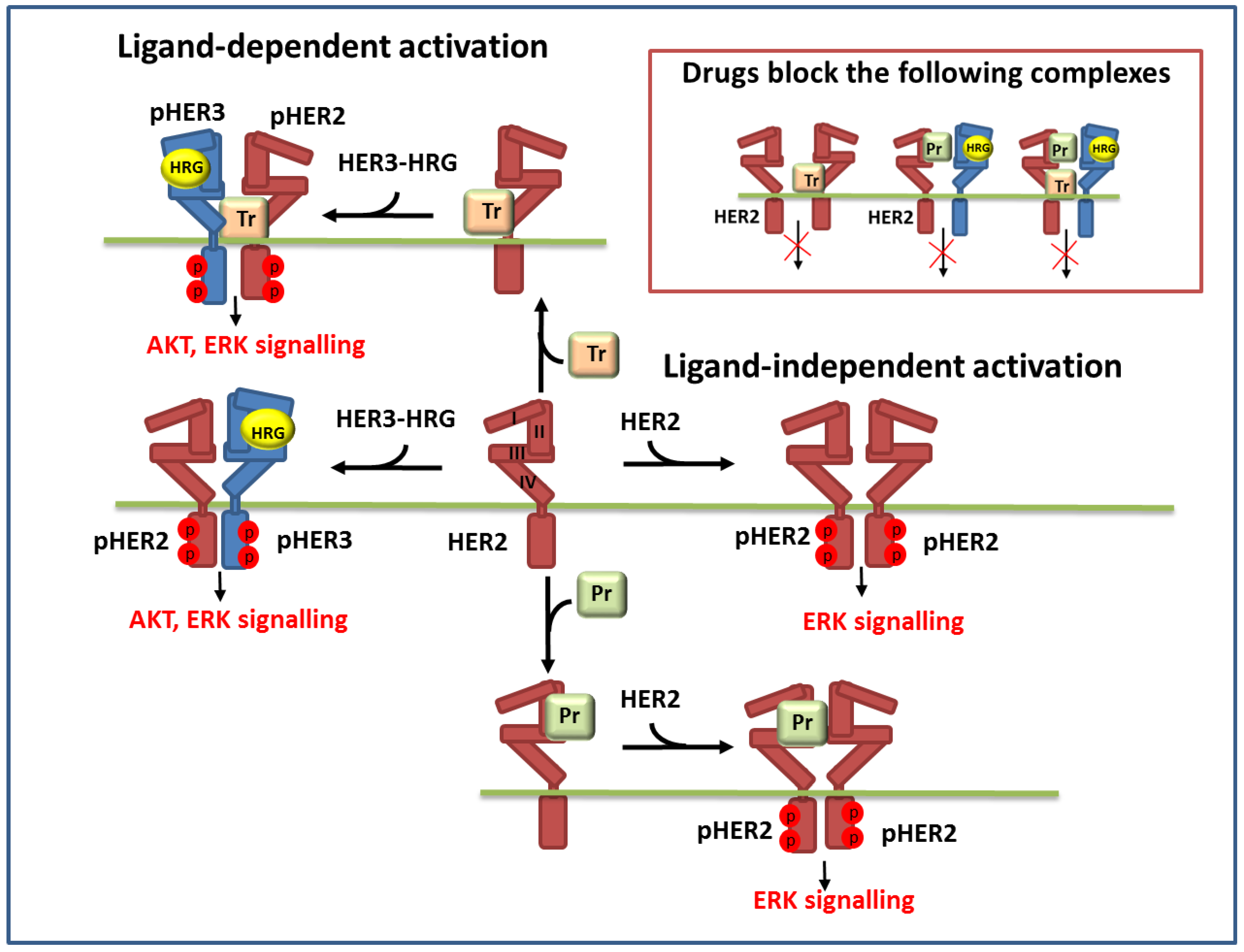
2. Experimental
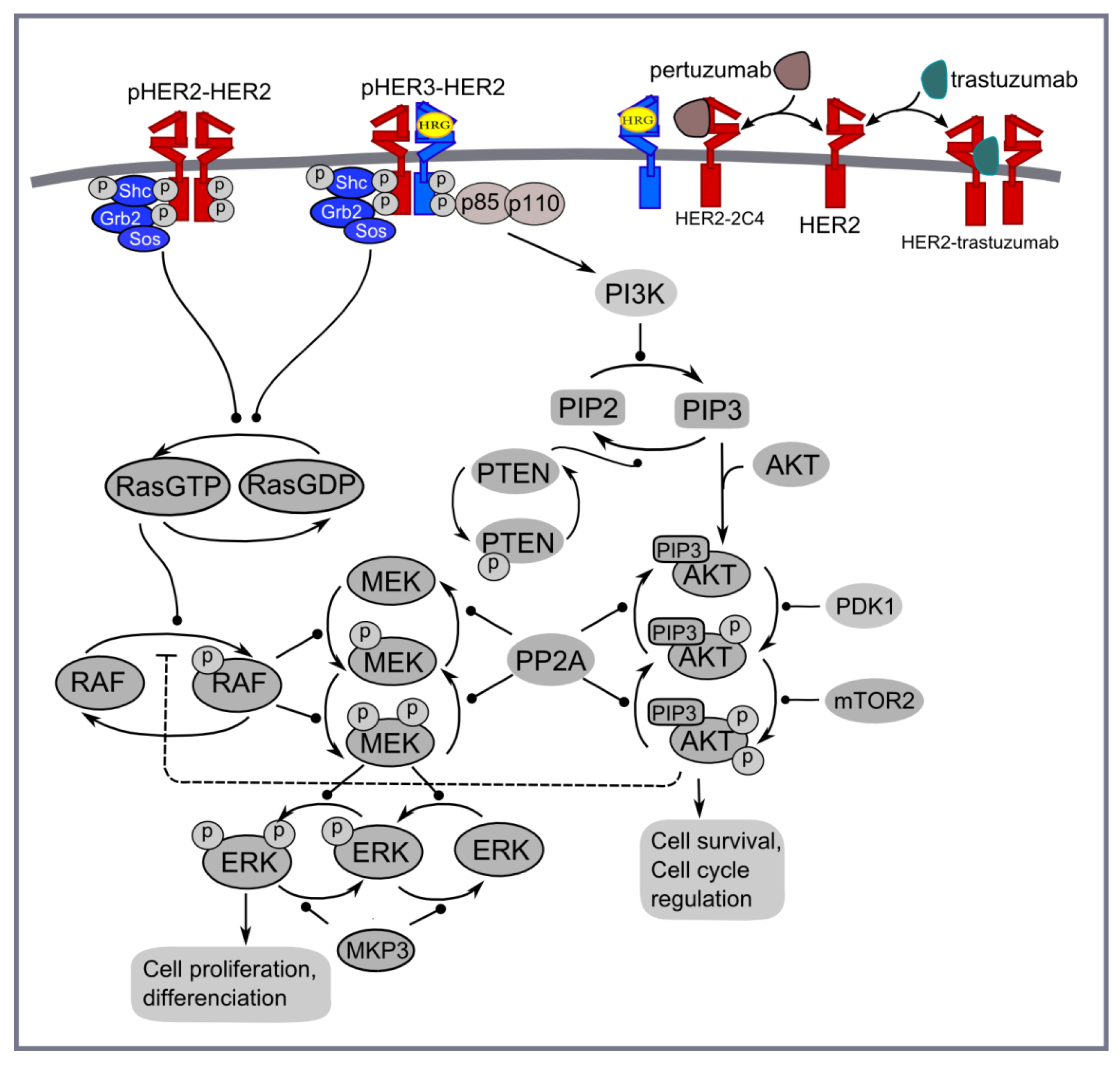
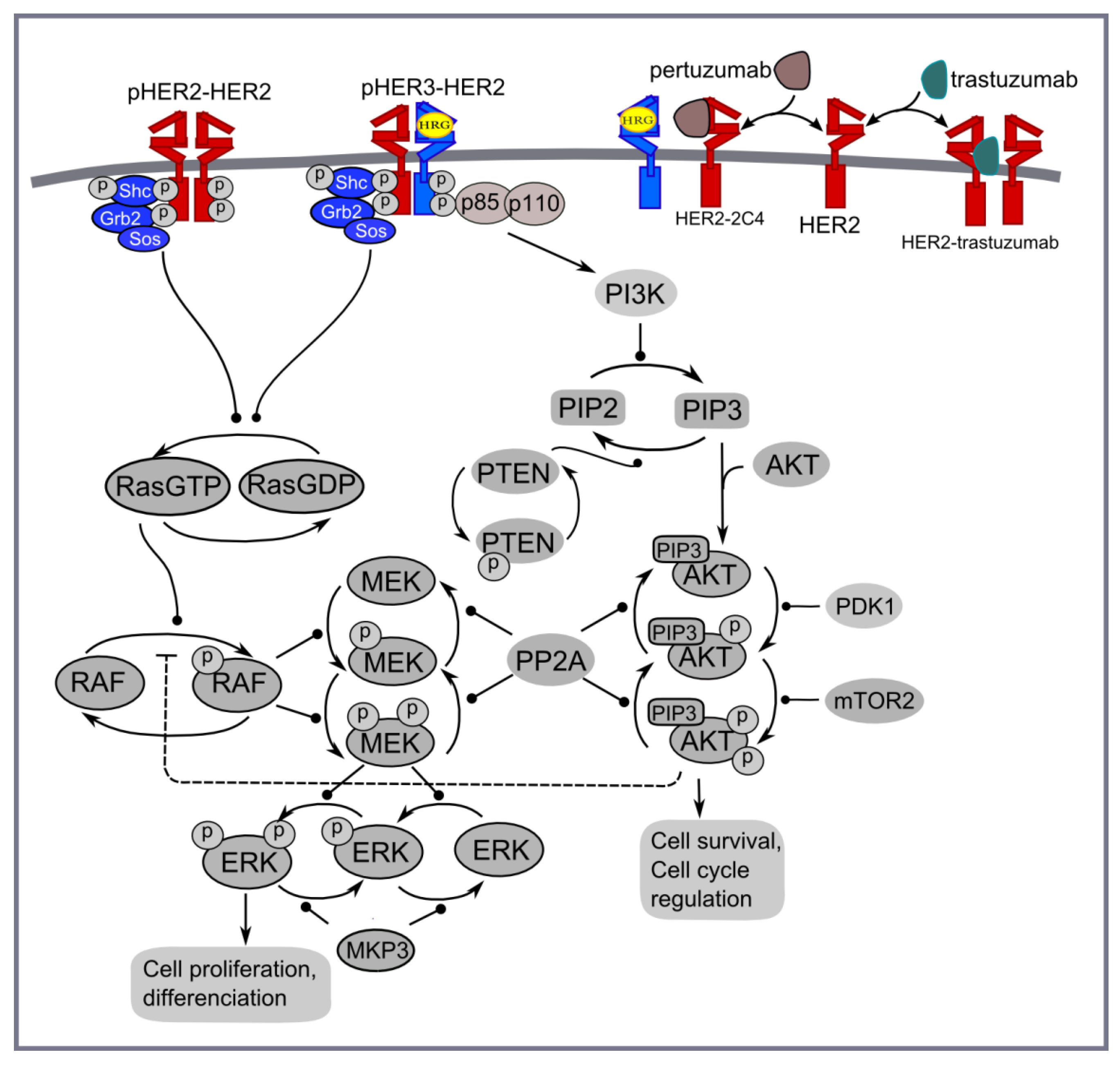
2.1. Computational Modelling
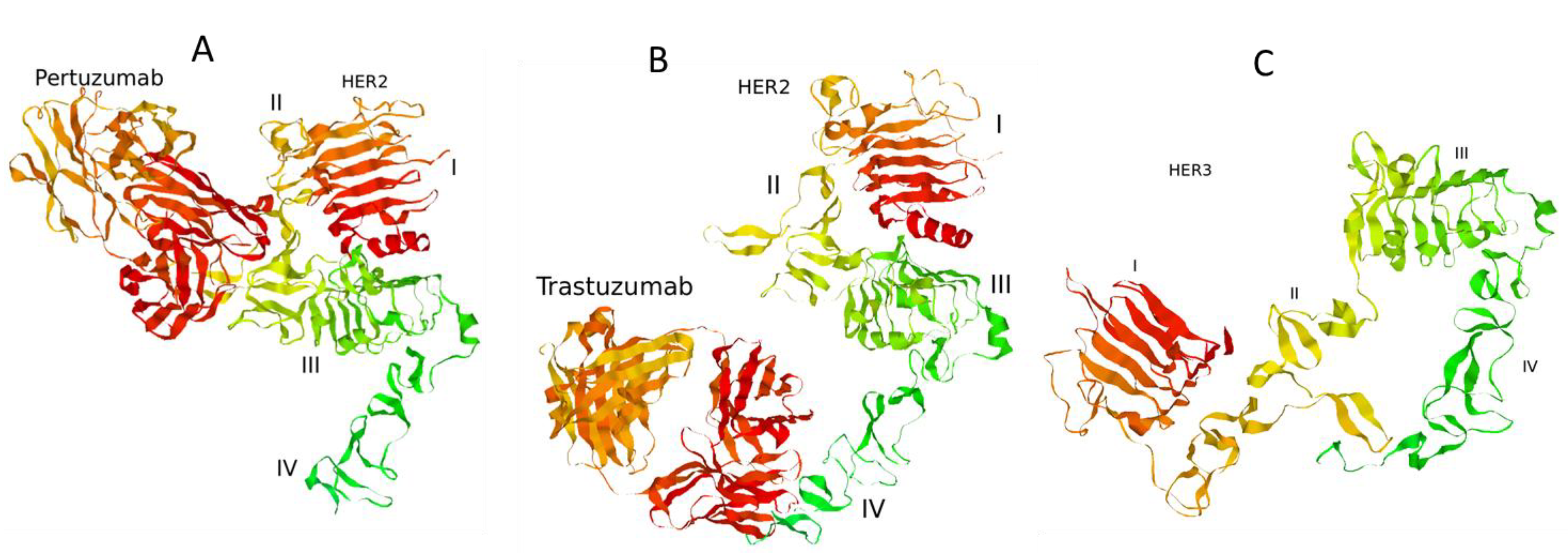
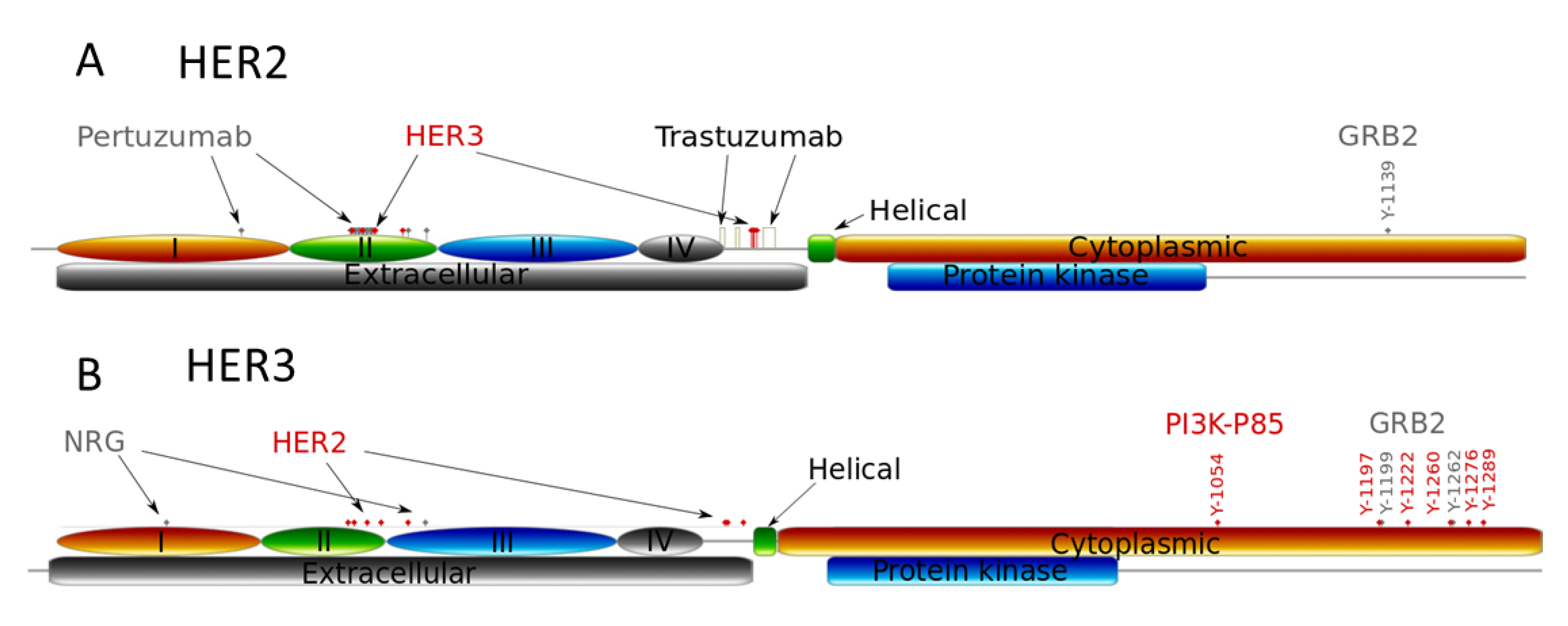

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Receptor 1 | Receptor 2 | Ligand | Effect | Ref. |
|---|---|---|---|---|---|
| Trastuzumab | HER2 | HER3 | yes | no/minor | [46] |
| Trastuzumab | HER2 | HER3 | no | yes | [31] |
| Trastuzumab | p95HER2 | no | yes | [30] | |
| Trastuzumab | HER2 | HER2 | no | yes | [2] |
| Pertuzumab | HER2 | HER3 | yes | yes | [14] |
| Pertuzumab | HER2 | HER3 | no | no/minor | [31] |
| Pertuzumab | HER2 | HER2 | no | no | [2] |
2.2. Bioinformatics Method
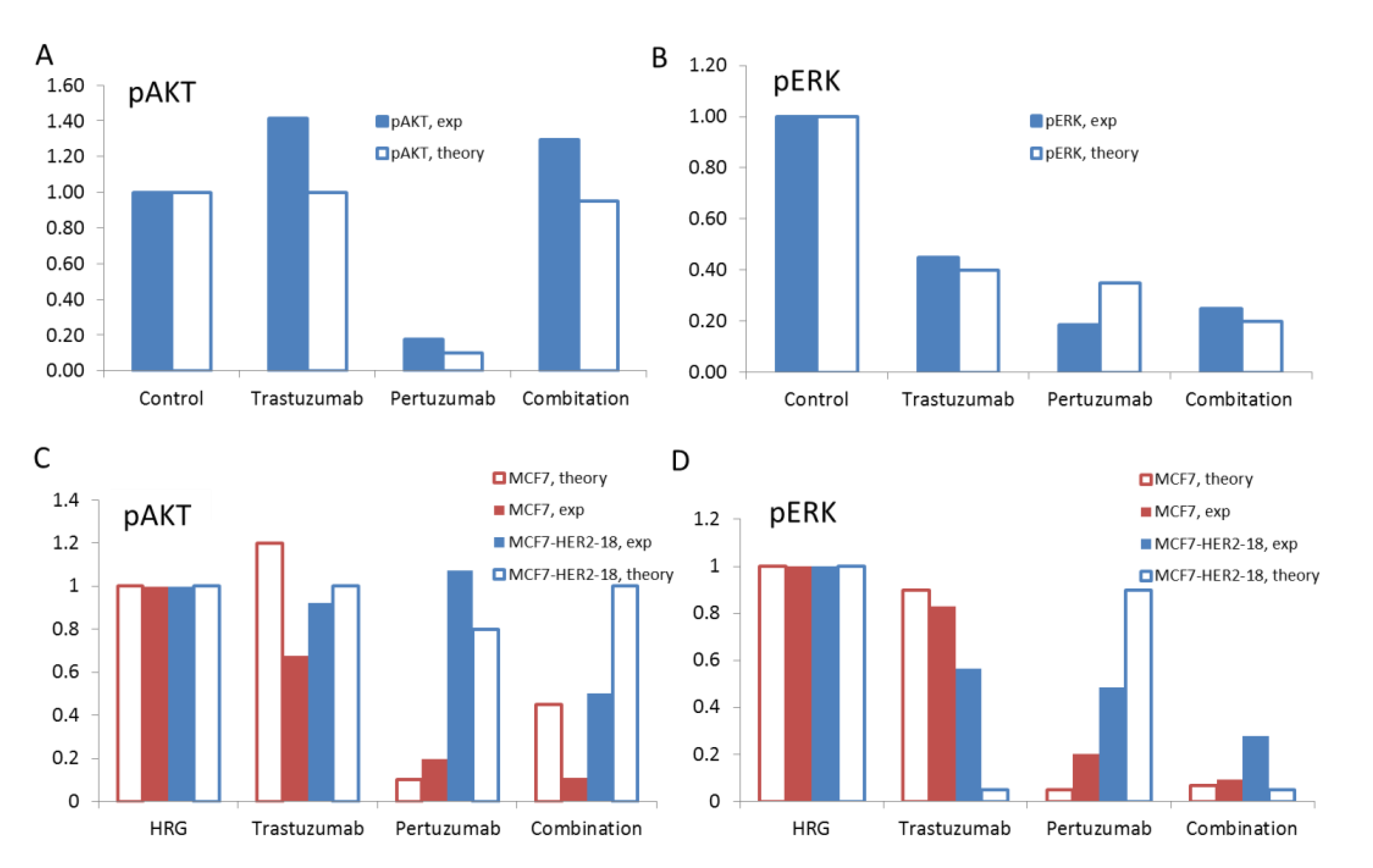
2.3. Cell Lines and Treatment with Stimuli and/or Inhibitors
3. Results and Discussion
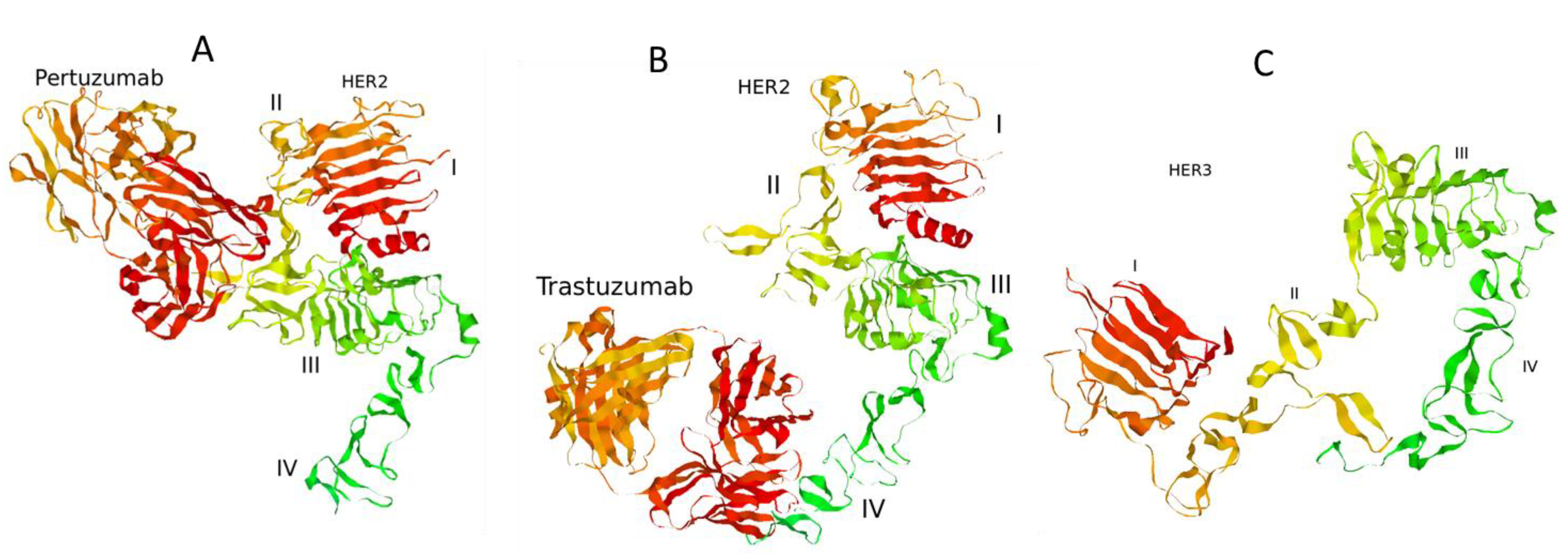
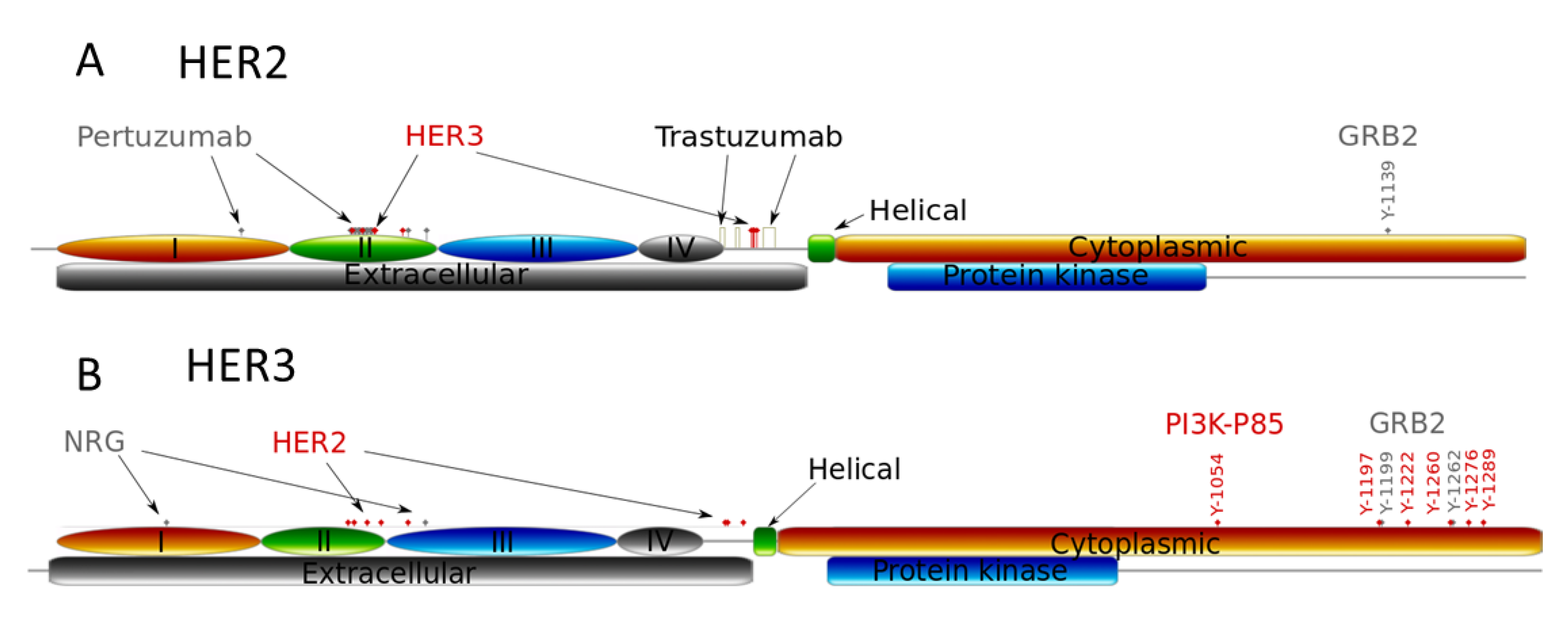
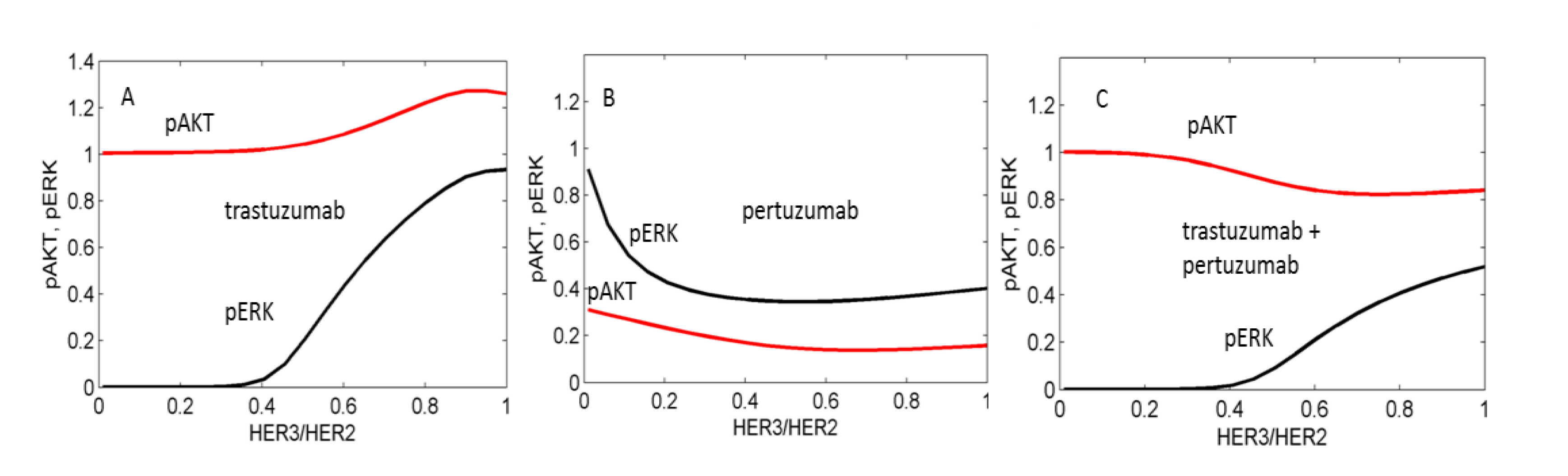
3.1. Computational Modelling of Mono- and Combination Anti-HER2 Therapies at Different Co-Expression Levels of HER3/HER2 Receptors
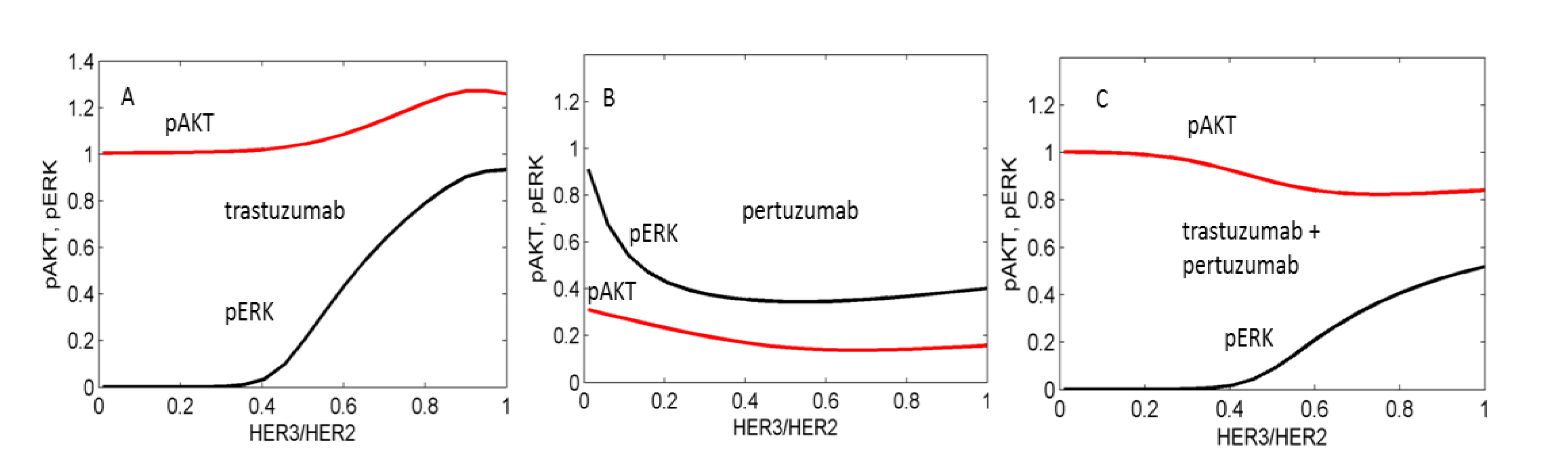
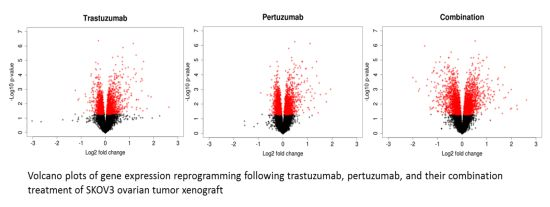
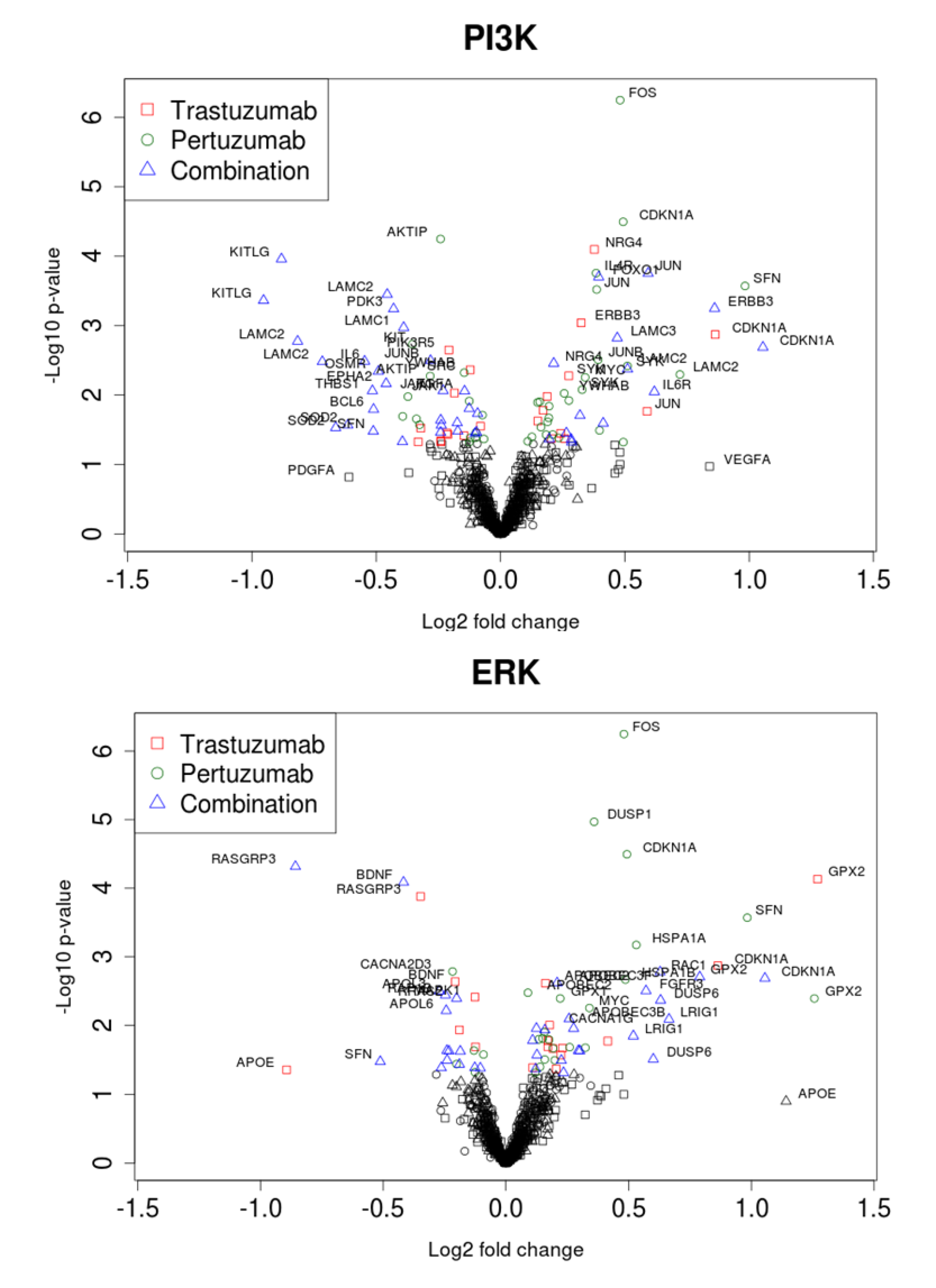
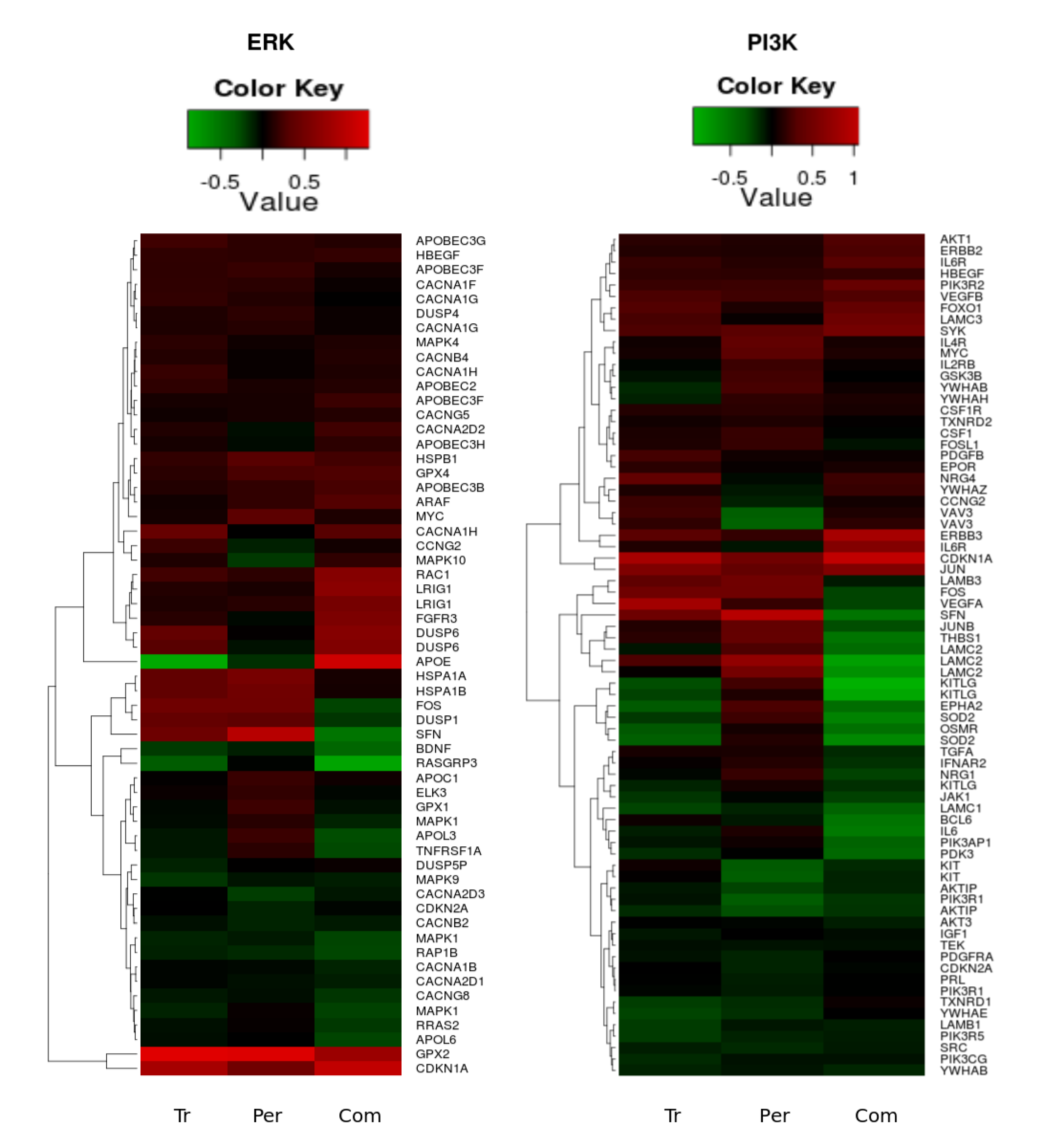
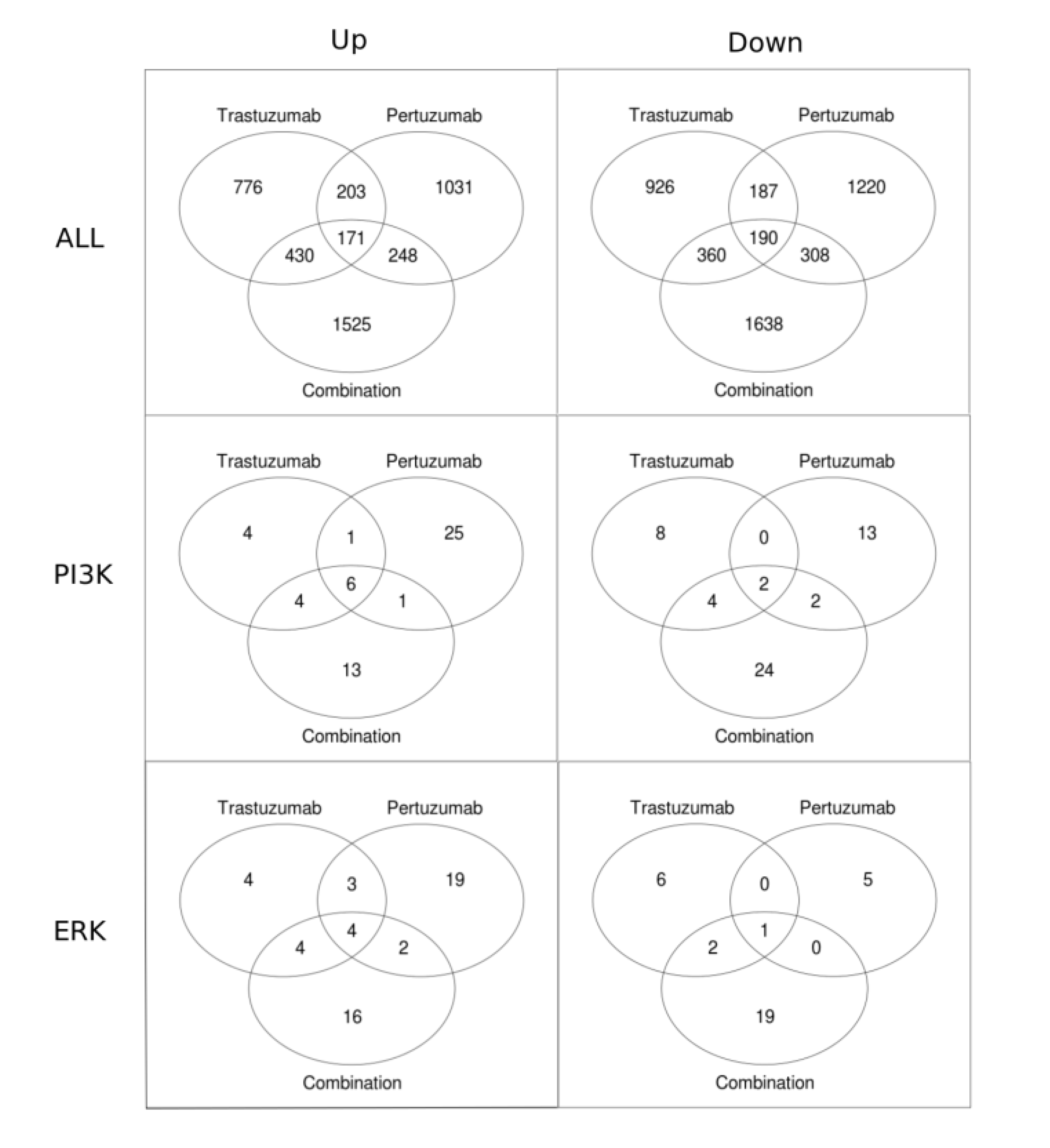
3.2. Genetic Reprogramming of HER2/HER3 Signalling Following Anti-HER2 Therapy
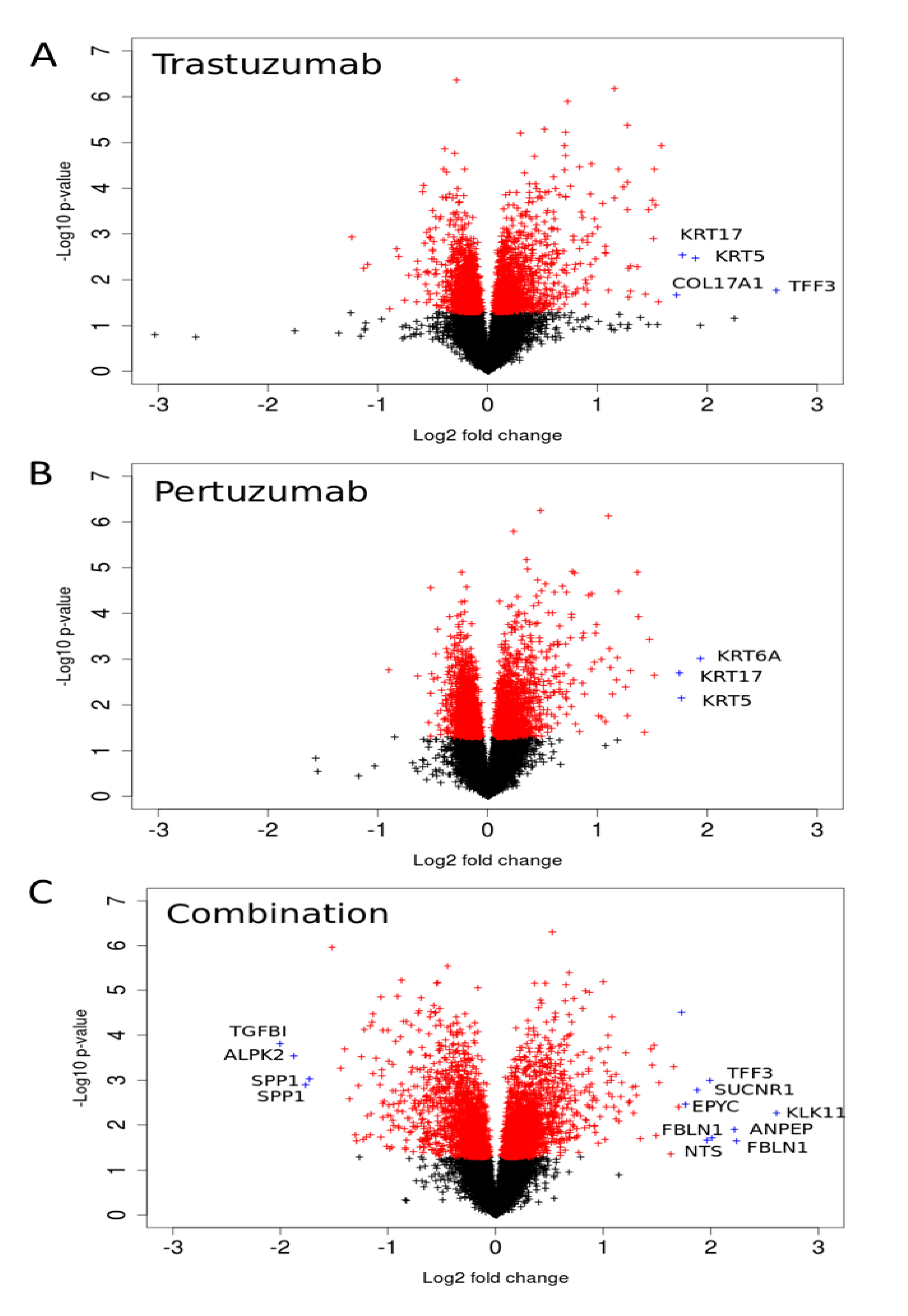
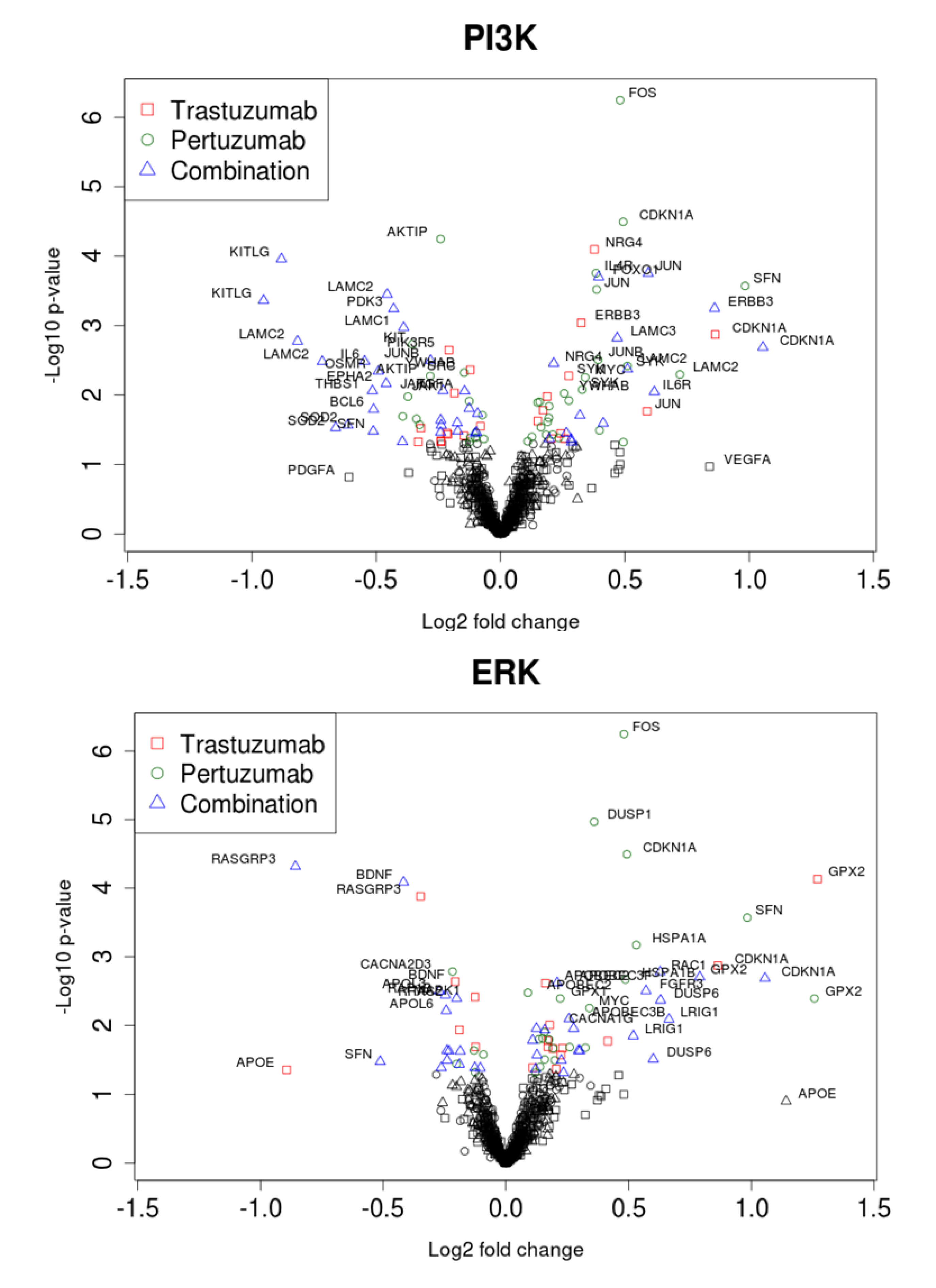
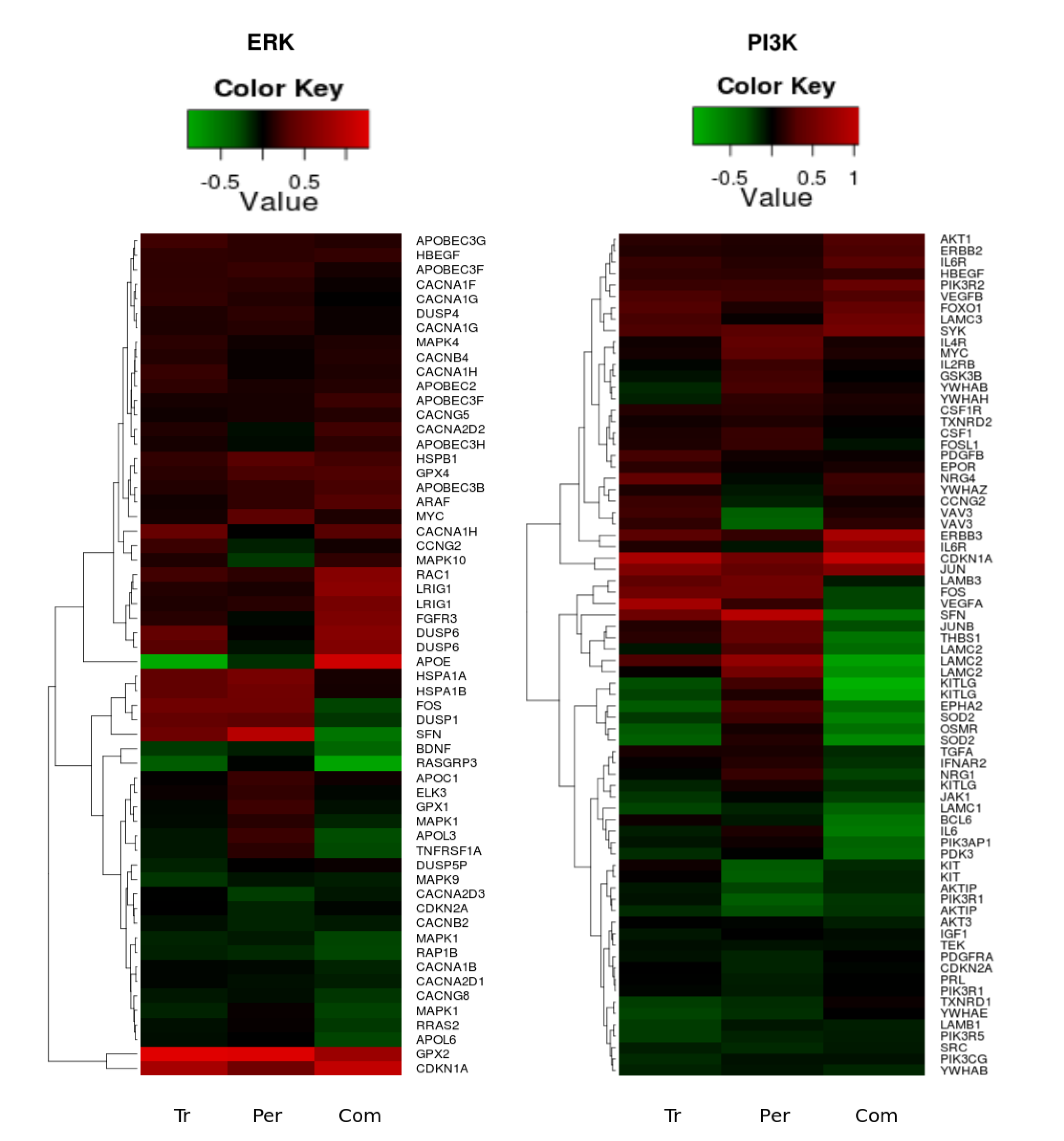
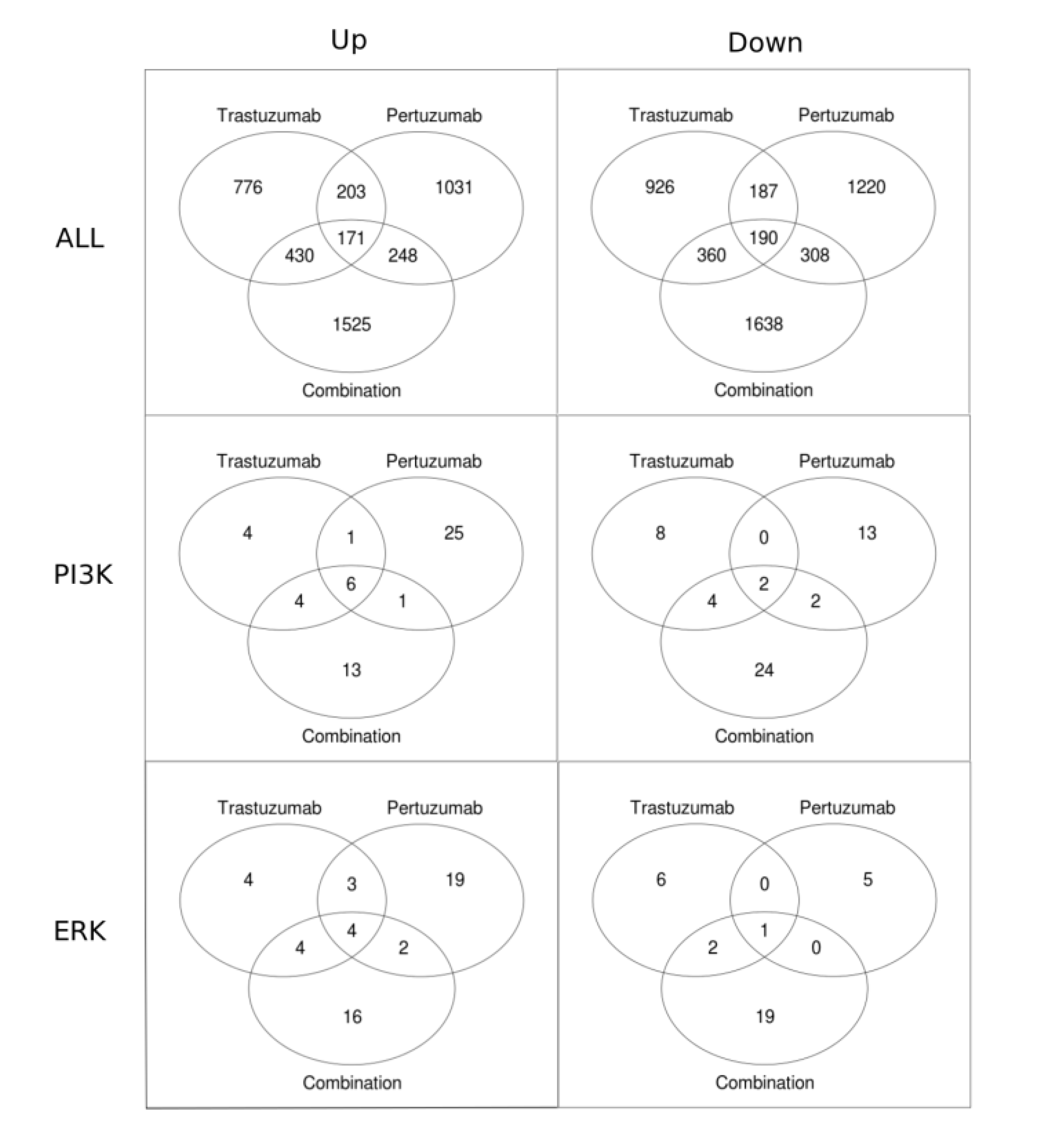
3.3. Linking the Efficacy of Mono- and Combination Therapy to ErbB Co-Expression Level
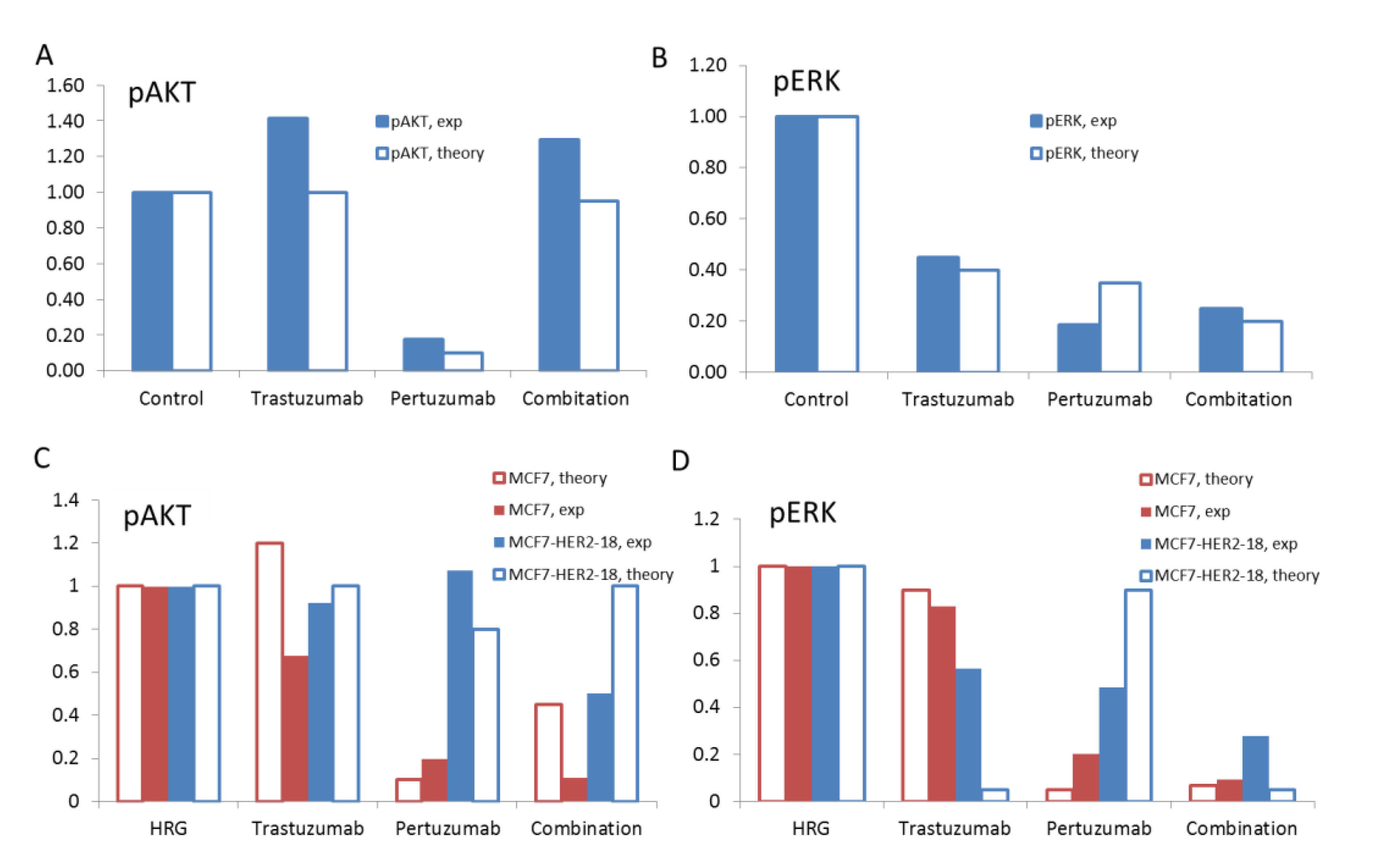
3.4. Discussion
4. Conclusion
Supplementary Files
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell. Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; González-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef]
- Landgraf, R. HER2 therapy. HER2 (ERBB2): Functional diversity from structurally conserved building blocks. Breast Cancer Res. 2007, 9, 202. [Google Scholar] [CrossRef]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar]
- Alroy, I.; Yarden, Y. The ErbB signaling network in embryogenesis and oncogenesis: Signal diversification through combinatorial ligand-receptor interactions. FEBS Lett. 1997, 410, 83–86. [Google Scholar] [CrossRef]
- Schulze, W.X.; Deng, L.; Mann, M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 2005. [CrossRef]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell. Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Negro, A.; Brar, B.K.; Lee, K.F. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog. Horm. Res. 2004, 59, 1–12. [Google Scholar] [CrossRef]
- Normanno, N.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; De Luca, A.; Caponigro, F.; Salomon, D.S. The ErbB receptors and their ligands in cancer: An overview. Curr. Drug Targets 2005, 6, 243–257. [Google Scholar] [CrossRef]
- Brix, D.; Clemmensen, K.; Kallunki, T. When Good Turns Bad: Regulation of Invasion and Metastasis by ErbB2 Receptor Tyrosine Kinase. Cells 2014, 3, 53–78. [Google Scholar] [CrossRef]
- Flågeng, M.H.; Knappskog, S.; Haynes, B.P.; Lønning, P.E.; Mellgren, G. Inverse regulation of EGFR/HER1 and HER2–4 in normal and malignant human breast tissue. PLoS One 2013, 8, e74618. [Google Scholar]
- Sassen, A.; Rochon, J.; Wild, P.; Hartmann, A.; Hofstaedter, F.; Schwarz, S.; Brockhoff, G. Cytogenetic analysis of HER1/EGFR, HER2, HER3 and HER4 in 278 breast cancer patients. Breast Cancer Res. 2008, 10, R2. [Google Scholar] [CrossRef]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; Scher, H.I.; Sliwkowski, M.X. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002, 2, 127–137. [Google Scholar] [CrossRef]
- Sharial, M.; Crown, J.; Hennessy, B.T. Overcoming resistance and restoring sensitivity to HER2-targeted therapies in breast cancer. Ann. Oncol. 2012, 23, 3007–3016. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef]
- Narayan, M.; Wilken, J.A.; Harris, L.N.; Baron, A.T.; Kimbler, K.D.; Maihle, N.J. Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res. 2009, 69, 2191–2194. [Google Scholar] [CrossRef]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5021–5026. [Google Scholar] [CrossRef]
- Grovdal, L.M.; Kim, J.; Holst, M.R.; Knudsen, S.L.; Grandal, M.V; van Deurs, B. EGF receptor inhibitors increase ErbB3 mRNA and protein levels in breast cancer cells. Cell. Signal. 2012, 24, 296–301. [Google Scholar] [CrossRef]
- Jørgensen, J.T. Companion diagnostics in oncology - current status and future aspects. Oncology 2013, 85, 59–68. [Google Scholar] [CrossRef]
- Yap, T.A.; Omlin, A.; de Bono, J.S. Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef]
- Swain, S.M.; Kim, S.B.; Cortés, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; de Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887. [Google Scholar] [CrossRef]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar] [CrossRef]
- Yamashita-Kashima, Y.; Iijima, S.; Yorozu, K.; Furugaki, K.; Kurasawa, M.; Ohta, M.; Fujimoto-Ouchi, K. Pertuzumab in combination with trastuzumab shows significantly enhanced antitumor activity in HER2-positive human gastric cancer xenograft models. Clin. Cancer Res. 2011, 17, 5060–5070. [Google Scholar] [CrossRef]
- Faratian, D.; Zweemer, A.J.M.; Nagumo, Y.; Sims, A.H.; Muir, M.; Dodds, M.; Mullen, P.; Um, I.; Kay, C.; Hasmann, M.; et al. Trastuzumab and pertuzumab produce changes in morphology and estrogen receptor signaling in ovarian cancer xenografts revealing new treatment strategies. Clin. Cancer Res. 2011, 17, 4451–4461. [Google Scholar] [CrossRef]
- Sims, A.H.; Zweemer, A.J.M.; Nagumo, Y.; Faratian, D.; Muir, M.; Dodds, M.; Um, I.; Kay, C.; Hasmann, M.; Harrison, D.J.; et al. Defining the molecular response to trastuzumab, pertuzumab and combination therapy in ovarian cancer. Br. J. Cancer 2012, 106, 1779–1789. [Google Scholar] [CrossRef]
- Fuentes, G.; Scaltriti, M.; Baselga, J.; Verma, C.S. Synergy between trastuzumab and pertuzumab for human epidermal growth factor 2 (Her2) from colocalization: An in silico based mechanism. Breast Cancer Res. 2011, 13, R54. [Google Scholar] [CrossRef]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Kholodenko, B.; Yaffe, M.B.; Kolch, W. Computational approaches for analyzing information flow in biological networks. Sci. Signal. 2012, 5, re1. [Google Scholar]
- Kleiman, L.B.; Maiwald, T.; Conzelmann, H.; Lauffenburger, D.A.; Sorger, P.K. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell. 2011, 43, 723–737. [Google Scholar] [CrossRef]
- Birtwistle, M.R.; Hatakeyama, M.; Yumoto, N.; Ogunnaike, B.A.; Hoek, J.B.; Kholodenko, B.N. Ligand-dependent responses of the ErbB signaling network: Experimental and modeling analyses. Mol. Syst. Biol. 2007, 3, 144. [Google Scholar]
- Schoeberl, B.; Pace, E.A.; Fitzgerald, J.B.; Harms, B.D.; Xu, L.; Nie, L.; Linggi, B.; Kalra, A.; Paragas, V.; Bukhalid, R.; et al. Therapeutically targeting ErbB3: A key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci. Signal. 2009, 2, ra31. [Google Scholar]
- Joslin, E.J.; Shankaran, H.; Opresko, L.K.; Bollinger, N.; Lauffenburger, D.A.; Wiley, H.S. Structure of the EGF receptor transactivation circuit integrates multiple signals with cell context. Mol. Biosyst. 2010, 6, 1293–1306. [Google Scholar] [CrossRef]
- Xu, A.M.; Huang, P.H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef]
- Shankaran, H.; Wiley, H.S.; Resat, H. Modeling the effects of HER/ErbB1–3 coexpression on receptor dimerization and biological response. Biophys. J. 2006, 90, 3993–4009. [Google Scholar] [CrossRef]
- Shankaran, H.; Zhang, Y.; Tan, Y.; Resat, H. Model-based analysis of HER activation in cells co-expressing EGFR, HER2 and HER3. PLoS Comput. Biol. 2013, 9, e1003201. [Google Scholar] [CrossRef]
- Zhang, Y.; Opresko, L.; Shankaran, H.; Chrisler, W.B.; Wiley, H.S.; Resat, H. HER/ErbB receptor interactions and signaling patterns in human mammary epithelial cells. BMC Cell. Biol. 2009, 10, 78. [Google Scholar] [CrossRef]
- Samaga, R.; Saez-Rodriguez, J.; Alexopoulos, L.G.; Sorger, P.K.; Klamt, S. The logic of EGFR/ErbB signaling: Theoretical properties and analysis of high-throughput data. PLoS Comput. Biol. 2009, 5, e1000438. [Google Scholar] [CrossRef]
- Nagumo, Y.; Faratian, D.; Mullen, P.; Harrison, D.J.; Hasmann, M.; Langdon, S.P. Modulation of HER3 is a marker of dynamic cell signaling in ovarian cancer: Implications for pertuzumab sensitivity. Mol. Cancer Res. 2009, 7, 1563–1571. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank-RCSB PDB. Available online: http://www.rcsb.org/pdb/home/home.do (accessed 30 May, 2014).
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Cho, H.-S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Austin, C.D.; de Mazière, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell. 2004, 15, 5268–5282. [Google Scholar] [CrossRef]
- Garrett, J.T.; Sutton, C.R.; Kuba, M.G.; Cook, R.S.; Arteaga, C.L. Dual blockade of HER2 in HER2-overexpressing tumor cells does not completely eliminate HER3 function. Clin. Cancer Res. 2013, 19, 610–619. [Google Scholar] [CrossRef]
- Faratian, D.; Goltsov, A.; Lebedeva, G.; Sorokin, A.; Moodie, S.; Mullen, P.; Kay, C.; Um, I.H.; Langdon, S.; Goryanin, I.; et al. Systems biology reveals new strategies for personalizing cancer medicine and confirms the role of PTEN in resistance to trastuzumab. Cancer Res. 2009, 69, 6713–6720. [Google Scholar] [CrossRef]
- Goltsov, A.; Faratian, D.; Langdon, S.P.; Bown, J.; Goryanin, I.; Harrison, D.J. Compensatory effects in the PI3K/PTEN/AKT signaling network following receptor tyrosine kinase inhibition. Cell. Signal. 2011, 23, 407–416. [Google Scholar] [CrossRef]
- Goltsov, A.; Faratian, D.; Langdon, S.P.; Mullen, P.; Harrison, D.J.; Bown, J. Features of the reversible sensitivity-resistance transition in PI3K/PTEN/AKT signalling network after HER2 inhibition. Cell. Signal. 2012, 24, 493–504. [Google Scholar] [CrossRef]
- Goltsov, A.; Langdon, S.P.; Goltsov, G.; Harrison, D.J.; Bown, J. Customizing the therapeutic response of signaling networks to promote antitumor responses by drug combinations. Front. Oncol. 2014, 4, 13. [Google Scholar]
- Lebedeva, G.; Sorokin, A.; Faratian, D.; Mullen, P.; Goltsov, A.; Langdon, S.P.; Harrison, D.J.; Goryanin, I. Model-based global sensitivity analysis as applied to identification of anti-cancer drug targets and biomarkers of drug resistance in the ErbB2/3 network. Eur. J. Pharm. Sci. 2012, 46, 244–258. [Google Scholar] [CrossRef]
- Cui, X.; Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003, 4, 210. [Google Scholar]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed 30 May 2014).
- Cytoscape: An Open Source Platform for Complex Network Analysis and Visualization. Available online: http://www.cytoscape.org/ (accessed 30 May 2014).
- Chen, W.W.; Schoeberl, B.; Jasper, P.J.; Niepel, M.; Nielsen, U.B.; Lauffenburger, D.A.; Sorger, P.K. Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol. Syst. Biol. 2009, 5, 239. [Google Scholar]
- Hu, H. HER Receptor-Mediated Dynamic Signalling in Breast Cancer Cells. PhD Thesis, University of Edinburgh, Edinburgh, UK, 2011; pp. 144–157. [Google Scholar]
- Hu, H.; Goltsov, A.; Bown, J.L.; Sims, A.H.; Langdon, S.P.; Harrison, D.J.; Faratian, D. Feedforward and feedback regulation of the MAPK and PI3K oscillatory circuit in breast cancer. Cell. Signal. 2013, 25, 26–32. [Google Scholar] [CrossRef]
- Pickl, M.; Ries, C.H. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2009, 28, 461–468. [Google Scholar] [CrossRef]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 1992, 24, 85–95. [Google Scholar] [CrossRef]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef]
- Wilken, J.A.; Webster, K.T.; Maihle, N.J. Trastuzumab sensitizes ovarian cancer cells to EGFR-targeted therapeutics. J. Ovarian Res. 2010, 3, 7. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2718–2723. [Google Scholar]
- Sergina, N. V; Moasser, M.M. The HER family and cancer: Emerging molecular mechanisms and therapeutic targets. Trends Mol. Med. 2007, 13, 527–534. [Google Scholar] [CrossRef]
- Shapiro, G.; Kwak, E.; Baselga, J.; Rodon, J.; Scheffold, C.; Laird, A.D.; Bedell, C.; Edelman, G. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors. ASCO Meet. Abstr. 2009, 27, 3500. [Google Scholar]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Guo, S.; Sonenshein, G.E. Forkhead box transcription factor FOXO3a regulates estrogen receptor alpha expression and is repressed by the Her-2/neu/phosphatidylinositol 3-kinase/Akt signaling pathway. Mol. Cell. Biol. 2004, 24, 8681–8690. [Google Scholar] [CrossRef]
- Singh, A.; Ye, M.; Bucur, O.; Zhu, S.; Tanya Santos, M.; Rabinovitz, I.; Wei, W.; Gao, D.; Hahn, W.C.; Khosravi-Far, R. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14–3-3 and AKT. Mol. Biol. Cell. 2010, 21, 1140–1152. [Google Scholar] [CrossRef]
- Jiao, P.; Feng, B.; Xu, H. Mapping MKP-3/FOXO1 interaction and evaluating the effect on gluconeogenesis. PLoS One 2012, 7, e41168. [Google Scholar] [CrossRef]
- Ouyang, W.; Liao, W.; Luo, C.T.; Yin, N.; Huse, M.; Kim, M.V; Peng, M.; Chan, P.; Ma, Q.; Mo, Y.; et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 2012, 491, 554–559. [Google Scholar] [CrossRef]
- Chow, K.T.; Timblin, G.A.; McWhirter, S.M.; Schlissel, M.S. MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 2013, 210, 1621–1634. [Google Scholar] [CrossRef]
- Cossa, G.; Lanzi, C.; Cassinelli, G.; Carenini, N.; Arrighetti, N.; Gatti, L.; Corna, E.; Zunino, F.; Zaffaroni, N.; Perego, P. Differential outcome of MEK1/2 inhibitor-platinum combinations in platinum-sensitive and -resistant ovarian carcinoma cells. Cancer Lett. 2014, 347, 212–224. [Google Scholar] [CrossRef]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14–3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef]
- Karger, S.; Weidinger, C.; Krause, K.; Sheu, S.Y.; Aigner, T.; Gimm, O.; Schmid, K.W.; Dralle, H.; Fuhrer, D. FOXO3a: A novel player in thyroid carcinogenesis? Endocr. Relat. Cancer 2009, 16, 189–199. [Google Scholar]
- Ju, Y.; Xu, T.; Zhang, H.; Yu, A. FOXO1-dependent DNA damage repair is regulated by JNK in lung cancer cells. Int. J. Oncol. 2014, 44, 1284–1292. [Google Scholar]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Shats, I.; Gatza, M.L.; Liu, B.; Angus, S.P.; You, L.; Nevins, J.R. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013, 73, 6056–6067. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal. 2010, 5, 10. [Google Scholar]
- Burris, H.A. Dual kinase inhibition in the treatment of breast cancer: Initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist 2004, 9, 10–15. [Google Scholar] [CrossRef]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; Defazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110α inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goltsov, A.; Deeni, Y.; Khalil, H.S.; Soininen, T.; Kyriakidis, S.; Hu, H.; Langdon, S.P.; Harrison, D.J.; Bown, J. Systems Analysis of Drug-Induced Receptor Tyrosine Kinase Reprogramming Following Targeted Mono- and Combination Anti-Cancer Therapy. Cells 2014, 3, 563-591. https://doi.org/10.3390/cells3020563
Goltsov A, Deeni Y, Khalil HS, Soininen T, Kyriakidis S, Hu H, Langdon SP, Harrison DJ, Bown J. Systems Analysis of Drug-Induced Receptor Tyrosine Kinase Reprogramming Following Targeted Mono- and Combination Anti-Cancer Therapy. Cells. 2014; 3(2):563-591. https://doi.org/10.3390/cells3020563
Chicago/Turabian StyleGoltsov, Alexey, Yusuf Deeni, Hilal S. Khalil, Tero Soininen, Stylianos Kyriakidis, Huizhong Hu, Simon P. Langdon, David J. Harrison, and James Bown. 2014. "Systems Analysis of Drug-Induced Receptor Tyrosine Kinase Reprogramming Following Targeted Mono- and Combination Anti-Cancer Therapy" Cells 3, no. 2: 563-591. https://doi.org/10.3390/cells3020563
APA StyleGoltsov, A., Deeni, Y., Khalil, H. S., Soininen, T., Kyriakidis, S., Hu, H., Langdon, S. P., Harrison, D. J., & Bown, J. (2014). Systems Analysis of Drug-Induced Receptor Tyrosine Kinase Reprogramming Following Targeted Mono- and Combination Anti-Cancer Therapy. Cells, 3(2), 563-591. https://doi.org/10.3390/cells3020563