Abstract
The Gram-negative bacterium Aggregatibacter actinomycetemcomitans is part of the HACEK group that causes infective endocarditis, a constituent of the oral flora that promotes some forms of periodontal disease and a member of the family of species that secrete a cytolethal distending toxin (Cdt). The family of bacteria that express the cdt genes participate in diseases that involve the disruption of a mucosal or epithelial layer. In vitro studies have shown that human gingival epithelial cells (HGEC) are native targets of the Cdt that typically induces DNA damage that signals growth arrest at the G2/M interphase of the cell cycle. The gingival epithelium is an early line of defense in the oral cavity against microbial assault. When damaged, bacteria collectively gain entry into the underlying connective tissue where microbial products can affect processes and pathways in infiltrating inflammatory cells culminating in the destruction of the attachment apparatus of the tooth. One approach has been the use of an ex vivo gingival explant model to assess the effects of the Cdt on the morphology and integrity of the tissue. The goal of this review is to provide an overview of these studies and to critically examine the potential contribution of the Cdt to the breakdown of the protective gingival barrier.
1. Introduction
Aggregatibacter actinomycetemcomitans is a member of the taxonomic family Pasteurellaceae that also includes the genera Actinobacillus/Aggregatibacter [1], Haemophilus, Mannheimia and Pasteurella. This species along with Actinobacillus segnis, Actinobacillus aphophilus, Haemophilus influenzae, H. parainfluenzae, Cardiobacterium hominis, Eikenella corrodens and Kingella kingae, form the HACEK group of bacteria that cause rare cases of infective endocarditis in children [2]. Aggregatibacter actinomycetemcomitans has also been strongly implicated in the development of localized aggressive periodontitis (LAP) and possibly contributes to chronic periodontitis (CP), two derivatives of periodontal disease. The disease is initiated by a persistent polymicrobial infection [3,4] and sustained by interactions between the microbial antagonists and host immune system [5]. This bacterium, along with other members of the pathogenic periodontal microflora, produces a variety of products that directly interact with or damage cells and tissues. However, A. actinomycetemcomitans is the only indigenous member of the human oral flora identified to date that expresses complex operons for two cytotoxins—a leukotoxin (Lkt) [6] and cytolethal distending toxin (Cdt) [7]. These toxins have significant potential to contribute to the pathogenesis of periodontal diseases [8].
The A. actinomycetemcomitans Cdt is a member of a family of related toxins present in a group of Gram-negative bacterial species that are facultative or microaerophilic and key pathogens in diseases that involve the perturbation of a mucosal (enteritis, gastric ulcers, chancroid) or epithelial (periodontal diseases) layer. By convention the various Cdts are identified by an abbreviated genus and species prefix such as AaCdt [9]. Studies in my laboratory have been focused on the AaCdt and the possible role of this toxin in the breakdown of the human gingival epithelium. Perturbation of this tissue is thought to be an early event in periodontal disease. Bacterial sampling of global and country-specific patient populations has shown that a significant proportion of oral isolates of A. actinomycetemcomitans carry the cdt genetic locus. Strains that have cdt gene sequences and exhibit associated cytotoxic activity have been recovered with reasonable frequency from subjects diagnosed with periodontal disease [10,11,12,13,14,15,16]. Systemic Cdt antibodies have been found in periodontitis patients indicating infection with Cdt+ strains [17,18,19]. In our studies, all fresh clinical isolates of A. actinomycetemcomitans obtained from a large geographically homogeneous population of LAP families contain a chromosomal locus for the Cdt [20,21]. Although some of these isolates have cdt gene deletions of various lengths, all members of one restriction fragment length polymorphism (RFLP) cluster group contain a complete cdt operon [7]. There was a high statistical correlation between this RFLP group II and conversion of young children from a healthy to diseased periodontal status [22]. More recently, a study of 249 isolates of A. actinomycetemcomitans from 200 Ghanian adolescents were screened for serotype, the presence of cdt gene sequences and the ability to induce cell cycle arrest of HL-60 cells [23]. Complete cdt gene sequences were found in 79% of the isolates examined and all of these isolates exhibited Cdt activity. Fifty-three percent of the Cdt+ isolates correlated with attachment loss indicative of LAP. In another recent study, A. actinomycetemcomitans isolated from 255 subgingival samples from aggressive and chronic periodontitis and clinically healthy sites in 30 Chinese subjects were screened for only the cdtB gene sequence [24]. The cdtB gene was detected in isolates from 78% of the aggressive sites, 74% of the chronic sites and none of the healthy sites. Although that study concluded that Cdt+ strains may correlate with disease, no attempt was made to confirm that the cdtB+ isolates produced active toxin. In total these studies support an indirect and less than 100% association between the Cdt and LAP. Thus, an elusive goal has been to link Cdt activity with the manifestation of specific in vivo events characteristic of the disease. As best stated in a recent review, “One of the true challenges in the CDT field is to understand the in vivo consequences of CDT action during infection” [25]. The goal of this review is to present and critically analyze current information supporting the hypothesis that the AaCdt plays a role in early events associated with periodontal disease.
2. Aggregatibacter actinomycetemcomitans Cdt Structure and Function
2.1. Cell Surface Recognition
The cdt operon resides on the A. actinomycetemcomitans chromosome [7]. The three structural genes, cdtA, cdtB and cdtC, are expressed from a polycistronic operon [26,27,28]. The sizes of the AaCdt gene products are approximately 18–25 kDa (CdtA), 31 kDa (CdtB) and 21 kDa (CdtC). All three proteins are required to produce a toxin that can intoxicate cells and inhibit their proliferation.
Based on the crystal structure of the AaCdt [29], the products of the cdtA and cdtC genes are predicted to form a heterodimer partially separated by a deep groove which functions as a binding site for a receptor on the target cell surface (Figure 1). Studies using the EcCdt indicated that CdtA and CdtC recognize N-linked fucose moieties on the surface of HeLa cells [30]. This binding specificity was exploited to employ an ELISA assay, based on the fucose-containing glycoprotein thyroglobulin, to study binding kinetics of the AaCdtA and AaCdtC subunits [29]. Recombinant CdtA alone can bind to cells in culture, based on the results of immunofluorescence experiments [27,30,31] and in an enzyme-linked immunosorbent assay on live cells (CELISA) [32,33]. CdtC has limited amino acid sequence similarity with CdtA [32,33] suggesting, but not proving, a related function. Some studies have shown that recombinant CdtC binds to HeLa and HEp-2 cells in culture and in the CELISA [30,33,34]. In contrast, other studies have found that CdtC binds poorly to CHO cells [32]. In addition to these data which showed strong binding of the Cdt to membrane glycoproteins, a separate study found that recombinant AaCdt binds to gangliosides GM1 [Galβ(1,3)GalNAcβ(1,4)[NeuAcα(2,3)]-Galβ(1,4)Glc-ceramide], GM2 [GalNAcβ(1,4)[NeuAcα(2,3)]-Galβ(1,4)Glc-ceramide] and GM3 [NeuAcα(2,3)Galβ(1,4)Glc-ceramide] [35]. GM3 is a component of membrane lipid rafts [36]. Lipid rafts are areas of the cell membrane that are enriched for cholesterol and have been implicated in Cdt binding primarily via a cholesterol recognition/interaction amino acid consensus (CRAC) motif in CdtC [37,38]. Application of a novel haploid genetic screening method, using a derivative of a chronic myeloid leukemia cell line, found that recombinant EcCdt binds to the G-protein-coupled receptor TMEM181 [39]. In spite of the information obtained from these binding kinetic studies, the identity of a specific cell surface receptor for the AaCdt remains elusive. It is becoming apparent that the species-specific Cdts, or subgroups containing the Aggregatibacter/Haemophilus and Escherichia/Salmonella/Campylobacter/Helicobacter Cdts, may have distinct host cell receptors and mechanisms of intoxication [38]. However, the reason why the various species-specific Cdts have different properties has not yet been deciphered at the molecular level.
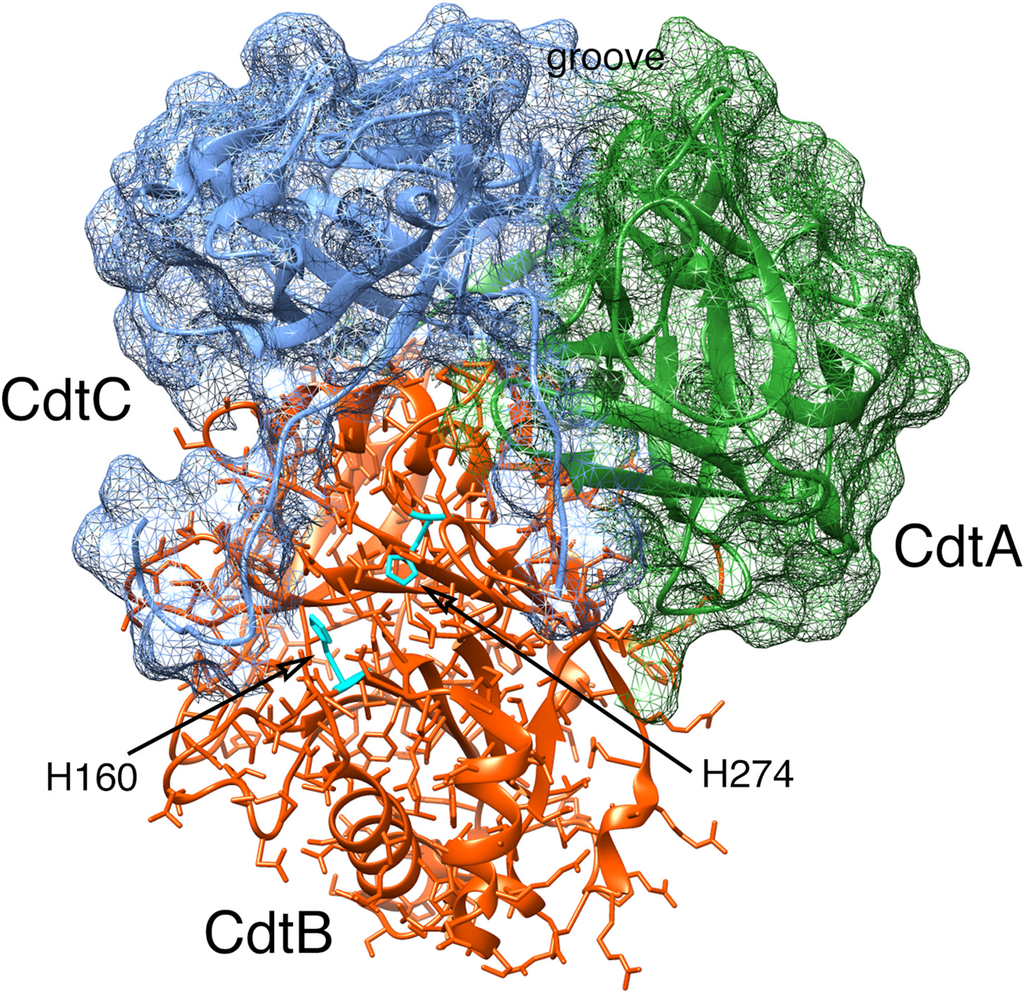
Figure 1.
Computer model of the AaCdt showing the structural relationship of the three subunits: CdtA, CdtB and CdtC. The subunits are depicted as ribbon backbone models using the Protein Data Bank (PDB) file 2F2F, deposited by Yamada et al. [29], and UCSF Chimera 1.8.1. Side chains are shown only in CdtB. The surfaces of CdtA and CdtC are shown as mesh models. Two residues, H160 and H274, in the active site of CdtB are required for toxin activity. Abbreviations: Cdt, cytolethal distending toxin.
2.2. Cytotoxicity
The third subunit, or product of the cdtB gene, represents the biologically active component and has to enter cells to elicit virulent effects. A broad comparison of deduced amino acid sequences shows that CdtB belongs to a superfamily of enzymes that includes the endonucleases, exonucleases, sphingomyelinases, and inositol polyphosphate 5-phosphatases [40]. The phylogenetic relationships imply that CdtB appears to be most closely related to prokaryotic and bacteriophage deoxyribonuclease I and sphingomyelinase C (Figure 2; subclusters 3a, 3b and 3c) with broader similarities to phosphatidylinositol-3,4,5-triphosphate phosphatase and eukaryotic deoxyribonuclease I (clusters 1 and 2). Based on these phylogenetic relationships CdtB has the potential to exhibit multiple enzymatic activities in vitro and in vivo.
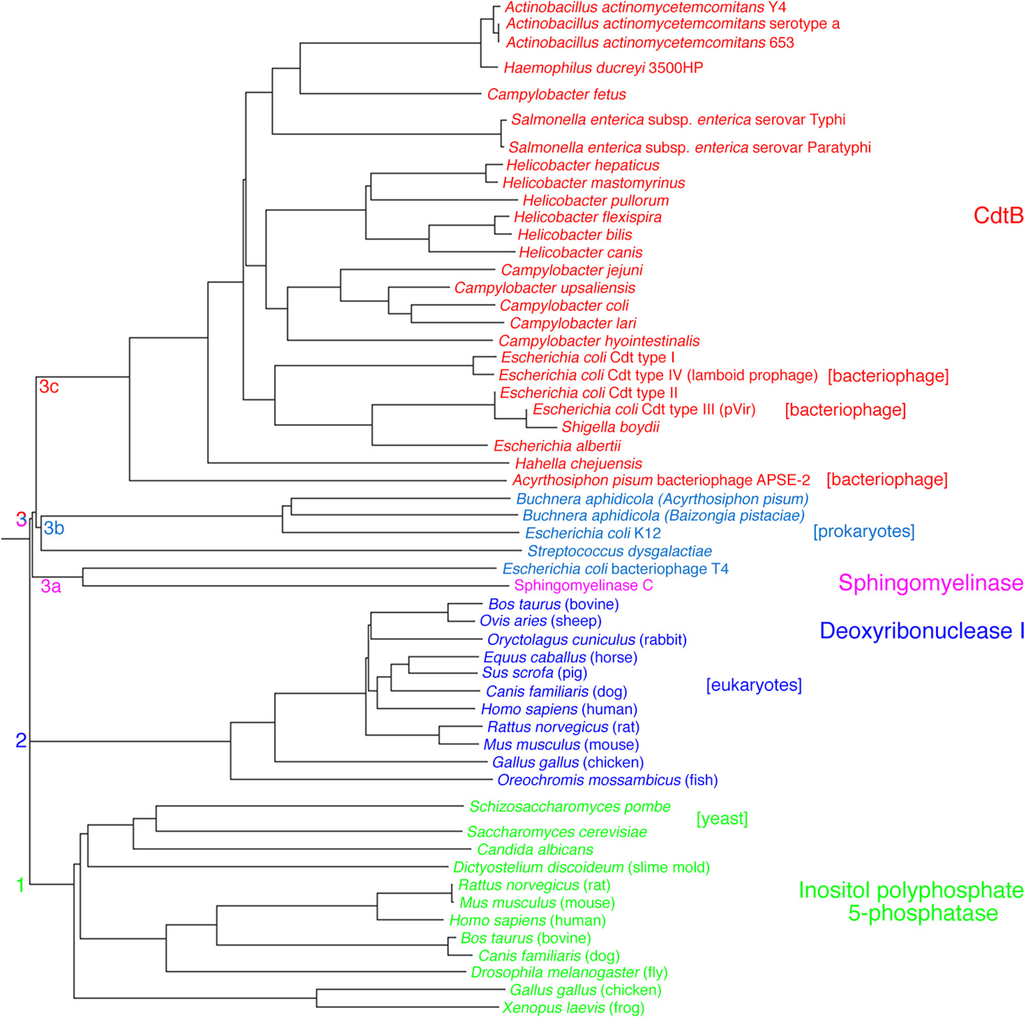
Figure 2.
Rooted phylogenetic tree of the deduced amino acid sequences of a representative group of deoxyribonucleases (DNase I), inositol polyphosphate 5-phosphatases and sphingomyelinase. The tree was constructed with PHYLIP 3.6. The lengths of the lines show relative genetic distances.
2.2.1. Primary Activities Associated with CdtB
DNA damage in the form of double-strand breaks, as assessed by pulsed-field gel electrophoresis, is one of the CdtB-related activities observed in sensitive cell types exposed to the heterotrimer [31,41,42]. CdtB requires divalent cations for DNA-nicking activity [43,44] with an optimum concentration of 50 mM MgCl2, CaCl2 or MnCl2 for the AaCdtB [45]. Mutating conserved deoxyribonuclease active site residues His160 and His274 in the AaCdt [46], or their equivalent His residues in the other species-specific CdtBs, result in the loss of both in vitro nuclease activity and cell cycle arrest [44,47]. Therefore, CdtB can act as a neutral, cation-dependent deoxyribonuclease I-like nuclease in most susceptible cell types examined [31,45]. This DNA-damaging activity is unusual for a bacterial cytotoxin and classifies the Cdt as a genotoxin [48].
The fact that CdtB is a member of the large metaloenzyme superfamily led to the proposal that the toxin acted upon susceptible cells or cell lines by dephosphorylation of the Wee1 kinase or Cdc25 phosphatase [49]. These two enzymes are important for regulating the phosphorylation of tyrosine residues in Cdc2 kinase. An increase in Cdc2 phosphorylation leads to cell cycle arrest. This rationale was used to form the hypothesis that the primary mode of action of CdtB is that of a phosphatase rather than that of a nuclease. Additional support for the phosphatase mode of action came from studies showing that recombinant AaCdtB behaved as a phosphatidylinositol-3,4,5-triphosphate phosphatase in lymphoid cell lines [50]. It was proposed that the action of the Cdt in lymphocytes causes a depletion of phosphatidylinositol-3,4,5-triphosphate which in turn inactivates the Akt pathway leading to cell cycle arrest and apoptosis. Human macrophages exposed to the AaCdt experience inhibition of phosphatidylinositol-4,5-bisphosphate 3-kinase signaling pathway and an associated increase in glycogen synthase kinase 3β affecting production and secretion of the cytokines interleukin (IL)-1β), IL-6 and tumor necrosis factor (TNF)-α [51]. However, the macrophages did not exhibit Cdt-induced apoptosis.
2.2.2. Cell Signaling Activities
It has been reported that the AaCdt stimulates the synthesis of IL-1β, IL-6, and IL-8 in human peripheral blood mononuclear cells (PBMC) [26] and IL-8 in CjCdt-treated human embryo intestinal epithelial cells (INT407) [52]. Nitric oxide production was altered in macrophage cultures due to the presence of the AaCdt [53]. Exposure of macrophages to the toxin also resulted in increased levels of IL-1β, IL-10, IL-20 and TNF-α. These findings indicated that the AaCdt may modulate macrophage function in vivo by upsetting the equilibrium between pro-inflammatory and anti-inflammatory cytokines. However, recombinant AaCdt failed to stimulate IL-1β in human periodontal ligament fibroblasts but up-regulated IL-6 and the receptor activator of NF-κB ligand (RANKL) in these cells [54,55]. Another report showed that HGF challenged with whole A. actinomycetemcomitans bacteria exhibited increased expression of IL-6 and IL-8 [56]. This increase in expression was not attributed to a specific product but the Cdt is a reasonable candidate. RANKL was also up-regulated in response to the HdCdt in a T-lymphocyte leukemia cell line (Jurkat) [57]. RANKL is a member of the TNF ligand superfamily and binds to its associated receptor RANK on osteoclast progenitor cells [58]. This action leads to differentiation of the progenitor cells into bone-resorbing osteoclasts [59]. Since expression of these molecules is involved in the induction of osteoclast differentiation they have a central role in the regulation of bone resorption that is a key feature of periodontitis [60,61].
2.2.3. Cell Cycle Arrest as a Result of DNA Damage
Early studies of the Cdt showed that the toxin was active against HeLa, Hep-2, CHO and Vero cells [26,62,63,64,65,66]. Caco-2, a well-known human colon carcinoma cell line [67], and two human keratinocyte cell lines, HaCat [68,69] and Henle-407 (intestinal epithelial cells) [47], experience cell cycle arrest when exposed to the Cdt. DNA damage induced by CdtB typically leads to cell cycle arrest due to activation of checkpoint responses. Cell cycle checkpoints guard the faithful progression to cell division when DNA damage is detected. Subsequent growth arrest results from the block in cell cycle progression that occurs in response to the Cdt at the either the G1/S or G2/M phase transitions. The pathways for cell cycle arrest in response to the Cdt appear to be cell-type specific and have been outlined in various reviews [70,71,72]. Epithelial-like cell lines such as HeLa [66,67,73], HEp-2 [26] and Caco-2 [67] as well as immortalized human gingival keratinocytes [74] are arrested at the G2/M phase transition (Figure 3). The key inhibitory step in this Cdt-altered pathway is the failure of protein phosphatase Cdc25 to dephosphorylate checkpoint protein kinase Cdc2 (Cdk1). Toxins that modulate the eukaryotic cell cycle have been termed cyclomodulins [72,75].
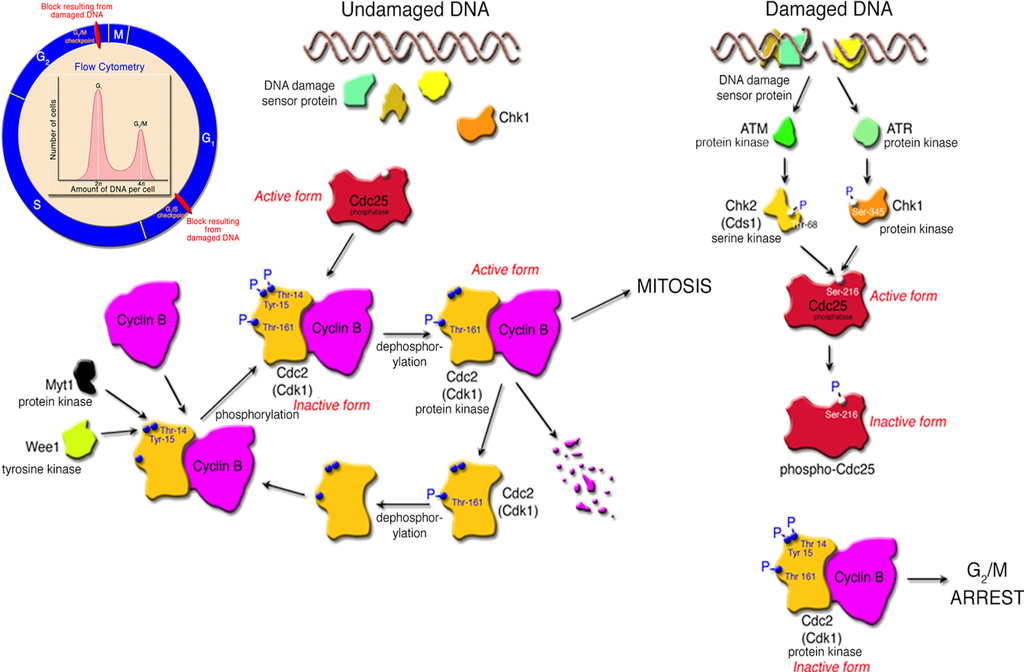
Figure 3.
Checkpoint pathway, in response to DNA damage, that leads to growth arrest at the G2/M interphase of the cell cycle in epithelioid cells. The inset illustrates the standard DNA damage checkpoints at G1/S and G2/M in the cell cycle. Cell cycle arrest at G2/M can be measured by flow cytometry or cell sorting. Populations that are arrested at G2/M accumulate cells that have a 4n DNA content.
HEp-2 and KB cell lines, generally thought to have originated from epidermoid carcinomas of the mouth and larynx, respectively, are sensitive to the AaCDT [76]. This is particularly significant since the KB cell line had been used extensively in A. actinomycetemcomitans invasion studies [77,78,79]. Furthermore, an immortalized human orolabial cell line, GMSM-K [simian virus (SV40) transformed] [80], is particularly sensitive to the Cdt and served as an early epithelial cell model for AaCdt studies prior to our ability to culture primary human gingival epithelial cells (HGEC) [31,81]. Interestingly, this cell line was arrested at the S phase of the cell cycle in response to the toxin.
Primary cells isolated from the epithelial layers of human gingival tissue, obtained from routine crown-lengthening procedures, had an epithelioid morphology, bound a monoclonal antibody specific for epithelial cell adhesion molecule (Ep-CAM) and failed to bind an anti-fibroblast CD90/Thy-1 antigen (Ab-1) antibody [82]. These cells arrested at the G2/M interphase of the cell cycle when exposed to recombinant AaCdt for 18 h. There was no effect on the cell cycle when these cells were exposed to toxin containing the mutated CdtB subunit (CdtABH160AC) for up to 36 h.
In our hands, oral cells of mesenchymal origin such as human periodontal ligament fibroblasts (HPLF) [31], human gingival fibroblasts (HGF) [82] cementoblasts [81] and osteoblasts (unpublished observations) appear to be resistant to the DNA-damaging and cell cycle arresting effects of the Cdt. Cells cultured from the connective tissue layer of gingival tissue exhibited morphology typical of fibroblasts and bound the CD90/Thy-1 (Ab-1) antibody. Cultures of these HGF failed to accumulate cells having a 4n DNA content upon continuous exposure to recombinant AaCdt for up to 96 h [82]. Others have reported a non-lethal inhibition of proliferation of HPLF attributed to the AaCdt [83] and, in a subsequent study, cell cycle arrest at both G1 and G2/M in HPLF and HGF [84]. It is unclear why there are conflicting results. However, the reproducible ability of the AaCdt to inhibit the proliferation of primary HGEC suggests that a predominant activity of the toxin in vivo is in damaging the protective epithelial barrier that is important in preventing the initial development of periodontal disease.
3. Breakdown of the Gingival Epithelial Barrier
3.1. Gingival Tissue
Since it was observed that epithelial cells are highly susceptible to Cdt-mediated intoxication they are likely to be natural targets of the AaCdt in the noninflamed or healthy oral cavity. Gingival epithelium is made up of three components: oral (OE), sulcular (SE) and junctional epithelium (JE) [85]. The OE is a keratinizing form of epithelium and provides a very effective physical barrier to microbial invasion of the underlying gingival connective tissue (CT). In contrast, junctional epithelium, and in some instances sulcular epithelium, are non-keratinizing forms of epithelium. Therefore, the junctional and sulcular epithelium are semi-permeable allowing the transport of soluble macromolecules from the gingival sulcus into the underlying connective tissue. The basal cell layers of all three types of gingival epithelia are composed of rapidly proliferating cells that migrate towards the outer surface of the tissue. In this process the cells leave the cell cycle and start to differentiate. Relative to oral epithelium and sulcular epithelium this process is associated with loss of expression of the cytokeratins K5, K14 and K19 found in basal cells. Suprabasal junctional epithelial cells continue to express K19 [85]. Thus, junctional epithelium is considered to be a less well-differentiated type of epithelium. Subsequently, the suprabasal cells of all three layers start to express different patterns of alternative cytokeratins. Terminally differentiated cells found in keratinizing epithelia also express a unique group of proteins that includes filaggrin, involucrin and loricrin [86,87].
The gingival epithelial surface is under constant assault by biofilm-forming bacteria, including A. actinomycetemcomitans, which colonize the human oral cavity. Colonization of the subgingival microenvironment places A. actinomycetemcomitans in close proximity to junctional and sulcular epithelium cells. This tissue layer creates an early line of defense against microorganisms by forming a barrier that blocks or, at least minimizes, bacterial invasion of the underlying connective tissue [88,89]. Bacterial products, along with factors released from recruited inflammatory cells, induce degradation of connective tissue and bone resulting in damage to the supporting structures of the teeth. Therefore, breakdown of this epithelial barrier early in the development of periodontal disease has significant consequences for oral health. Two key observations provided the rationale to test the effects of the AaCdt in a human ex vivo model. (i) All members of the Cdt family are produced by bacterial species associated with diseases that involve tissue lined with epithelial cells. Additionally, (ii) epithelioid cells are particularly sensitive to the anti-proliferative related activities of the toxin.
3.2. Damaging Effects of Cdt on Human Gingival Tissue
Previous studies had shown that human gingival explants (HGX) could survive in the laboratory with the basal epithelial cells exhibiting intense proliferative activity for up to 48 h [90]. No evidence of degenerative changes or de-differentiation of cells in the explants was detected for up to four days of incubation [91]. We found that clinically healthy tissue originating from routine crown lengthening surgeries conducted on physically healthy adults maintained their morphological integrity in tissue culture medium for up to 36 h ex vivo [82]. Histological staining of untreated tissue sections with hematoxylin and eosin (H&E) showed that the oral epithelium, rete pegs (RP) and connective tissue appeared normal for a minimum of 24 h. However, in some sections a slight mechanical separation of the keratinized surface layer (KL) was observed after 18 h of incubation.
3.2.1. Changes in Tissue Morphology and Cellular Organization
The epithelial cell layers of gingival explants exposed to recombinant AaCdt for 18 h exhibited pronounced separation of the keratinized surface layer, extensive disruption of the epithelial layers marked by a swollen appearance or thickening and a loss of structural integrity of the rete pegs [82]. Cells in the oral epithelium were dramatically distended and interstitial spacing between the cells increased dramatically indicating an apparent loss of cell junctions. The morphological changes were reminiscent of the histology of gingival tissue obtained from patients who had clinical signs of periodontal disease. Very similar effects were observed when recombinant AaCdt was topically applied to the palatal gingival sulcus of the maxillary molars of rats [92].
To confirm that the Cdt-induced changes in morphology were specific to the toxin, gingival explants were also treated with Cdt reconstituted with the mutated CdtB subunit (CdtABH160AC). Gingival explants exposed to CdtABH160AC for up to 36 h appeared to be unaffected. Both the wild-type and mutated CdtB subunit were found predominantly in the oral epithelium as detected by immunofluorescence. A minimal amount of CdtB appeared to reach the connective tissue that most likely was due to the epithelial damage induced by the toxin.
3.2.2. Disruption of Cell Junctions
Abnormal physiological processes associated with disease can disrupt cell-cell contact leading to degradation of tissue organization and structure [93]. Perturbations in the regulation of cell-cell junction stability and dynamics impact the barrier properties of epithelial layers and can have detrimental effects on tissue remodeling [94].
Changes in tissue morphology were observed following exposure to the AaCdt that upon general appearance could be predicted to be due to a loss of cell-cell contact. Contact between epithelial cells, essential for tissue integrity, is maintained by complexes consisting of transmembrane, cytosolic and cytoskeletal proteins. The two major types of junctions relevant to epithelial cells located in oral epithelium are zonula occludens or tight junctions which contain the transmembrane proteins claudin and occludin and the cytoplasmic protein ZO-1 [95] and zonula adherens or adherens junctions which are complexes that interact with intracellular scaffolding and signaling molecules [95,96,97,98]. Two other types of cell junctions include macula adherens or desmosomes which are transmembrane glycoproteins (desmogleins) found in stratified squamous epithelia such as that found in the soft tissue lining of the mouth, surface layer of the skin, the esophagus and vagina [93] and gap junctions which are made up of paracrystalline connexin channels in a multitude of tissues [93,99].
The presence of tight junctions in human gingival epithelium was suggested previously but not unequivocally demonstrated [100,101]. Microscopic evidence of tight junctions in human gingival biopsies was reported [102]. However, there were areas in the tissue where tight junctions were not detected. In another study using cultured human gingival keratinocytes only a minimal increase in transmembrane electrical resistance (TER) was detected as the cells formed multilayers and this increase was not altered by exposure of the cells to either Porphyromonas gingivalis or A. actinomycetemcomitans for up to 24 h [103]. It has been reported that P. gingivalis is capable of degrading epithelial cell-cell junction complexes [104]. Attempts were made to use changes in TER to measure the effect of the Cdt on tight junction maintenance in post-confluent cultures of HGEC isolated from the HGX [105]. However, this approach was unsuccessful because the HGEC apparently failed to form tight junctions in vitro. This was confirmed by the low concentration of claudin 1, a primary component of tight junction filaments [106], in the cells as determined by immunofluorescence. Attempts to identify tight junctions in the gingival explants were also unsuccessful since technical limitations preclude the measurement of TER in tissue and claudin 1 was not readily detected.
Ye et al. [107] reported a relatively high level of expression of E-cadherin in the basal cell layer of healthy human gingiva. Therefore, E-cadherin was used as a marker to examine the effects of the recombinant AaCdt on adherens junctions in situ [105]. E-cadherin localized predominantly at the plasma membrane, outlining epithelial cell-cell contact zones in the oral epithelium of untreated HGX. E-cadherin staining was absent on the surface of those epithelial cells that lined the leading edge of the rete pegs that were in contact with the basement membrane. The absence of E-cadherin at this site was most likely due to the fact that these cells attach to the basement membrane via hemidesmosomes. Staining with wheat germ agglutinin confirmed the presence of intact epithelial cell membranes in those cells that were adjacent to the basement membrane. These results supported previous observations that normal oral epithelium showed strong pericellular staining of E-cadherin in the basal and suprabasal cell layers but not in the basal aspect of keratinocytes adjacent to the basement membrane [108].
Exposure of the HGX to the AaCdt resulted in a statistically significant increase in the E-cadherin mRNA expression level in 50% of HGX and 66% of HGEC cultures obtained from 19 clinically healthy subjects. A pronounced increase in the relative fluorescence of E-cadherin in the oral epithelium of the tissue correlated with an increase in E-cadherin mRNA expression in those samples examined. The protein was detected throughout the cytosol indicating extensive intracellular redistribution. E-cadherin was now detected on the surface of those cells that lined the rete peg-basement membrane border in the Cdt-treated HGX. Not only did this observation confirm that Cdt induces redistribution of E-cadherin within the epithelial cells but also indicates that the toxin causes detachment of basal cells from the basement membrane. Exposure of HGX to CdtABH160AC minimally affected the intensity of E-cadherin staining but not to the extent of that observed in HGX incubated with wild type toxin. HGX from patients that had clinical signs of periodontal disease exhibited relatively high levels of E-cadherin in the spinous layer.
3.2.3. Changes in Expression of Intracellular Scaffolding Proteins
Adherens junction structure relies on dynamic interactions among E-cadherin, catenin complexes and the actin cytoskeleton [94,97,109,110,111,112]. The accumulation of F-actin assemblies having a morphology similar to that of actin stress fibers were observed in CHO cells treated with recombinant EcCdt [113]. This change in the structural apparatus of the cells correlated with an inhibition of cell division and, therefore, further demonstrated the influence of the Cdt on the regulation or function of cell cycle-dependent events leading to cytokinesis.
We found that 25% of HGX and 50% of 19 HGEC cultures examined showed a statistically significant increase in β-catenin mRNA expression after exposure to the recombinant AaCdt [105]. Actin mRNA expression increased in 63% of HGX and 83% of the HGEC cultures following Cdt-treatment. The β-catenin concentration in the spinous layer of selected HGX was elevated, relative to that in untreated tissue, following exposure to the recombinant Cdt for 18 h. There was also a marked increase in actin staining as early as 6 h after the addition of the toxin. HGX from patients that had clinical signs of periodontal disease exhibited relatively high levels of β-catenin in the spinous layer.
The Cdt induces significant changes in both the distribution and expression of the component proteins of the adherens junction complex in both HGEC and HGX from a majority of samples examined. These effects may be associated with the cellular distention caused by the toxin since stretching of the cells would be expected to exert physical effects on the membrane and cell surface. The variability detected among the tissue samples in response to the Cdt could be due to a multitude of environmental and/or donor-specific factors. The precise mechanism by which the Cdt affects the expression and redistribution of the adherens junction complex is not known at this time.
3.3. Gingival Breakdown Model
Our studies provide compelling evidence for the contribution of the AaCdt to the breakdown of the human gingival epithelium. As noted in a recent review, “These results, showing a direct damage of human oral epithelium and mucosa by CDT, indicate a clear role for AaCDT in the pathogenesis of periodontitis.” [114]. Based on the ex vivo effects of the toxin on HGX a model is proposed detailing the role of the Cdt in breakdown of the gingival epithelial barrier and subsequent contribution to events leading to the development of periodontal disease (Figure 4).
There is experimental evidence indicating that the Cdt is secreted as an active heterotrimer by A. actinomycetemcomitans [115]. This is indirectly supported by a myriad of studies that have used cell-free medium from exponential or stationary phase cultures of A. actinomycetemcomitans as a source of toxin for cell culture experiments and activity assays. Therefore, A. actinomycetemcomitans can colonize the oral cavity and continuously release the Cdt. It is assumed that the cdt genes are constitutively expressed because there is no direct evidence to show either transcriptional or post-transcriptional regulation. It is possible that the splicing of intervening sequences from cdt transcripts may be a mechanism of regulating the expression of the AaCdt proteins [116]. However, there is no proof of such a mechanism of post-transcriptional control. It has been proposed, based on the analysis of mutations in the luxS gene, that the CjCdt is under the regulation of autoinducer AI-2 (furanosyl borate diester) [117]. However, there is no further evidence to support this mechanism in the regulation of the AaCdt.
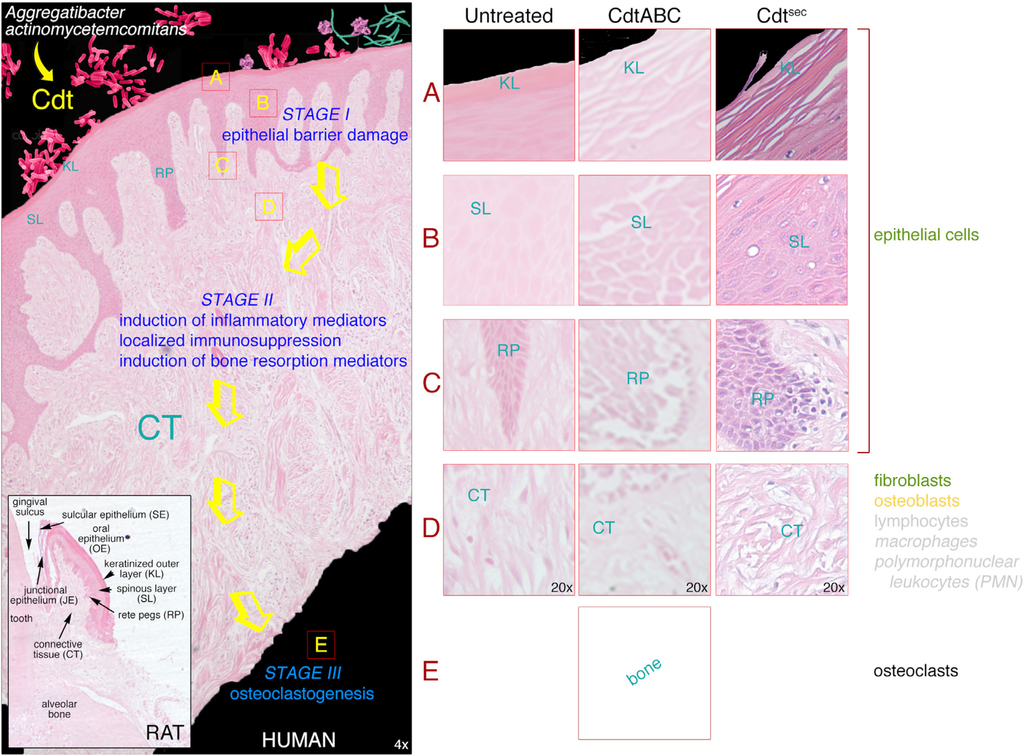
Figure 4.
Human gingival explant (HGX) model of the effects of secreted AaCdt on tissue obtained from clinically healthy subjects. HGX were untreated or exposed to recombinant wild-type AaCdt (CdtABC) or native toxin secreted by the bacterium (Cdtsec). Tissue sections were stained with hematoxylin and eosin. Abbreviations: KL, keratinized layer; SL, spinous layer; RP, rete peg; CT, connective tissue. Inset shows the overall histology of the gingival tissues and various types of epithelium (junctional, sulcular and oral) in the upper jaw of a rat. The tissue was decalcified, sectioned and stained with hematoxylin and eosin.
Since the Cdt is a secreted product the bacterium does not have to be in direct contact with the gingival epithelium or epithelial cells in the tissue to impart damage. The secreted toxin can compromise the barrier function of the gingival epithelium by binding to and intoxicating the keratinized epithelial cells at the surface of the tissue, thereby disrupting the keratinized surface layer (Stage IA). The specific molecular mechanism of damage to the keratinized cells in this layer is not known at this time because these are non-proliferating cells. As the cells in the spinous layer start to differentiate their rate of proliferation slows and they eventually exit the cell cycle as they go through terminal differentiation becoming part of the keratinized layer. The keratinized layer appears to slough off the HGX in sections or thin strips in response to the toxin. Disruption of the keratinized layer creates an environment in which the Cdt has access to the highly proliferating cells in the oral epithelium. Cells within the deeper layers of epithelium should therefore be good targets for the toxin since they continuously proliferate prior to terminal differentiation as they migrate towards the epithelial surface [118].
Based on the analysis of the effects of the Cdt on HGEC isolated from the same gingival tissue as that used in the ex vivo experiments, the AaCdt most likely induces irreversible cell cycle arrest (G2/M) of the epithelial cells, in situ, in the spinous layer and rete pegs (Stage IB and IC). This is evidenced by pronounced swelling of the tissue and enlargement or distention of the epithelial cells. Cell-cell contact in the tissue appears to be significantly disrupted by an increase in the interstitial spaces between cells most likely due to the redistribution of adherens junction and scaffolding proteins (Figure 5). The Cdt-induced changes related to cell proliferation within the spinous layer and rete pegs may be secondary to effects on cell-cell junctions. Distention of the HGEC and dissolution of cell junctions could promote more wide-spread penetration of the toxin throughout the tissue. Breakdown of the oral epithelium would also facilitate entry of other members of the periodontal microflora and a host of bacterial products into the connective tissue.
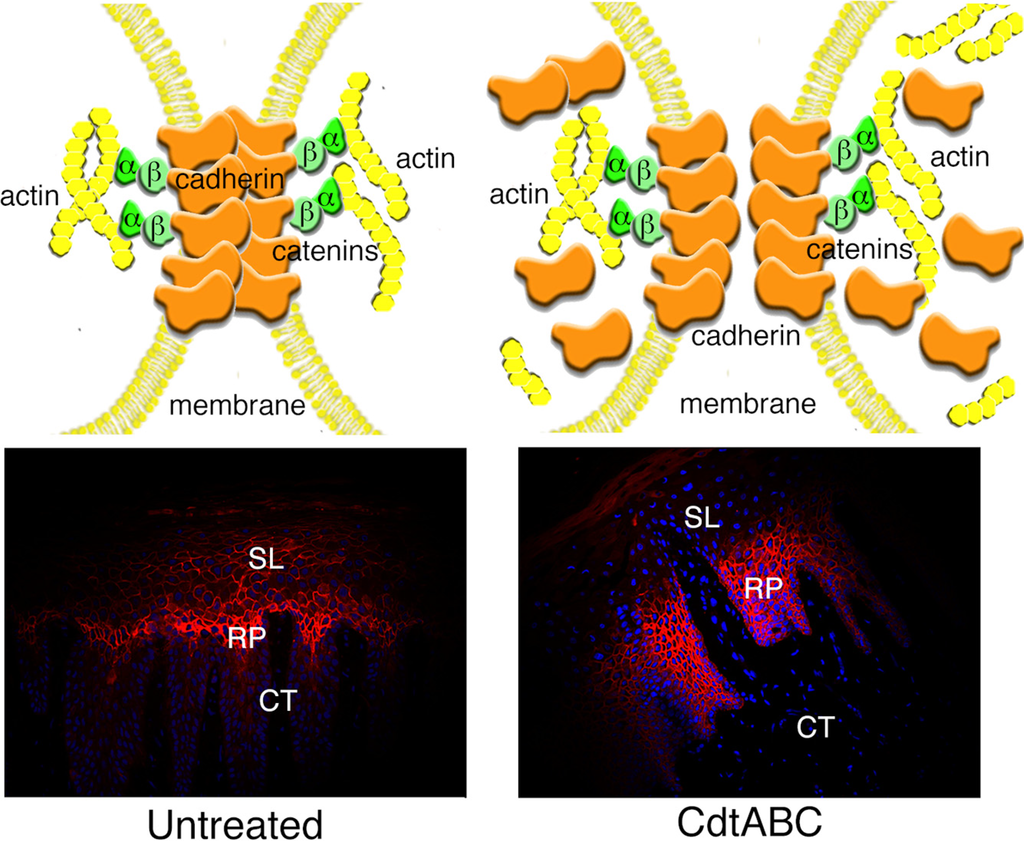
Figure 5.
Model of the effects of the AaCdt on gingival epithelial cell adherens junctions in situ. E-cadherin was detected with a polyclonal antibody conjugated to fluorescein (red fluorescence) in untreated HGX and explants exposed to recombinant AaCdt. Cell nuclei were stained with DAPI (blue fluorescence). Abbreviations: SL, spinous layer; RP, rete peg; CT, connective tissue.
Based on the assessment of the affects of the AaCdt on cultured HPLF and HGF in our studies [31,82], the toxin may not significantly affect the proliferation of the resident fibroblasts and osteoblasts in the extracellular matrix. Our results differed from those of Belibasakis et al. [83,84]. However, the toxin may induce the fibroblasts to release pro-inflammatory cytokines such as IL-6 and mediators of bone resorption such as RANKL in Stage II as shown by that group in in vitro studies [57].
Epithelial cells also function as part of a signaling network that alerts inflammatory cells to a microbial assault [119,120]. In response to the epithelial cell damage and cell signaling induced by the Cdt, inflammatory cells such as lymphocytes, macrophages and polymorphonuclear leukocytes (PMN) can infiltrate and increase in number in the connective tissue. The Cdt stimulates these cells to increase cytokine production and may lead to localized immune suppression by inhibiting the proliferation of the infiltrating T-lymphocytes and macrophages exacerbating the immune response. Human CD4+ and CD8+ T lymphocytes as well as monocytes undergo cell cycle arrest, without cell distention in the G2 phase when treated with extracts containing the AaCdt [121,122]. Lymphocytes treated with recombinant AaCDT undergo apoptosis via activation of the caspase cascade [123]. Furthermore, there is evidence for a role for the apoptosis regulator Bcl-2 [124] in the apoptotic process. These results were confirmed by Nalbant et al. [121] who also found that Cdt-induced apoptosis involves the up-regulation of Fas and FasL, down-regulation of Bcl-2, activation of caspase-3 and was dependent on the presence of monocytes. Binding of the Fas ligand (FasL) to the Fas receptor (CD95) on target cells is one of the pathways by which apoptosis is initiated [125]. Use of a human leukemic cell line (MOLT-4) indicated that two independent pathways, caspase-dependent (early) apoptotic cell death and caspase-independent (late) cell death, may be involved in Cdt-induced death in some types of T-cells [126,127]. Inhibition of immune cell proliferation by cell cycle arrest and/or apoptosis may be a contributing factor to immune suppression that would allow the continued colonization of infected sites by A. actinomycetemcomitans or other periodontal pathogens in periodontal disease. In addition, results of in vitro experiments with U937 cells, a macrophage-like cell line, suggested that macrophages are potential in vivo targets of the toxin [128]. The AaCdt may disrupt macrophages by inhibiting phagocytic activity, modulating nitric oxide production and altering the expression of pro-inflammatory and anti-inflammatory cytokines [53,129]. However, it should also be noted that A. actinomycetemcomitans resides in a biofilm external to the tissue and may be protected to some degree from immune surveillance and clearance by phagocytosis.
Finally, the Cdt-induced mediators, released in Stage II, that stimulate differentiation of progenitor cells into osteobclasts and endotoxin or lipopolysaccharide (LPS) produced by Gram-negative bacteria can lead to osteoclastogenesis (Stage III) which is the defining event in periodontal disease.
4. Concluding Statements
Attempting to provide unequivocal proof that a particular microbial product makes a major contribution to human infectious disease is a daunting undertaking. Periodontal diseases pose a particularly challenging problem due to the complex polymicrobial etiology and the uniqueness of this disease to the human condition. The use of gene knock-out mutants in animals has been employed to study some oral microbial virulence factors. However, existing animal models do not seem to be sufficient to address the unique genotoxic effects of the Cdt. Although the HGX model for characterizing the possible role(s) of the AaCdt in disease-associated tissue damage is not a perfect one, it provides a novel experimental approach to begin to address these important questions. In addition, there are unique advantages in being able to correlate data from an examination of the effects of the toxin on cells in situ with data from experiments that characterize the inhibitory pathways affected by the toxin in the same cells in culture.
The characteristics of the Cdt make a compelling case that this bacterial product is a relevant virulence factor in A. actinomycetemcomitans-associated diseases. Clearly, the AaCdt has the potential to affect cells important to the protection and integrity of the periodontium. In conjunction with the ability of some strains of A. actinomycetemcomitans to invade epithelial cells, the Cdt could greatly enhance the ability of this bacterium, as well as others, to bypass the physical barrier established by the gingival epithelium. Damage to a protective epithelial layer makes the Cdt a key participant in the earliest stages of the development of some forms of periodontal disease and serves as a paradigm for the other members of the Cdt family.
Acknowledgments
Recent experiments in my laboratory were performed by Monica Damek-Poprawa, Linsen Cao and Alla Volgina. I also thank Jonathan Korostoff for contributions to the cell and tissue studies and for helpful discussions.
Research performed in my laboratory as discussed in this review was supported by National Institutes of Health Grants DE012593 and DE017679 from the National Institute of Dental and Craniofacial Research.
Conflict of Interest
The author declares no potential conflict of interest with respect to the authorship and/or publication of this article.
References
- Norskov-Lauritsen, N.; Kilian, M. Reclassification of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, Haemophilus paraphrophilus; Haemophilus segnis as Aggregatibacter actinomycetemcomitans gen. nov., comb. nov., Aggregatibacter aphrophilus comb. nov. and Aggregatibacter segnis comb. nov., and emended description of Aggregatibacter aphrophilus to include V factor-dependent and V factor-independent isolates. Int. J. Syst. Evol. Microbiol. 2006, 56, 2135–2146. [Google Scholar] [CrossRef]
- Feder, H.M.J.; Roberts, J.C.; Salazar, J.; Leopold, H.B.; Toro-Salazar, O. HACEK endocarditis in infants and children: Two cases and a literature review. Pediatr. Infect. Dis. J. 2003, 22, 557–562. [Google Scholar]
- Armitage, G.C. Comparison of the microbiological features of chronic and aggressive periodontitis. Periodontology 2010, 53, 70–88. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune response: The key to bone resorption in periodontal disease. J. Periodontol. 2005, 76, 2033–2041. [Google Scholar] [CrossRef]
- Lally, E.T.; Golub, E.E.; Kieba, I.R.; Taichman, N.S.; Rosenbloom, J.; Rosenbloom, J.C.; Gibson, C.W.; Demuth, D.R. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J. Biol. Chem. 1989, 264, 15451–15456. [Google Scholar]
- Mayer, M.P.; Bueno, L.C.; Hansen, E.J.; DiRienzo, J.M. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect. Immun. 1999, 67, 1227–1237. [Google Scholar]
- Feng, Z.; Weinberg, A. Role of bacteria in health and disease of periodontal tissues. Periodontol 2000, 40, 50–76. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911. [Google Scholar] [CrossRef]
- Aberg, C.H.; Sjodin, B.; Lakio, L.; Pussinen, P.J.; Johansson, A.; Claesson, R. Presence of Aggregatibacter actinomycetemcomitans in young individuals: A 16-year clinical and microbiological follow-up study. J. Clin. Periodontol. 2009, 36, 815–822. [Google Scholar] [CrossRef]
- Ahmed, H.J.; Svensson, L.A.; Cope, L.D.; Latimer, J.L.; Hansen, E.J.; Ahlman, K.; Bayat-Turk, J.; Klamer, D.; Lagergård, T. Prevalence of cdtABC genes encoding cytolethal distending toxin among Haemophilus ducreyi and Actinobacillus actinomycetemcomitans strains. J. Med. Microbiol. 2001, 50, 860–864. [Google Scholar]
- Bandhaya, P.; Saraithong, P.; Likittanasombat, K.; Hengprasith, B.; Torrungruang, K. Aggregatibacter actinomycetemcomitans serotypes, the JP2 clone and cytolethal distending toxin genes in a Thai population. J. Clin. Periodontol. 2012, 39, 519–525. [Google Scholar] [CrossRef]
- Fabris, A.S.; DiRienzo, J.M.; Wikstrom, M.; Mayer, M.P. Detection of cytolethal distending toxin activity and cdt genes in Actinobacillus actinomycetemcomitans isolates from geographically diverse populations. Oral Microbiol. Immunol. 2002, 17, 231–238. [Google Scholar] [CrossRef]
- Nishikubo, S.; Ohara, M.; Ikura, M.; Katayanagi, K.; Fujiwara, T.; Komatsuzawa, H.; Kurihara, H.; Sugai, M. Single nucleotide polymorphism in the cytolethal distending toxin B gene confers heterogeneity in the cytotoxicity of Actinobacillus actinomycetemcomitans. Infect. Immun. 2006, 74, 7014–7020. [Google Scholar] [CrossRef]
- Tan, K.S.; Song, K.P.; Ong, G. Cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Occurrence and association with periodontal disease. J. Periodontal. Res. 2002, 37, 268–272. [Google Scholar]
- Yamano, R.; Ohara, M.; Nishikubo, S.; Fujiwara, T.; Kawamoto, T.; Ueno, Y.; Komatsuzawa, H.; Okuda, K.; Kurihara, H.; Suginaka, H.; et al. Prevalence of cytolethal distending toxin production in periodontopathogenic bacteria. J. Clin. Microbiol. 2003, 41, 1391–1398. [Google Scholar] [CrossRef]
- Ando, E.S.; De-Gennaro, L.A.; Faveri, M.; Feres, M.; DiRienzo, J.M.; Mayer, M.P. Immune response to cytolethal distending toxin of Aggregatibacter actinomycetemcomitans in periodontitis patients. J. Periodontal. Res. 2010, 45, 471–480. [Google Scholar]
- Johansson, A.; Buhlin, K.; Koski, R.; Gustafsson, A. The immunoreactivity of systemic antibodies to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in adult periodontitis. Eur. J. Oral Sci. 2005, 113, 197–202. [Google Scholar]
- Xynogala, I.; Volgina, A.; DiRienzo, J.M.; Korostoff, J. Evaluation of the humoral immune response to the cytolethal distending toxin of Aggregatibacter actinomycetemcomitans Y4 in subjects with localized aggressive periodontitis. Oral Microbiol. Immunol. 2009, 24, 116–123. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Slots, J.; Sixou, M.; Sol, M.A.; Harmon, R.; McKay, T.L. Specific genetic variants of Actinobacillus actinomycetemcomitans correlate with disease and health in a regional population of families with localized juvenile periodontitis. Infect. Immun. 1994, 62, 3058–3065. [Google Scholar]
- DiRienzo, J.M.; McKay, T.L. Identification and characterization of genetic cluster groups of Actinobacillus actinomycetemcomitans isolated from the human oral cavity. J. Clin. Microbiol. 1994, 32, 75–81. [Google Scholar]
- Bueno, L.C.; Mayer, M.P.; DiRienzo, J.M. Relationship between conversion of localized juvenile periodontitis-susceptible children from health to disease and Actinobacillus actinomycetemcomitans leukotoxin promoter structure. J. Periodontol. 1998, 69, 998–1007. [Google Scholar] [CrossRef]
- Hoglund Aberg, C.; Antonoglou, G.; Haubek, D.; Kwamin, F.; Claesson, R.; Johansson, A. Cytolethal distending toxin in isolates of Aggregatibacter actinomycetemcomitans from Ghanaian adolescents and association with serotype and disease progression. PLoS One 2013, 8, e65781. [Google Scholar]
- Wang, X.; Li, L.; Yang, M.; Geng, Y.; Chen, H.; Xu, Y.; Sun, Y. Prevalence and distribution of Aggregatibacter actinomycetemcomitans and its cdtB gene in subgingival plaque of Chinese periodontitis patients. BMC Oral Health 2014, 14, 37. [Google Scholar] [CrossRef]
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124. [Google Scholar]
- Akifusa, S.; Poole, S.; Lewthwaite, J.; Henderson, B.; Nair, S.P. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect. Immun. 2001, 69, 5925–5930. [Google Scholar] [CrossRef]
- Mao, X.; DiRienzo, J.M. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell. Microbiol. 2002, 4, 245–255. [Google Scholar]
- Saiki, K.; Konishi, K.; Gomi, T.; Nishihara, T.; Yoshikawa, M. Reconstitution and purification of cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiol. Immunol. 2001, 45, 497–506. [Google Scholar] [CrossRef]
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci. 2006, 15, 362–372. [Google Scholar] [CrossRef]
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar] [CrossRef]
- Kanno, F.; Korostoff, J.; Volgina, A.; DiRienzo, J.M. Resistance of human periodontal ligament fibroblasts to the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. J. Periodontol. 2005, 76, 1189–1201. [Google Scholar] [CrossRef]
- Cao, L.; Volgina, A.; Huang, C.M.; Korostoff, J.; DiRienzo, J.M. Characterization of point mutations in the cdtA gene of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Mol. Microbiol. 2005, 58, 1303–1321. [Google Scholar] [CrossRef]
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890. [Google Scholar] [CrossRef]
- Akifusa, S.; Heywood, W.; Nair, S.P.; Stenbeck, G.; Henderson, B. Mechanism of internalization of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiology 2005, 151, 1395–1402. [Google Scholar]
- Mise, K.; Akifusa, S.; Watarai, S.; Ansai, T.; Nishihara, T.; Takehara, T. Involvement of ganglioside GM3 in G2/M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2005, 73, 4846–4852. [Google Scholar] [CrossRef]
- Prokazova, N.V.; Samovilova, N.N.; Gracheva, E.V.; Golovanova, N.K. Ganglioside GM3 and its biological functions. Biochemistry (Moscow) 2009, 74, 235–249. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Brown, A.; Walker, L.; Besack, D.; Zekavat, A.; Wrenn, S.; Krummenacher, C.; Shenker, B.J. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 2009, 284, 10650–10658. [Google Scholar] [CrossRef]
- Eshraghi, A.; Maldonado-Arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J. Biol. Chem. 2010, 285, 18199–18207. [Google Scholar]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5895–5900. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar] [CrossRef]
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlow, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar] [CrossRef]
- Dassanayake, R.P.; Griep, M.A.; Duhamel, G.E. The cytolethal distending toxin B sub-unit of Helicobacter hepaticus is a Ca2+- and Mg2+-dependent neutral nuclease. FEMS Microbiol. Lett. 2005, 251, 219–225. [Google Scholar] [CrossRef]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Cao, L.; Volgina, A.; Bandelac, G.; Korostoff, J. Functional and structural characterization of chimeras of a bacterial genotoxin and human type I DNAse. FEMS Microbiol. Lett. 2009, 291, 222–231. [Google Scholar] [CrossRef]
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434. [Google Scholar]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar]
- Dlakic, M. Is CdtB a nuclease or a phosphatase? Science 2001, 291, 547. [Google Scholar] [CrossRef]
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; Labelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108. [Google Scholar]
- Shenker, B.J.; Walker, L.P.; Zekavat, A.; Dlakic, M.; Boesze-Battaglia, K. Blockade of the PI-3K signaling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell. Microbiol. 2014. [Google Scholar] [CrossRef]
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar] [CrossRef]
- Ando-Suguimoto, E.S.; da Silva, M.P.; Kawamoto, D.; Chen, C.; DiRienzo, J.M.; Mayer, M.P. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine 2014, 66, 46–53. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Kalfas, S.; Lerner, U.H. The cytolethal distending toxin induces receptor activator of NF-κB ligand expression in human gingival fibroblasts and periodontal ligament cells. Infect. Immun. 2005, 73, 342–351. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Lagergård, T.; Kalfas, S.; Lerner, U.H. Cytokine responses of human gingival fibroblasts to Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cytokine 2005, 30, 56–63. [Google Scholar] [CrossRef]
- Uchida, Y.; Shiba, H.; Komatsuzawa, H.; Takemoto, T.; Sakata, M.; Fujita, T.; Kawaguchi, H.; Sugai, M.; Kurihara, H. Expression of IL-1β and IL-8 by human gingival epithelial cells in response to Actinobacillus actinomycetemcomitans. Cytokine 2001, 14, 152–161. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Brage, M.; Lagergård, T.; Johansson, A. Cytolethal distending toxin upregulates RANKL expression in Jurkat T-cells. Apmis 2008, 116, 499–506. [Google Scholar] [CrossRef]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3540–3545. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Liu, D.; Xu, J.K.; Figliomeni, L.; Huang, L.; Pavlos, N.J.; Rogers, M.; Tan, A.; Price, P.; Zheng, M.H. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int. J. Mol. Med. 2003, 11, 17–21. [Google Scholar]
- Mogi, M.; Ozeki, N.; Nakamura, H.; Togari, A. Dual roles for NF-κB activation in osteoblastic cells by serum deprivation: Osteoblastic apoptosis and cell-cycle arrest. Bone 2004, 35, 507–516. [Google Scholar] [CrossRef]
- Escalas, N.; Davezac, N.; de Rycke, J.; Baldin, V.; Mazars, R.; Ducommun, B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: Involvement of the CDC25 phosphatase. Exp. Cell Res. 2000, 257, 206–212. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb. Pathog. 1988, 4, 115–126. [Google Scholar] [CrossRef]
- Pérès, S.Y.; Marches, O.; Daigle, F.; Nougayrede, J.P.; Herault, F.; Tasca, C.; de Rycke, J.; Oswald, E. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 1997, 24, 1095–1107. [Google Scholar]
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-independent expression of p21(CIP1/WAF1) in plasmacytic cells during G2 cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534. [Google Scholar] [CrossRef]
- Sugai, M.; Kawamoto, T.; Pérès, S.Y.; Ueno, Y.; Komatsuzawa, H.; Fujiwara, T.; Kurihara, H.; Suginaka, H.; Oswald, E. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 1998, 66, 5008–5019. [Google Scholar]
- Whitehouse, C.A.; Balbo, P.B.; Pesci, E.C.; Cottle, D.L.; Mirabito, P.M.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect. Immun. 1998, 66, 1934–1940. [Google Scholar]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. The cytolethal distending toxin from the chancroid bacterium Haemophilus ducreyi induces cell-cycle arrest in the G2 phase. J. Clin. Invest. 1999, 103, 107–115. [Google Scholar] [CrossRef]
- Stevens, M.K.; Latimer, J.L.; Lumbley, S.R.; Ward, C.K.; Cope, L.D.; Lagergård, T.; Hansen, E.J. Characterization of a Haemophilus ducreyi mutant deficient in expression of cytolethal distending toxin. Infect. Immun. 1999, 67, 3900–3908. [Google Scholar]
- Smith, J.L.; Bayles, D.O. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit. Rev. Microbiol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef]
- Oswald, E.; Nougayrede, J.P.; Taieb, F.; Sugai, M. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 2005, 8, 83–91. [Google Scholar] [CrossRef]
- Comayras, C.; Tasca, C.; Pérès, S.Y.; Ducommun, B.; Oswald, E.; de Rycke, J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect. Immun. 1997, 65, 5088–5095. [Google Scholar]
- Alaoui-El-Azher, M.; Mans, J.J.; Baker, H.V.; Chen, C.; Progulske-Fox, A.; Lamont, R.J.; Handfield, M. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 2010, 5, e11714. [Google Scholar]
- Nougayrede, J.P.; Taieb, F.; de Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Song, M.; Wan, L.S.; Ellen, R.P. Kinetics of KB and HEp-2 cell responses to an invasive, cytolethal distending toxin-producing strain of Actinobacillus actinomycetemcomitans. Oral Microbiol. Immunol. 2002, 17, 245–251. [Google Scholar] [CrossRef]
- Lepine, G.; Caudry, S.; DiRienzo, J.M.; Ellen, R.P. Epithelial cell invasion by Actinobacillus actinomycetemcomitans strains from restriction fragment-length polymorphism groups associated with juvenile periodontitis or carrier status. Oral Microbiol. Immunol. 1998, 13, 341–347. [Google Scholar] [CrossRef]
- Meyer, D.H.; Lippmann, J.E.; Fives-Taylor, P.M. Invasion of epithelial cells by Actinobacillus actinomycetemcomitans: A dynamic, multistep process. Infect. Immun. 1996, 64, 2988–2997. [Google Scholar]
- Meyer, D.H.; Sreenivasan, P.K.; Fives-Taylor, P.M. Evidence for invasion of a human oral cell line by Actinobacillus actinomycetemcomitans. Infect. Immun. 1991, 59, 2719–2726. [Google Scholar]
- Gilchrist, E.P.; Moyer, M.P.; Shillitoe, E.J.; Clare, N.; Murrah, V.A. Establishment of a human polyclonal oral epithelial cell line. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2000, 90, 340–347. [Google Scholar]
- Kang, P.; Korostoff, J.; Volgina, A.; Grzesik, W.; DiRienzo, J.M. Differential effect of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans on co-cultures of human oral cells. J. Med. Microbiol. 2005, 54, 785–794. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Haris, M.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Cytolethal distending toxin damages the oral epithelium of gingival explants. J. Dent. Res. 2011, 90, 874–879. [Google Scholar] [CrossRef]
- Belibasakis, G.; Johansson, A.; Wang, Y.; Claesson, R.; Chen, C.; Asikainen, S.; Kalfas, S. Inhibited proliferation of human periodontal ligament cells and gingival fibroblasts by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. Eur. J. Oral Sci. 2002, 110, 366–373. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Mattsson, A.; Wang, Y.; Chen, C.; Johansson, A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. Apmis 2004, 112, 674–685. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Lang, N.P. The junctional epithelium: From health to disease. J. Dent. Res. 2005, 84, 9–20. [Google Scholar] [CrossRef]
- Presland, R.B.; Dale, B.A. Epithelial structural proteins of the skin and oral cavity: Function in health and disease. Crit. Rev. Oral Biol. Med. 2000, 11, 383–408. [Google Scholar] [CrossRef]
- Bikle, D.D.; Oda, Y.; Xie, Z. Calcium and 1,25(OH)2D: Interacting drivers of epidermal differentiation. J. Steroid Biochem. Mol. Biol. 2004, 89, 355–360. [Google Scholar] [CrossRef]
- Christersson, L.A.; Albini, B.; Zambon, J.J.; Wikesjo, U.M.; Genco, R.J. Tissue localization of Actinobacillus actinomycetemcomitans in human periodontitis. I. Light, immunofluorescence and electron microscopic studies. J. Periodontol. 1987, 58, 529–539. [Google Scholar]
- Gibbons, R.J. Bacterial adhesion to oral tissues: A model for infectious diseases. J. Dent. Res. 1989, 68, 750–760. [Google Scholar] [CrossRef]
- Powell, R.N. Gingival tissue physiology in vitro. 3. Cell proliferation in gingival epithelium. J. Periodontal. Res. 1971, 6, 38–44. [Google Scholar] [CrossRef]
- Powell, R.N. Gingival tissue physiology in vitro. 1. Gingival organ culture. J. Periodontal. Res. 1967, 2, 290–296. [Google Scholar] [CrossRef]
- Ohara, M.; Miyauchi, M.; Tsuruda, K.; Takata, T.; Sugai, M. Topical application of Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces cell cycle arrest in the rat gingival epithelium in vivo. J. Periodontal. Res. 2011, 46, 389–395. [Google Scholar] [CrossRef]
- Franke, W.W. Discovering the molecular components of intercellular junctions-a historical view. Cold Spring Harb. Perspect. Biol. 2009, 1, a003061. [Google Scholar] [CrossRef]
- Cavey, M.; Lecuit, T. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 2009, 1, a002998. [Google Scholar]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. N.Y. Acad. Sci. 2012, 1258, 9–18. [Google Scholar]
- Miyaguchi, K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J. Struct. Biol. 2000, 132, 169–178. [Google Scholar] [CrossRef]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar]
- Kowalczyk, A.P.; Nanes, B.A. Adherens junction turnover: regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 2012, 60, 197–222. [Google Scholar] [CrossRef]
- Hirokawa, N.; Heuser, J.E. Quick-freeze, deep-etch visualization of the cytoskeleton beneath surface differentiations of intestinal epithelial cells. J. Cell Biol. 1981, 91, 399–409. [Google Scholar] [CrossRef]
- Thilander, H.; Bloom, G.D. Cell contacts in oral epithelia. J. Periodontal. Res. 1968, 3, 96–110. [Google Scholar] [CrossRef]
- Barnett, M.L.; Szabo, G. Gap junctions in human gingival keratinized epithelium. J. Periodontal. Res. 1973, 8, 117–126. [Google Scholar] [CrossRef]
- Meyle, J.; Gultig, K.; Rascher, G.; Wolburg, H. Transepithelial electrical resistance and tight junctions of human gingival keratinocytes. J. Periodontal. Res. 1999, 34, 214–222. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Moffatt, C.E.; Hagerty, D.; Whitmore, S.E.; Brown, T.A.; Graves, D.T.; Lamont, R.J. Interaction of oral bacteria with gingival epithelial cell multilayers. Mol. Oral Microbiol. 2011, 26, 210–220. [Google Scholar] [CrossRef]
- Katz, J.; Sambandam, V.; Wu, J.H.; Michalek, S.M.; Balkovetz, D.F. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect. Immun. 2000, 68, 1441–1449. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Korostoff, J.; Gill, R.; DiRienzo, J.M. Cell junction remodeling in gingival tissue exposed to a microbial toxin. J. Dent. Res. 2013, 92, 518–523. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Ye, P.; Chapple, C.C.; Kumar, R.K.; Hunter, N. Expression patterns of E-cadherin, involucrin, and connexin gap junction proteins in the lining epithelia of inflamed gingiva. J. Pathol. 2000, 192, 58–66. [Google Scholar] [CrossRef]
- Downer, C.S.; Speight, P.M. E-cadherin expression in normal, hyperplastic and malignant oral epithelium. Eur. J. Cancer B Oral Oncol. 1993, 29B, 303–305. [Google Scholar] [CrossRef]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Knudsen, K.A. Cadherins and associated proteins. In Vivo 1991, 5, 505–513. [Google Scholar]
- Brieher, W.M.; Yap, A.S. Cadherin junctions and their cytoskeleton(s). Curr. Opin. Cell Biol. 2012, 25, 1–8. [Google Scholar]
- Yamada, S.; Pokutta, S.; Drees, F.; Weis, W.I.; Nelson, W.J. Deconstructing the cadherin-catenin-actin complex. Cell 2005, 123, 889–901. [Google Scholar] [CrossRef]
- Aragon, V.; Chao, K.; Dreyfus, L.A. Effect of cytolethal distending toxin on F-actin assembly and cell division in Chinese hamster ovary cells. Infect. Immun. 1997, 65, 3774–3780. [Google Scholar]
- Lagergard, T.; Keith, J. Cytolethal distending toxin as virulence factor, protective antigen, and target for vaccine development. Vaccine: Dev. Ther. 2012, 2, 51–60. [Google Scholar] [CrossRef]
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the Actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487. [Google Scholar] [CrossRef]
- Tan, K.S.; Ong, G.; Song, K.P. Introns in the cytolethal distending toxin gene of Actinobacillus actinomycetemcomitans. J. Bacteriol. 2005, 187, 567–575. [Google Scholar] [CrossRef]
- Jeon, B.; Itoh, K.; Ryu, S. Promoter analysis of cytolethal distending toxin genes (cdtA, B, and C) and effect of a luxS mutation on CDT production in Campylobacter jejuni. Microbiol. Immunol. 2005, 49, 599–603. [Google Scholar] [CrossRef]
- Coburn, J.; Leong, J.M. Microbiology. Arresting features of bacterial toxins. Science 2000, 290, 287–288. [Google Scholar] [CrossRef]
- Handfield, M.; Baker, H.V.; Lamont, R.J. Beyond good and evil in the oral cavity: Insights into host-microbe relationships derived from transcriptional profiling of gingival cells. J. Dent. Res. 2008, 87, 203–223. [Google Scholar] [CrossRef]
- Kagnoff, M.F.; Eckmann, L. Epithelial cells as sensors for microbial infection. J. Clin. Invest. 1997, 100, 6–10. [Google Scholar] [CrossRef]
- Nalbant, A.; Chen, C.; Wang, Y.; Zadeh, H.H. Induction of T-cell apoptosis by Actinobacillus actinomycetemcomitans mutants with deletion of ltxA and cdtABC genes: possible activity of GroEL-like molecule. Oral Microbiol. Immunol. 2003, 18, 339–349. [Google Scholar] [CrossRef]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780. [Google Scholar]
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441. [Google Scholar]
- Reed, J.C.; Meister, L.; Tanaka, S.; Cuddy, M.; Yum, S.; Geyer, C.; Pleasure, D. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991, 51, 6529–6538. [Google Scholar]
- Griffith, T.S.; Brunner, T.; Fletcher, S.M.; Green, D.R.; Ferguson, T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995, 270, 1189–1192. [Google Scholar]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M. Caspase-2 and caspase-7 are involved in cytolethal distending toxin-induced apoptosis in Jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879. [Google Scholar] [CrossRef]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Nakachi, K.; Fujiwara, T.; Komatsuzawa, H.; Sugai, M. Cytolethal distending toxin induces caspase-dependent and -independent cell death in MOLT-4 cells. Infect. Immun. 2008, 76, 4783–4791. [Google Scholar] [CrossRef]
- Rabin, S.D.; Flitton, J.G.; Demuth, D.R. Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces apoptosis in nonproliferating macrophages by a phosphatase-independent mechanism. Infect. Immun. 2009, 77, 3161–3169. [Google Scholar] [CrossRef]
- Fernandes, K.P.; Mayer, M.P.; Ando, E.S.; Ulbrich, A.G.; Amarente-Mendes, J.G.; Russo, M. Inhibition of interferon-gamma-induced nitric oxide production in endotoxin-activated macrophages by cytolethal distending toxin. Oral Microbiol. Immunol. 2008, 23, 360–366. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).