Unlocking Doors without Keys: Activation of Src by Truncated C-terminal Intracellular Receptor Tyrosine Kinases Lacking Tyrosine Kinase Activity

Abstract
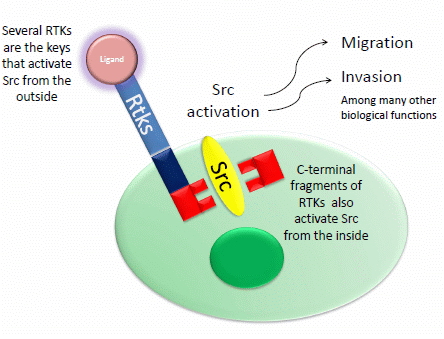
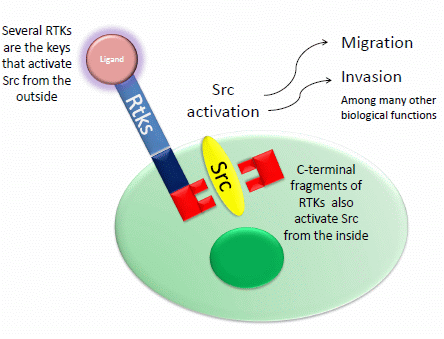
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. The VEGF Receptor Tyrosine Kinase Family
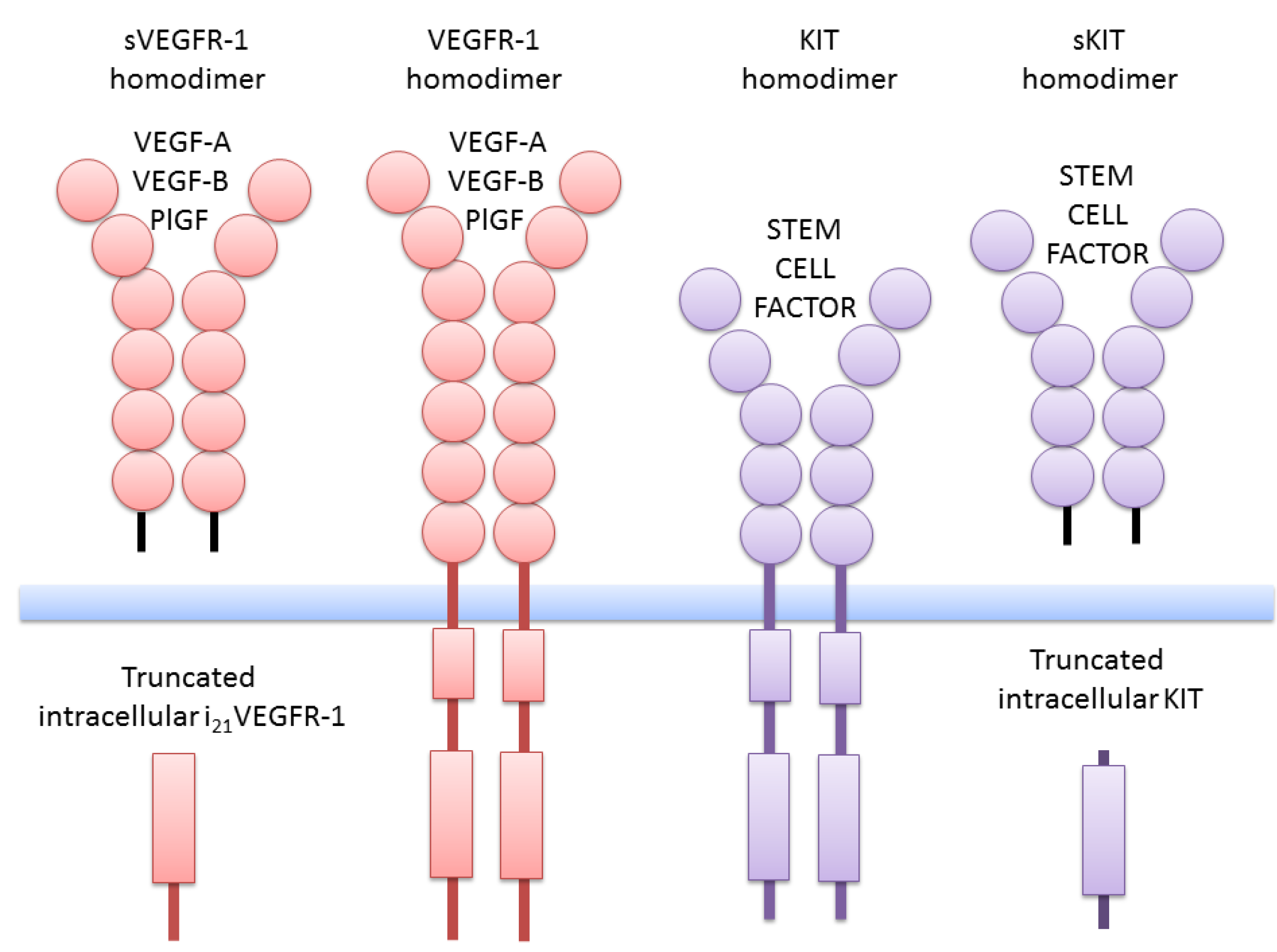
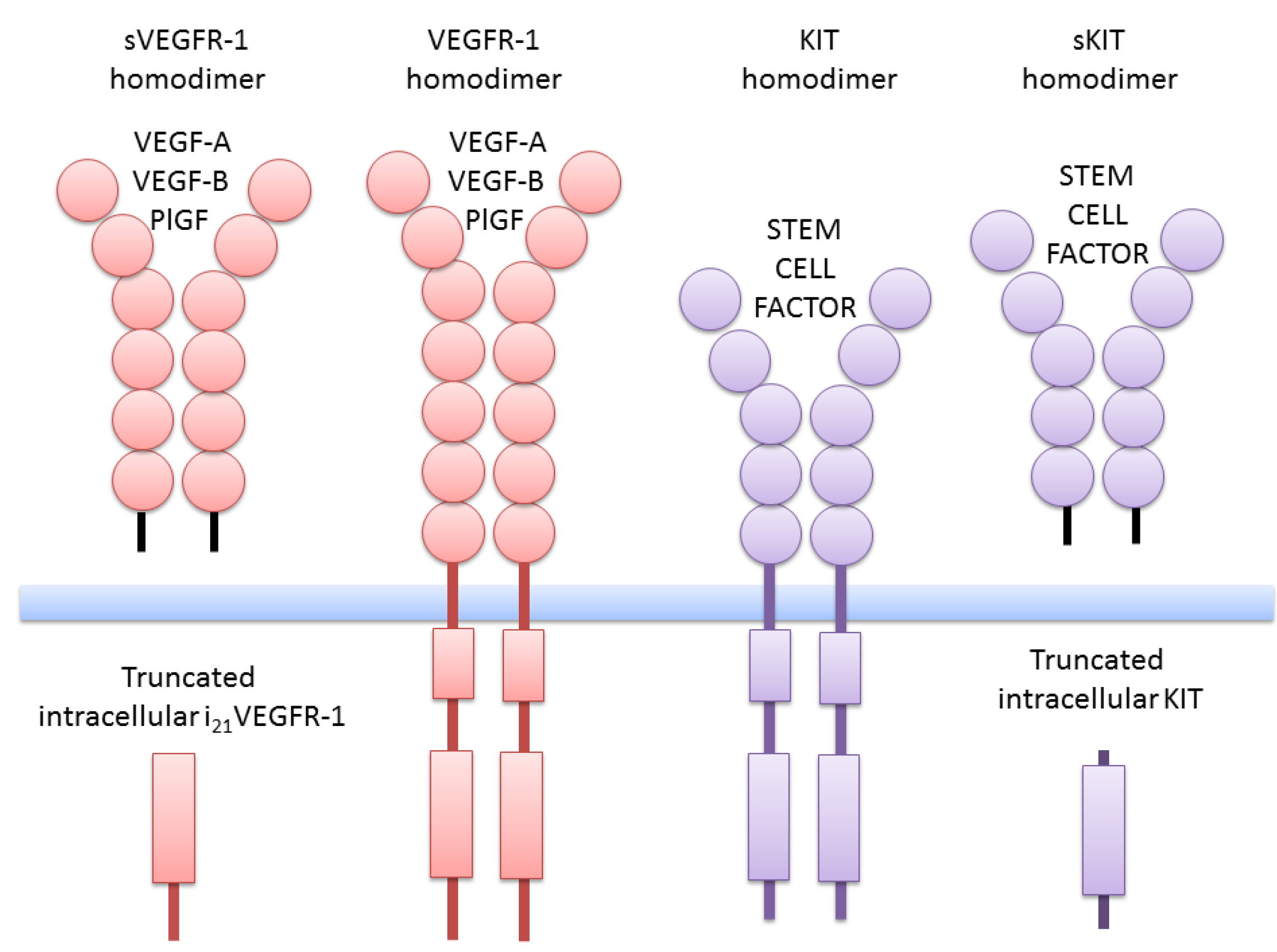
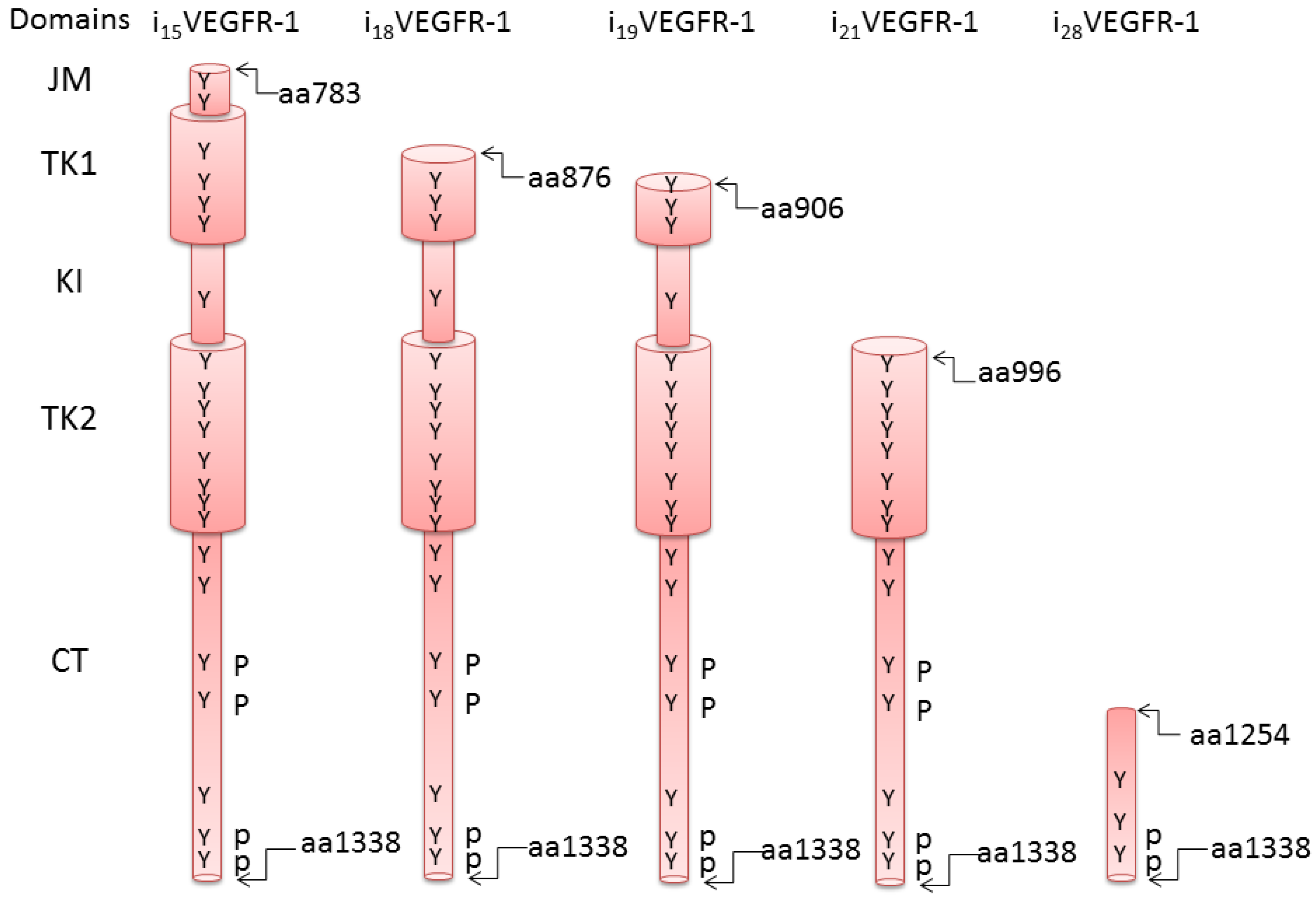
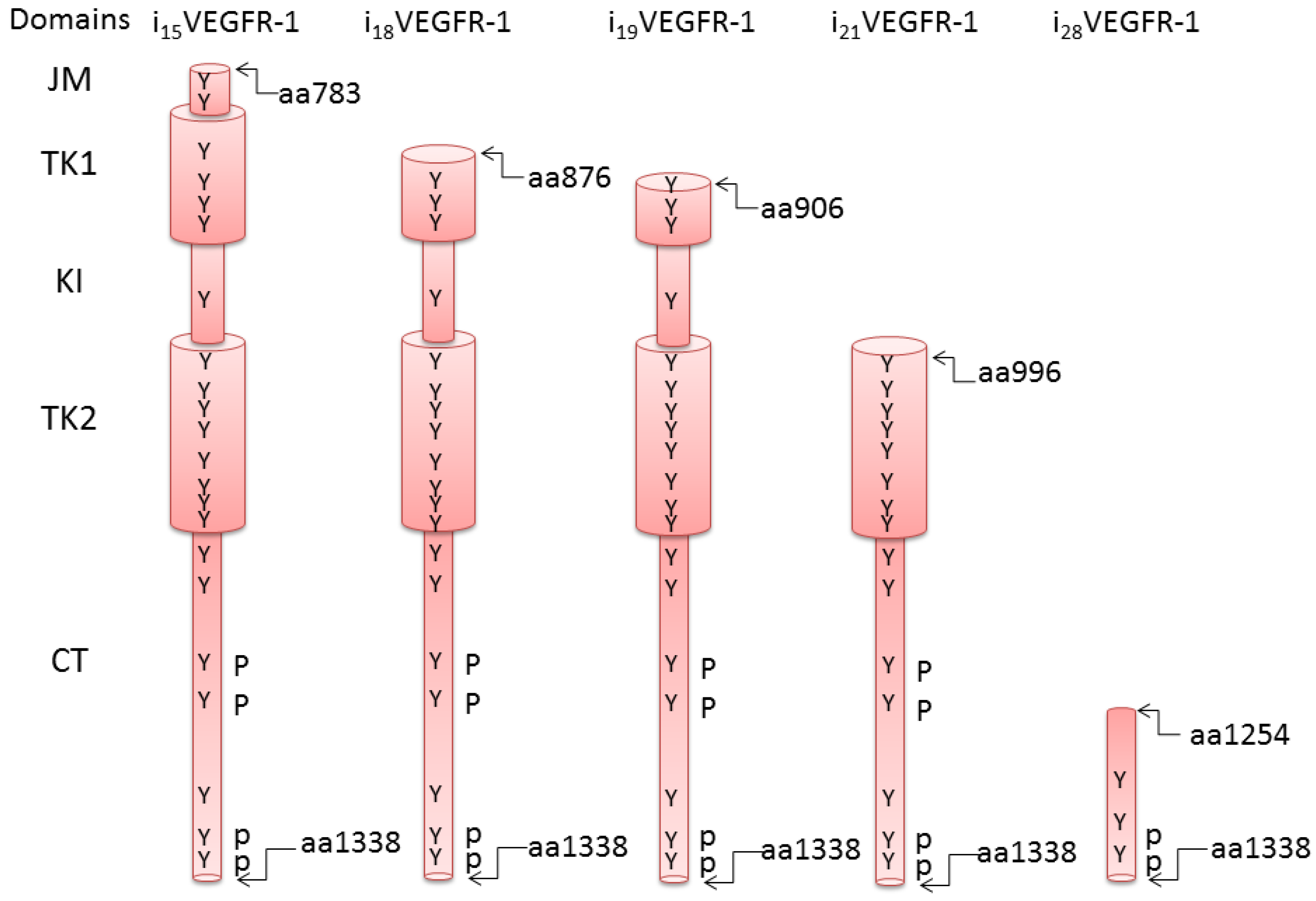
2. Truncated Isoforms of VEGFR-1
3. The KIT Receptor Tyrosine Kinase Family
4. Truncated KIT Isoforms
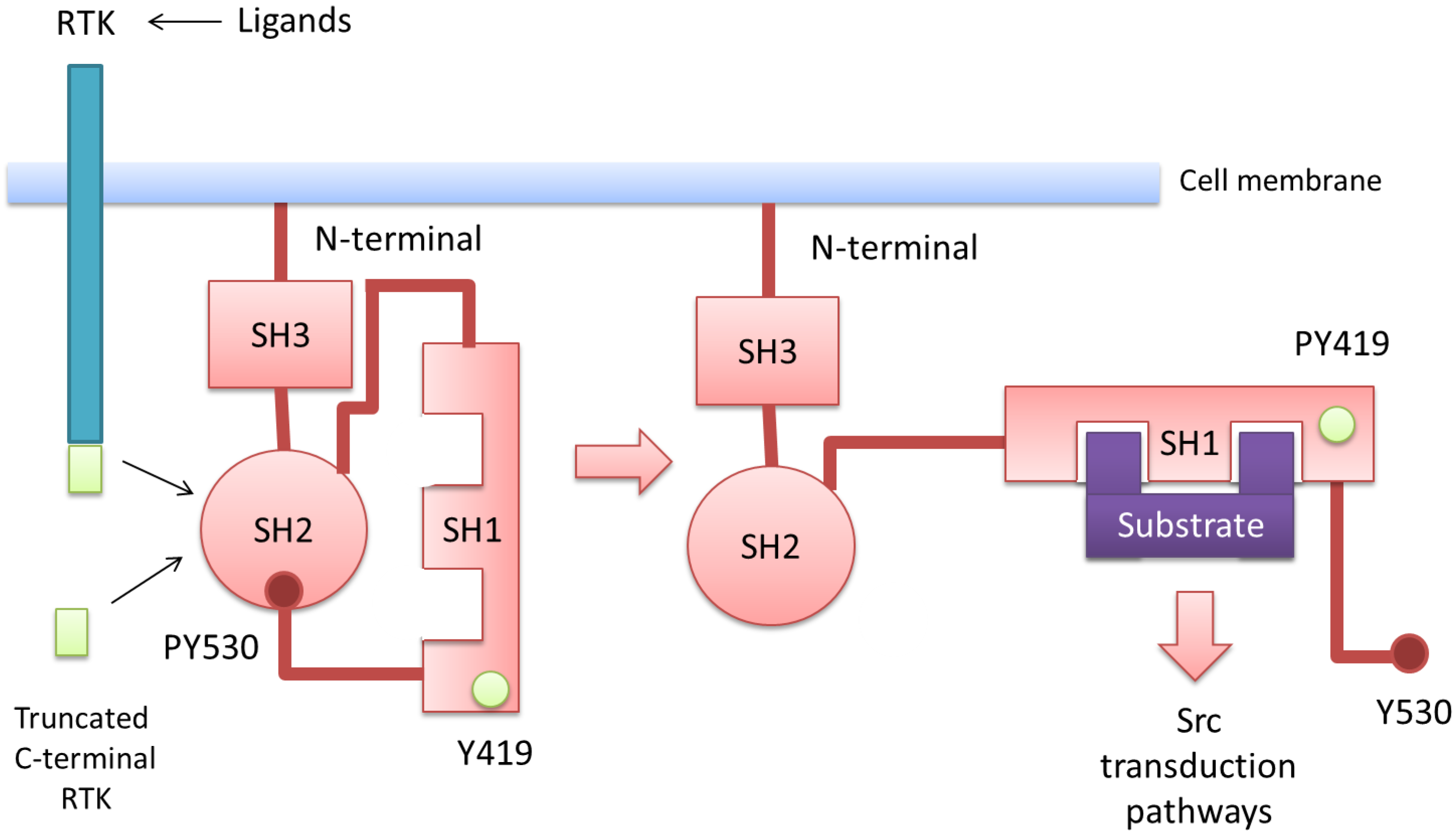
5. Src Activation through Receptor Tyrosine Kinases and Their Intracellular C-Terminal Truncated Isoforms


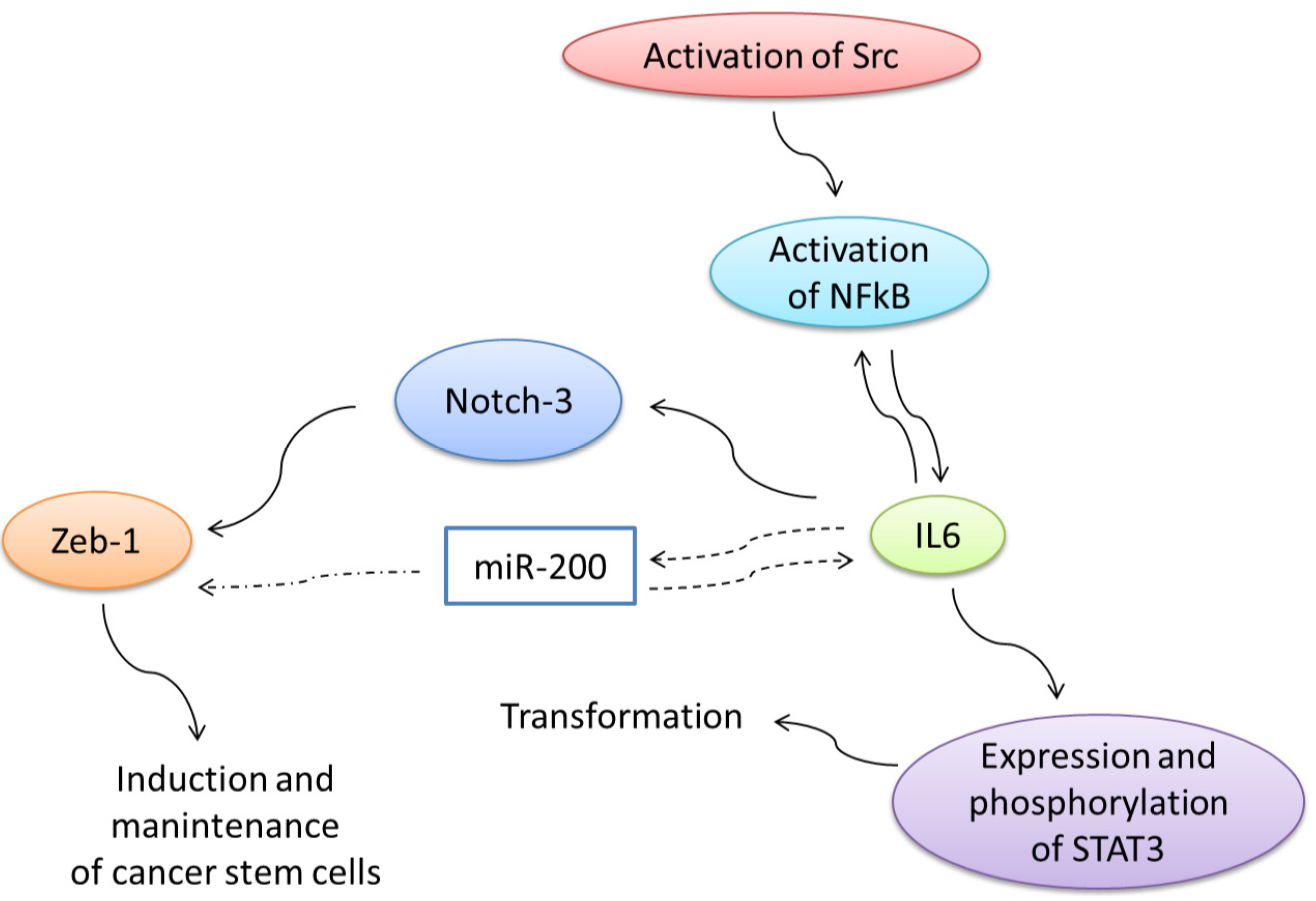
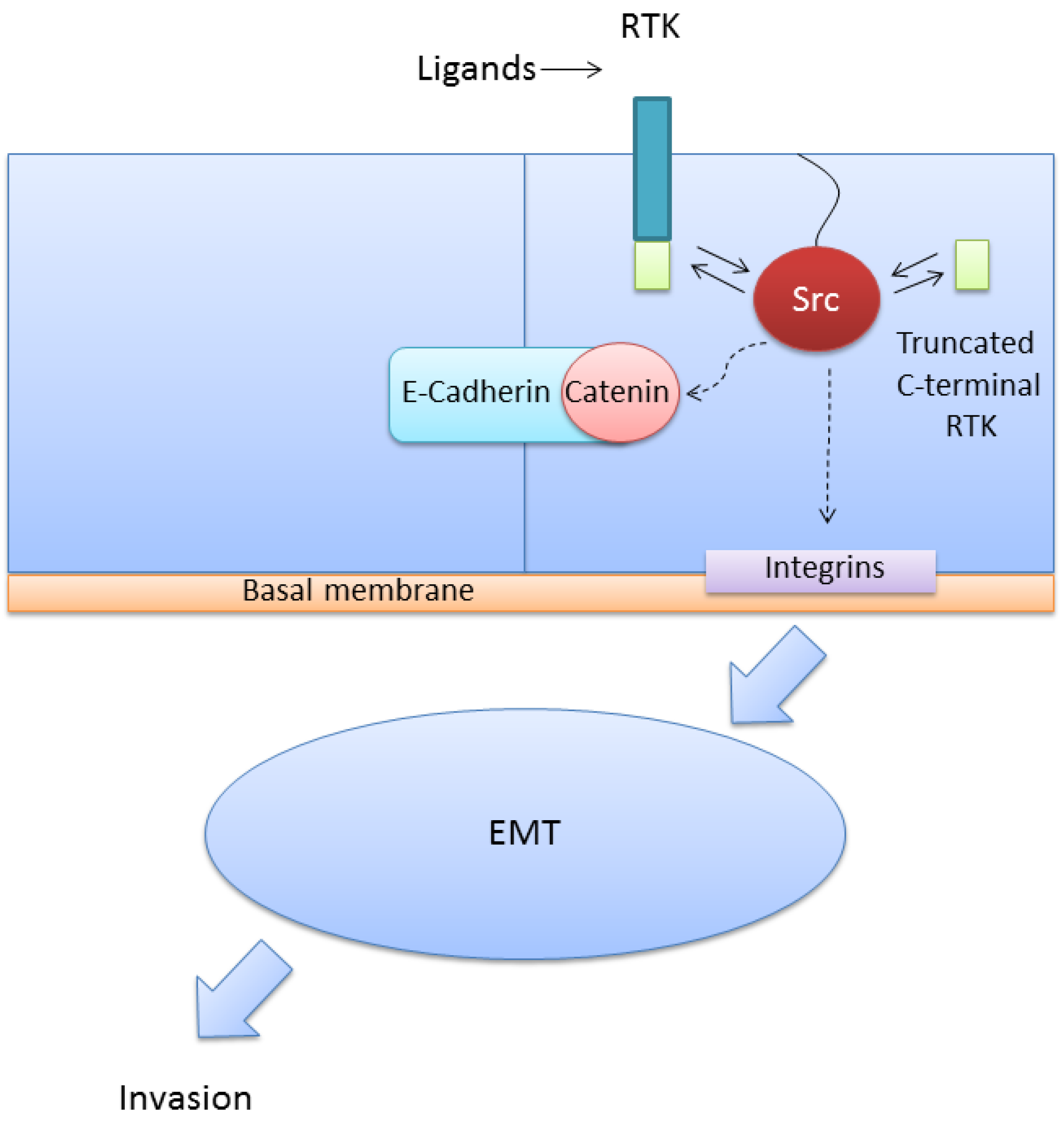
6. Expression of the Full-Length VEGFR-1 and the Truncated Intracellular Isoforms in Cancer Cells is Related with Increased Migration and Invasion through Activation of Src
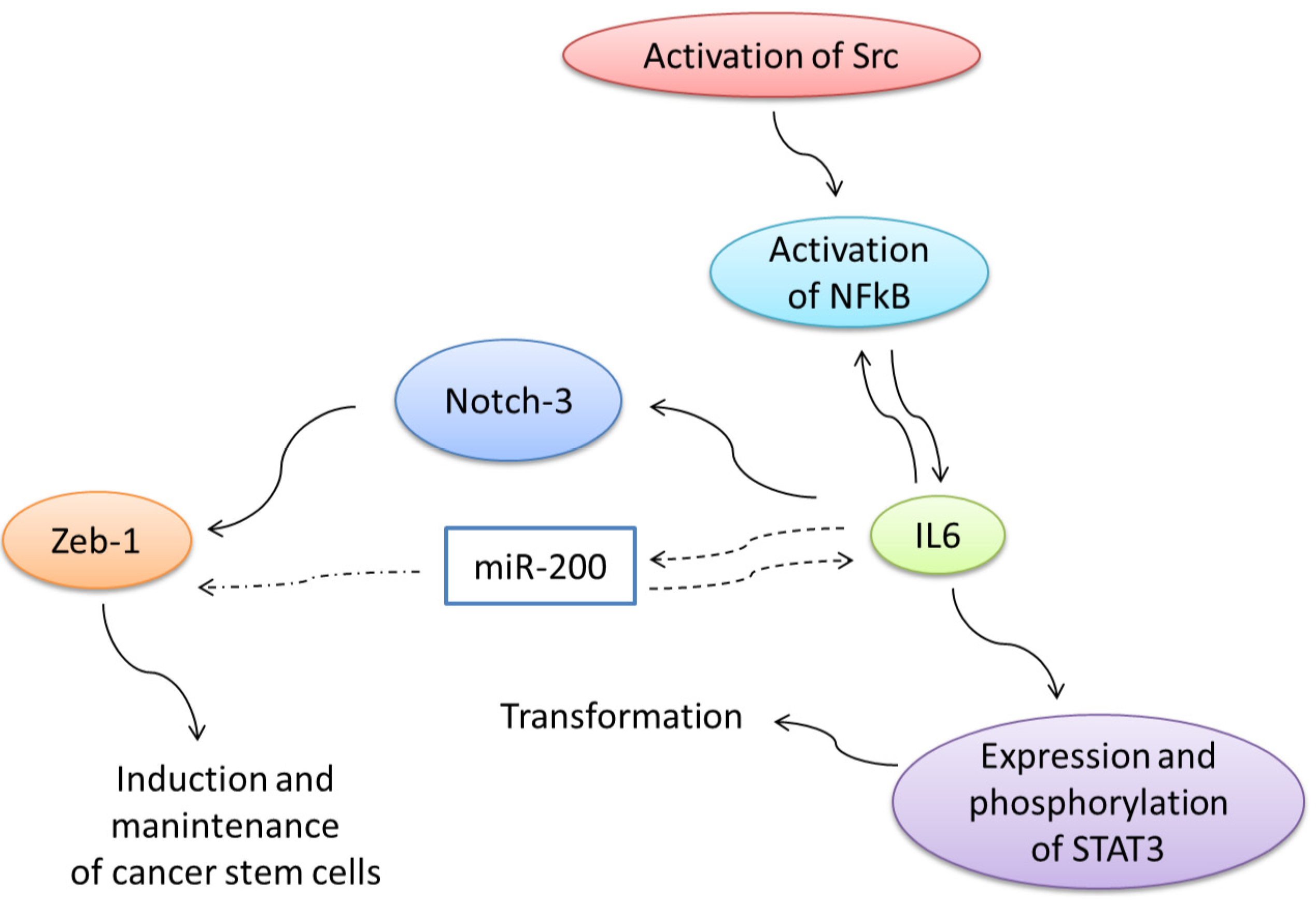
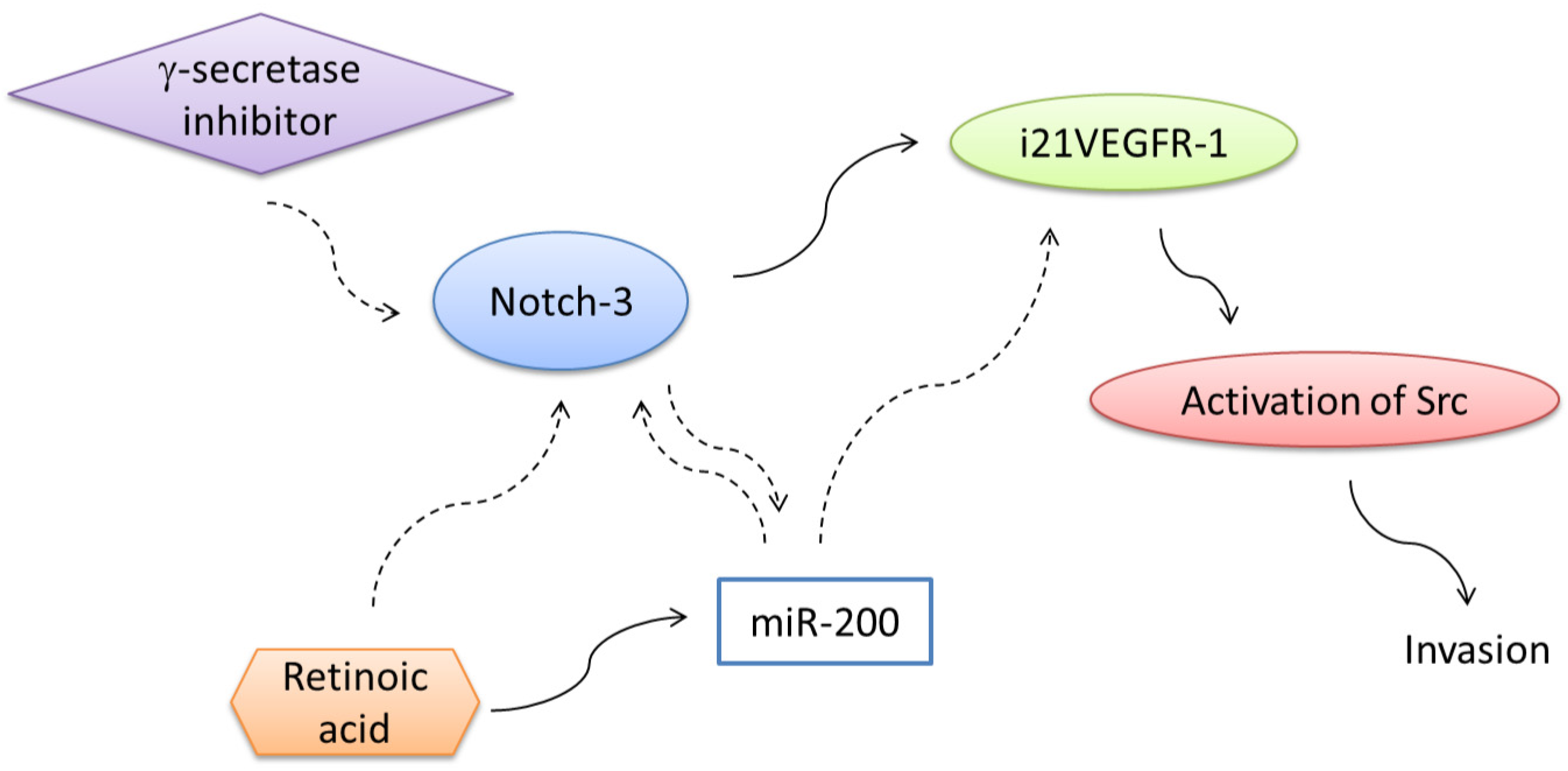
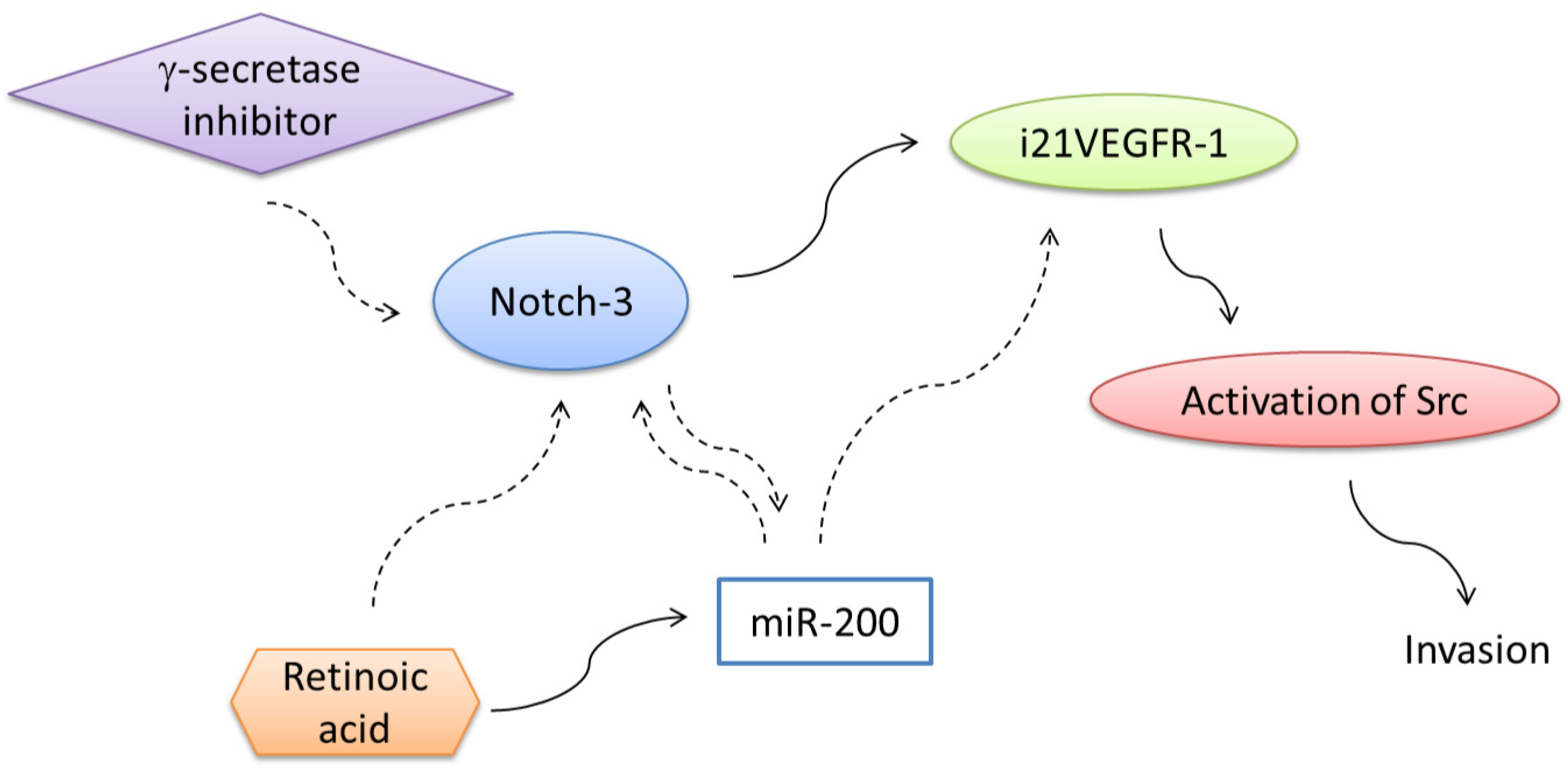
7. Regulation of Expression of Truncated Intracellular VEGFR-1 in Breast Cancer Cells
8. KIT, Truncated Intracellular KIT, and Cancer
9. Structural and Functional Similarities between Truncated Intracellular Isoforms of VEGFR-1 and KIT
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving nf-kappab, lin28, let-7 microrna, and il6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar]
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990, 5, 519–524. [Google Scholar]
- Terman, B.; Carrion, M.; Kovacs, E.; Rasmussen, B.; Eddy, R.; Shows, T. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991, 6, 1677–1683. [Google Scholar]
- Ito, N.; Wernstedt, C.; Engström, U.; Claesson-Welsh, L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of sh2 domain-containing molecules. J. Biol. Chem. 1998, 273, 23410–23418. [Google Scholar]
- Shinkai, A.; Ito, M.; Anazawa, H.; Yamaguchi, S.; Shitara, K.; Shibuya, M. Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J. Biol. Chem. 1998, 273, 31283–31288. [Google Scholar]
- Gille, H.; Kowalski, J.; Yu, L.; Chen, H.; Pisabarro, M.T.; Davis-Smyth, T.; Ferrara, N. A repressor sequence in the juxtamembrane domain of flt-1 (vegfr-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3'-kinase activation and endothelial cell migration. EMBO J. 2000, 19, 4064–4073. [Google Scholar] [CrossRef]
- Meyer, R.D.; Mohammadi, M.; Rahimi, N. A single amino acid substitution in the activation loop defines the decoy characteristic of vegfr-1/flt-1. J. Biol. Chem. 2006, 281, 867–875. [Google Scholar] [CrossRef]
- Huang, K.; Andersson, C.; Roomans, G.M.; Ito, N.; Claesson-Welsh, L. Signaling properties of vegf receptor-1 and -2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 2001, 33, 315–324. [Google Scholar] [CrossRef]
- Kondo, K.; Hiratsuka, S.; Subbalakshmi, E.; Matsushime, H.; Shibuya, M. Genomic organization of the flt-1 gene encoding for vascular endothelial growth factor (vegf) receptor-1 suggests an intimate evolutionary relationship between the 7-ig and the 5-ig tyrosine kinase receptors. Gene 1998, 208, 297–305. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; de Mol, M.; Bono, F.; et al. Role of plgf in the intra- and intermolecular cross talk between the vegf receptors flt1 and flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar]
- Cunningham, S.A.; Arrate, M.P.; Brock, T.A.; Waxham, M.N. Interactions of flt-1 and kdr with phospholipase c gamma: Identification of the phosphotyrosine binding sites. Biochem. Biophys. Res. Commun. 1997, 240, 635–639. [Google Scholar] [CrossRef]
- Yu, Y.; Hulmes, J.D.; Herley, M.T.; Whitney, R.G.; Crabb, J.W.; Sato, J.D. Direct identification of a major autophosphorylation site on vascular endothelial growth factor receptor flt-1 that mediates phosphatidylinositol 3'-kinase binding. Biochem. J. 2001, 358, 465–472. [Google Scholar]
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Shibuya, M. The phosphorylated 1169-tyrosine containing region of flt-1 kinase (vegfr-1) is a major binding site for plcgamma. Biochem. Biophys. Res. Commun. 1997, 238, 487–491. [Google Scholar] [CrossRef]
- Igarashi, K.; Isohara, T.; Kato, T.; Shigeta, K.; Yamano, T.; Uno, I. Tyrosine 1213 of flt-1 is a major binding site of nck and shp-2. Biochem. Biophys. Res. Commun. 1998, 246, 95–99. [Google Scholar] [CrossRef]
- Lesslie, D.; Summy, J.; Parikh, N.; Fan, F.; Trevino, J.; Sawyer, T.; Metcalf, C.; Shakespeare, W.; Hicklin, D.; Ellis, L.; et al. Vascular endothelial growth factor receptor-1 mediates migration of human colorectal carcinoma cells by activation of src family kinases. Br. J. Cancer 2006, 94, 1710–1717. [Google Scholar]
- Taylor, A.P.; Leon, E.; Goldenberg, D.M. Placental growth factor (plgf) enhances breast cancer cell motility by mobilising erk1/2 phosphorylation and cytoskeletal rearrangement. Br. J. Cancer 2010, 103, 82–89. [Google Scholar] [CrossRef]
- Kendall, R.; Thomas, K. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10705–10709. [Google Scholar] [CrossRef]
- Thomas, C.P.; Andrews, J.I.; Raikwar, N.S.; Kelley, E.A.; Herse, F.; Dechend, R.; Golos, T.G.; Liu, K.Z. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J. Clin. Endocrinol. Metab. 2009, 94, 2524–2530. [Google Scholar] [CrossRef]
- Thomas, C.P.; Andrews, J.I.; Liu, K.Z. Intronic polyadenylation signal sequences and alternate splicing generate human soluble flt1 variants and regulate the abundance of soluble flt1 in the placenta. FASEB J. 2007, 21, 3885–3895. [Google Scholar] [CrossRef]
- Raikwar, N.S.; Liu, K.Z.; Thomas, C.P. Protein kinase c regulates flt1 abundance and stimulates its cleavage in vascular endothelial cells with the release of a soluble plgf/vegf antagonist. Exp. Cell Res. 2013, 319, 2578–2587. [Google Scholar] [CrossRef]
- Rahimi, N.; Golde, T.E.; Meyer, R.D. Identification of ligand-induced proteolytic cleavage and ectodomain shedding of vegfr-1/flt1 in leukemic cancer cells. Cancer Res. 2009, 69, 2607–2614. [Google Scholar] [CrossRef]
- Mezquita, B.; Mezquita, J.; Pau, M.; Mezquita, C. A novel intracellular isoform of vegfr-1 activates src and promotes cell invasion in mda-mb-231 breast cancer cells. J. Cell. Biochem. 2010, 110, 732–742. [Google Scholar] [CrossRef]
- Mezquita, B.; Mezquita, J.; Barrot, C.; Carvajal, S.; Pau, M.; Mezquita, P.; Mezquita, C. A truncated-flt1 isoform of breast cancer cells is upregulated by notch and downregulated by retinoic acid. J. Cell. Biochem. 2014, 115, 52–61. [Google Scholar] [CrossRef]
- Besmer, P.; Lader, E.; George, P.C.; Bergold, P.J.; Qiu, F.H.; Zuckerman, E.E.; Hardy, W.D. A new acute transforming feline retrovirus with fms homology specifies a c-terminally truncated version of the c-fms protein that is different from sm-feline sarcoma virus v-fms protein. J. Virol. 1986, 60, 194–203. [Google Scholar]
- Yarden, Y.; Ullrich, A. Growth factor receptor tyrosine kinases. Annu. Rev. Biochem. 1988, 57, 443–478. [Google Scholar] [CrossRef]
- Merkwitz, C.; Lochhead, P.; Tsikolia, N.; Koch, D.; Sygnecka, K.; Sakurai, M.; Spanel-Borowski, K.; Ricken, A.M. Expression of kit in the ovary, and the role of somatic precursor cells. Prog. Histochem. Cytochem. 2011, 46, 131–184. [Google Scholar] [CrossRef]
- Sette, C.; Dolci, S.; Geremia, R.; Rossi, P. The role of stem cell factor and of alternative c-kit gene products in the establishment, maintenance and function of germ cells. Int. J. Dev. Biol. 2000, 44, 599–608. [Google Scholar]
- Reith, A.D.; Ellis, C.; Lyman, S.D.; Anderson, D.M.; Williams, D.E.; Bernstein, A.; Pawson, T. Signal transduction by normal isoforms and w mutant variants of the kit receptor tyrosine kinase. EMBO J. 1991, 10, 2451–2459. [Google Scholar]
- Crosier, P.S.; Ricciardi, S.T.; Hall, L.R.; Vitas, M.R.; Clark, S.C.; Crosier, K.E. Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood 1993, 82, 1151–1158. [Google Scholar]
- Broudy, V.C.; Kovach, N.L.; Bennett, L.G.; Lin, N.; Jacobsen, F.W.; Kidd, P.G. Human umbilical vein endothelial cells display high-affinity c-kit receptors and produce a soluble form of the c-kit receptor. Blood 1994, 83, 2145–2152. [Google Scholar]
- Turner, A.M.; Bennett, L.G.; Lin, N.L.; Wypych, J.; Bartley, T.D.; Hunt, R.W.; Atkins, H.L.; Langley, K.E.; Parker, V.; Martin, F. Identification and characterization of a soluble c-kit receptor produced by human hematopoietic cell lines. Blood 1995, 85, 2052–2058. [Google Scholar]
- Kasamatsu, S.; Hachiya, A.; Higuchi, K.; Ohuchi, A.; Kitahara, T.; Boissy, R.E. Production of the soluble form of kit, s-kit, abolishes stem cell factor-induced melanogenesis in human melanocytes. J. Invest. Dermatol. 2008, 128, 1763–1772. [Google Scholar] [CrossRef]
- Rossi, P.; Marziali, G.; Albanesi, C.; Charlesworth, A.; Geremia, R.; Sorrentino, V. A novel c-kit transcript, potentially encoding a truncated receptor, originates within a kit gene intron in mouse spermatids. Dev. Biol. 1992, 152, 203–207. [Google Scholar] [CrossRef]
- Zayas, J.; Spassov, D.; Nachtman, R.; Jurecic, R. Murine hematopoietic stem cells and multipotent progenitors express truncated intracellular form of c-kit receptor. Stem Cells Dev. 2008, 17, 343–353. [Google Scholar] [CrossRef]
- Paronetto, M.; Farini, D.; Sammarco, I.; Maturo, G.; Vespasiani, G.; Geremia, R.; Rossi, P.; Sette, C. Expression of a truncated form of the c-kit tyrosine kinase receptor and activation of src kinase in human prostatic cancer. Am. J. Pathol. 2004, 164, 1243–1251. [Google Scholar] [CrossRef]
- Sette, C.; Paronetto, M.; Barchi, M.; Bevilacqua, A.; Geremia, R.; Rossi, P. Tr-kit-induced resumption of the cell cycle in mouse eggs requires activation of a src-like kinase. EMBO J. 2002, 21, 5386–5395. [Google Scholar] [CrossRef]
- Yeatman, T. A renaissance for src. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Guarino, M. Src signaling in cancer invasion. J. Cell Physiol. 2010, 223, 14–26. [Google Scholar]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Martin, G.S. The hunting of the src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [Google Scholar] [CrossRef]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef]
- Rous, P. A transmissible avian neoplasm. (Sarcoma of the common Fowl.). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef]
- Parsons, J.T.; Weber, M.J. Genetics of src: Structure and functional organization of a protein tyrosine kinase. Curr. Top. Microbiol. Immunol. 1989, 147, 79–127. [Google Scholar]
- Roskoski, R. Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res. Commun 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-src. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef]
- Young, M.A.; Gonfloni, S.; Superti-Furga, G.; Roux, B.; Kuriyan, J. Dynamic coupling between the sh2 and sh3 domains of c-src and hck underlies their inactivation by c-terminal tyrosine phosphorylation. Cell 2001, 105, 115–126. [Google Scholar] [CrossRef]
- Sicheri, F.; Kuriyan, J. Structures of src-family tyrosine kinases. Curr. Opin. Struct. Biol. 1997, 7, 777–785. [Google Scholar] [CrossRef]
- Bromann, P.A.; Korkaya, H.; Courtneidge, S.A. The interplay between src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef]
- Behrens, J.; Vakaet, L.; Friis, R.; Winterhager, E.; van Roy, F.; Mareel, M.M.; Birchmeier, W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the e-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-src gene. J. Cell Biol. 1993, 120, 757–766. [Google Scholar] [CrossRef]
- Boyer, B.; Bourgeois, Y.; Poupon, M.F. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 2002, 21, 2347–2356. [Google Scholar] [CrossRef]
- Kanda, S.; Miyata, Y.; Kanetake, H.; Smithgall, T.E. Non-receptor protein-tyrosine kinases as molecular targets for antiangiogenic therapy (review). Int. J. Mol. Med. 2007, 20, 113–121. [Google Scholar]
- Mukhopadhyay, D.; Tsiokas, L.; Sukhatme, V.P. Wild-type p53 and v-src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res. 1995, 55, 6161–6165. [Google Scholar]
- Trevino, J.G.; Summy, J.M.; Gray, M.J.; Nilsson, M.B.; Lesslie, D.P.; Baker, C.H.; Gallick, G.E. Expression and activity of src regulate interleukin-8 expression in pancreatic adenocarcinoma cells: Implications for angiogenesis. Cancer Res. 2005, 65, 7214–7222. [Google Scholar] [CrossRef]
- Kim, Y.M.; Lee, Y.M.; Kim, H.S.; Kim, J.D.; Choi, Y.; Kim, K.W.; Lee, S.Y.; Kwon, Y.G. Tnf-related activation-induced cytokine (trance) induces angiogenesis through the activation of src and phospholipase c (plc) in human endothelial cells. J. Biol. Chem. 2002, 277, 6799–6805. [Google Scholar]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via il6 secretion. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1397–1402. [Google Scholar] [CrossRef]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. Il-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Rokavec, M.; Wu, W.; Luo, J.L. Il6-mediated suppression of mir-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol. Cell 2012, 45, 777–789. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.; Senior, R.; Shibuya, M. Mmp9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Marcellini, M.; de Luca, N.; Riccioni, T.; Ciucci, A.; Orecchia, A.; Lacal, P.M.; Ruffini, F.; Pesce, M.; Cianfarani, F.; Zambruno, G.; et al. Increased melanoma growth and metastasis spreading in mice overexpressing placenta growth factor. Am. J. Pathol. 2006, 169, 643–654. [Google Scholar] [CrossRef]
- Taylor, A.P.; Goldenberg, D.M. Role of placenta growth factor in malignancy and evidence that an antagonistic plgf/flt-1 peptide inhibits the growth and metastasis of human breast cancer xenografts. Mol. Cancer Ther. 2007, 6, 524–531. [Google Scholar] [CrossRef]
- Roybal, J.D.; Zang, Y.; Ahn, Y.H.; Yang, Y.; Gibbons, D.L.; Baird, B.N.; Alvarez, C.; Thilaganathan, N.; Liu, D.D.; Saintigny, P.; et al. Mir-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting flt1/vegfr1. Mol. Cancer Res. 2011, 9, 25–35. [Google Scholar] [CrossRef]
- Fan, F.; Wey, J.; McCarty, M.; Belcheva, A.; Liu, W.; Bauer, T.; Somcio, R.; Wu, Y.; Hooper, A.; Hicklin, D.; et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 2005, 24, 2647–2653. [Google Scholar] [CrossRef]
- Wey, J.; Fan, F.; Gray, M.; Bauer, T.; McCarty, M.; Somcio, R.; Liu, W.; Evans, D.; Wu, Y.; Hicklin, D.; et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 2005, 104, 427–438. [Google Scholar] [CrossRef]
- Yang, A.; Camp, E.; Fan, F.; Shen, L.; Gray, M.; Liu, W.; Somcio, R.; Bauer, T.; Wu, Y.; Hicklin, D.; et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006, 66, 46–51. [Google Scholar]
- Bates, R.; Mercurio, A. The epithelial-mesenchymal transition (emt) and colorectal cancer progression. Cancer Biol. Ther. 2005, 4, 365–370. [Google Scholar] [CrossRef]
- Kaplan, R.; Riba, R.; Zacharoulis, S.; Bramley, A.; Vincent, L.; Costa, C.; MacDonald, D.; Jin, D.; Shido, K.; Kerns, S.; et al. Vegfr1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Dawson, M.; Duda, D.; Fukumura, D.; Jain, R. Vegfr1-activity-independent metastasis formation. Nature 2009, 461, E4–E5. [Google Scholar] [CrossRef]
- Daenen, L.G.; Roodhart, J.M.; van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.; Voest, E.E. Chemotherapy enhances metastasis formation via vegfr-1-expressing endothelial cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, J.H.; Zhou, W.; Shin, J.; Noh, S.M.; Song, I.S.; Lee, S.H.; Kim, J. The expression of vegf receptor genes is concurrently influenced by epigenetic gene silencing of the genes and vegf activation. Epigenetics 2009, 4, 313–321. [Google Scholar] [CrossRef]
- Yamada, Y.; Watanabe, M.; Yamanaka, M.; Hirokawa, Y.; Suzuki, H.; Takagi, A.; Matsuzaki, T.; Sugimura, Y.; Yatani, R.; Shiraishi, T. Aberrant methylation of the vascular endothelial growth factor receptor-1 gene in prostate cancer. Cancer Sci. 2003, 94, 536–539. [Google Scholar] [CrossRef]
- Chen, J.; Imanaka, N.; Griffin, J.D. Hypoxia potentiates notch signaling in breast cancer leading to decreased e-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- Hughes, D.P. How the notch pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat. Res. 2009, 152, 479–496. [Google Scholar] [CrossRef]
- McGowan, P.M.; Simedrea, C.; Ribot, E.J.; Foster, P.J.; Palmieri, D.; Steeg, P.S.; Allan, A.L.; Chambers, A.F. Notch1 inhibition alters the cd44hi/cd24lo population and reduces the formation of brain metastases from breast cancer. Mol. Cancer Res. 2011, 9, 834–844. [Google Scholar] [CrossRef]
- Nam, D.H.; Jeon, H.M.; Kim, S.; Kim, M.H.; Lee, Y.J.; Lee, M.S.; Kim, H.; Joo, K.M.; Lee, D.S.; Price, J.E.; et al. Activation of notch signaling in a xenograft model of brain metastasis. Clin. Cancer Res. 2008, 14, 4059–4066. [Google Scholar] [CrossRef]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of colon cancer metastasis by aes through inhibition of notch signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef]
- Hur, K.; Toiyama, Y.; Takahashi, M.; Balaguer, F.; Nagasaka, T.; Koike, J.; Hemmi, H.; Koi, M.; Boland, C.R.; Goel, A. Microrna-200c modulates epithelial-to-mesenchymal transition (emt) in human colorectal cancer metastasis. Gut 2013, 62, 1315–1326. [Google Scholar] [CrossRef]
- Ahmad, A.; Aboukameel, A.; Kong, D.; Wang, Z.; Sethi, S.; Chen, W.; Sarkar, F.H.; Raz, A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by mir-200 in breast cancer cells. Cancer Res. 2011, 71, 3400–3409. [Google Scholar] [CrossRef]
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; de Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012, 19, 1208–1219. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The zeb1/mir-200 feedback loop controls notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A.; Pardanani, A. Kit: Molecule of interest for the diagnosis and treatment of mastocytosis and other neoplastic disorders. Curr. Cancer Drug Targets 2007, 7, 492–503. [Google Scholar] [CrossRef]
- Antonescu, C.R. The gist paradigm: Lessons for other kinase-driven cancers. J. Pathol. 2011, 223, 251–261. [Google Scholar] [CrossRef]
- Takaoka, A.; Toyota, M.; Hinoda, Y.; Itoh, F.; Mita, H.; Kakiuchi, H.; Adachi, M.; Imai, K. Expression and identification of aberrant c-kit transcripts in human cancer cells. Cancer Lett. 1997, 115, 257–261. [Google Scholar] [CrossRef]
- Mitchell, L.A.; Nixon, B.; Baker, M.A.; Aitken, R.J. Investigation of the role of src in capacitation-associated tyrosine phosphorylation of human spermatozoa. Mol. Hum. Reprod. 2008, 14, 235–243. [Google Scholar] [CrossRef]
- Mezquita, J.; Mezquita, B.; Pau, M.; Mezquita, C. Down-regulation of flt-1 gene expression by the proteasome inhibitor mg262. J. Cell Biochem. 2003, 89, 1138–1147. [Google Scholar] [CrossRef]
- Ginestier, C.; Wicinski, J.; Cervera, N.; Monville, F.; Finetti, P.; Bertucci, F.; Wicha, M.S.; Birnbaum, D.; Charafe-Jauffret, E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009, 8, 3297–3302. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mezquita, B.; Mezquita, P.; Pau, M.; Mezquita, J.; Mezquita, C. Unlocking Doors without Keys: Activation of Src by Truncated C-terminal Intracellular Receptor Tyrosine Kinases Lacking Tyrosine Kinase Activity. Cells 2014, 3, 92-111. https://doi.org/10.3390/cells3010092
Mezquita B, Mezquita P, Pau M, Mezquita J, Mezquita C. Unlocking Doors without Keys: Activation of Src by Truncated C-terminal Intracellular Receptor Tyrosine Kinases Lacking Tyrosine Kinase Activity. Cells. 2014; 3(1):92-111. https://doi.org/10.3390/cells3010092
Chicago/Turabian StyleMezquita, Belén, Pau Mezquita, Montserrat Pau, Jovita Mezquita, and Cristóbal Mezquita. 2014. "Unlocking Doors without Keys: Activation of Src by Truncated C-terminal Intracellular Receptor Tyrosine Kinases Lacking Tyrosine Kinase Activity" Cells 3, no. 1: 92-111. https://doi.org/10.3390/cells3010092
APA StyleMezquita, B., Mezquita, P., Pau, M., Mezquita, J., & Mezquita, C. (2014). Unlocking Doors without Keys: Activation of Src by Truncated C-terminal Intracellular Receptor Tyrosine Kinases Lacking Tyrosine Kinase Activity. Cells, 3(1), 92-111. https://doi.org/10.3390/cells3010092