
Early Development of an Innovative Nanoparticle-Based Multimodal Tool for Targeted Drug Delivery: A Step-by-Step Approach

 ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fluorescent Silica Nanoparticle Synthesis
2.2. Monoclonal Antibody Production
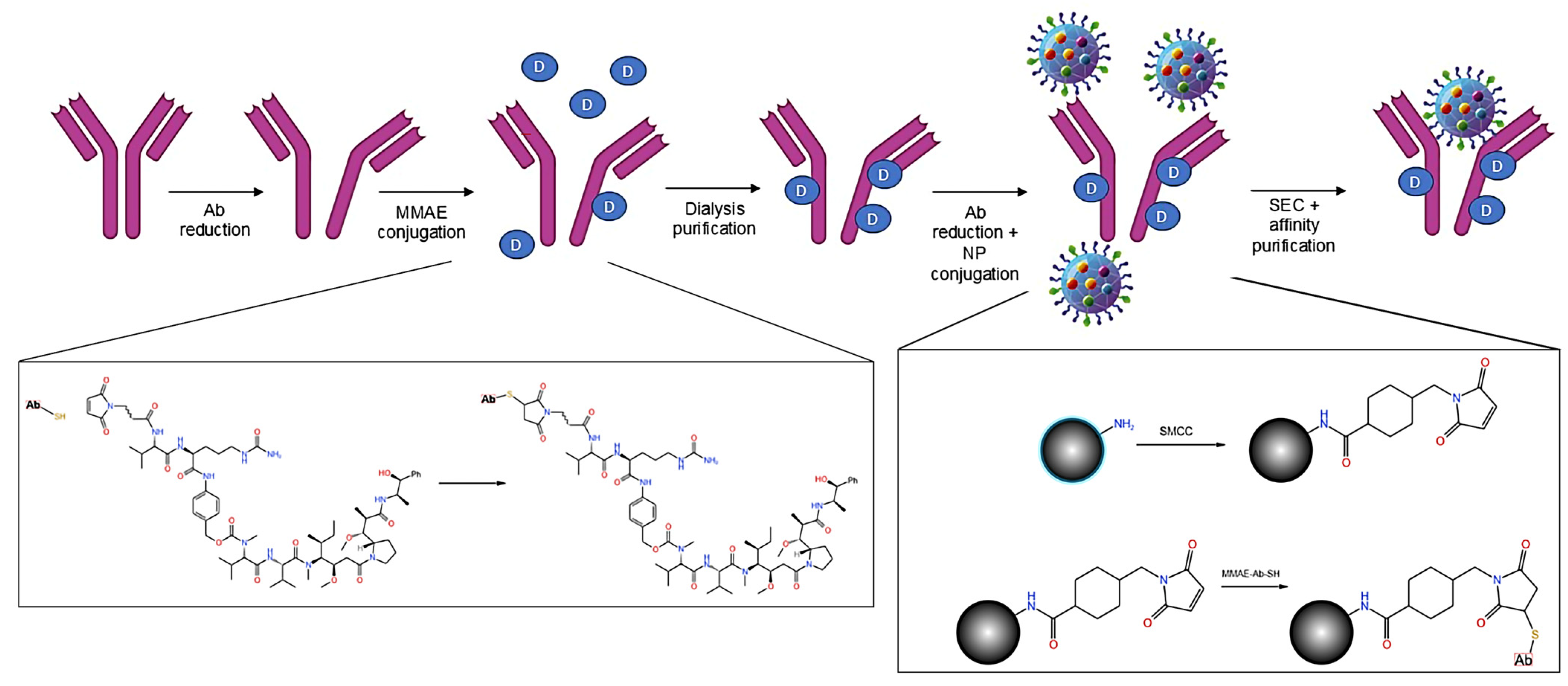
2.3. Antibody to Nanoparticle Conjugation
2.4. Antibody Drug Conjugation
2.5. Antibody Drug Nanoparticle Conjugation
2.6. Cell Culture
2.7. Cell Treatment
2.8. Viability Assay
3. Results
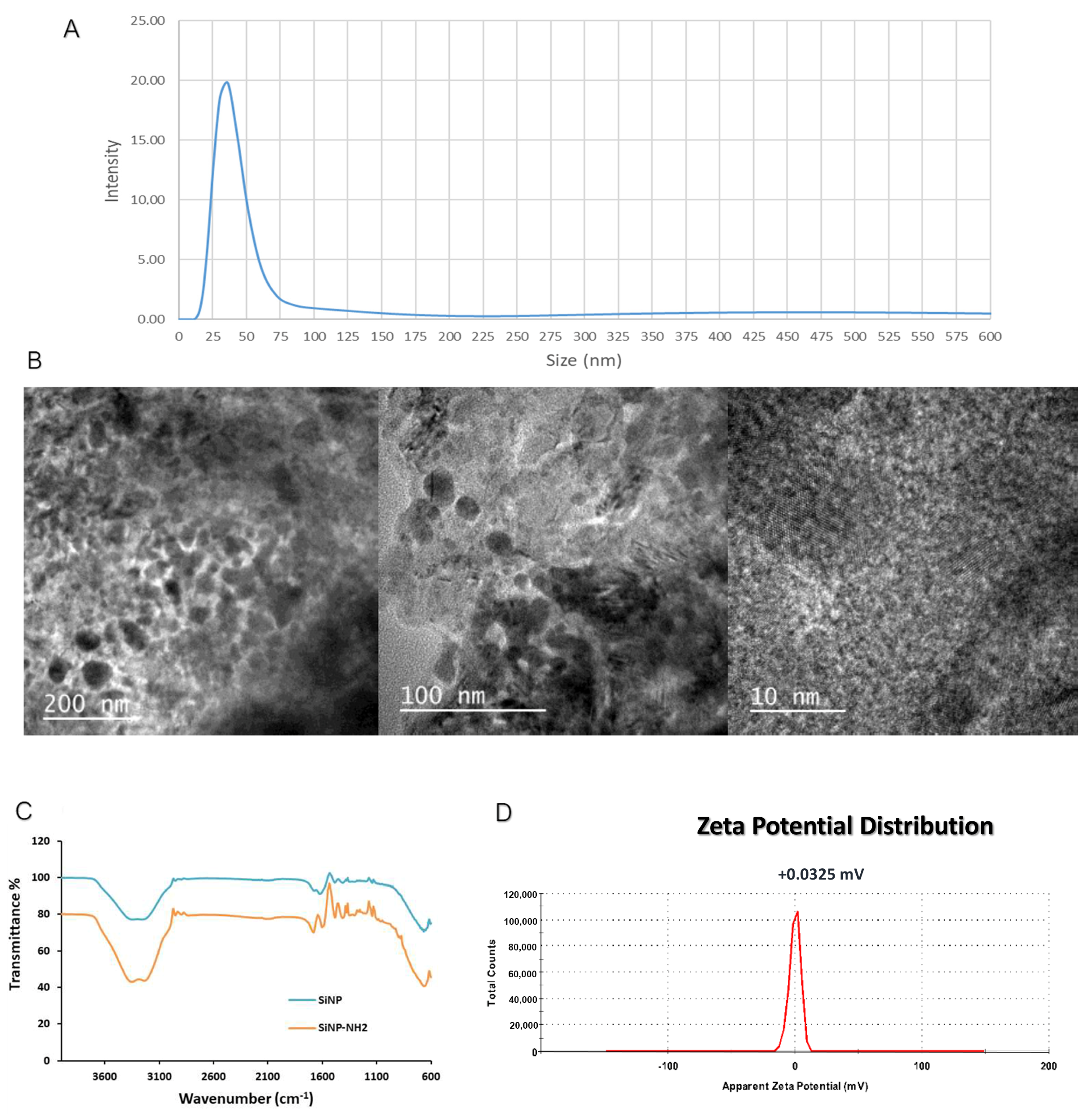
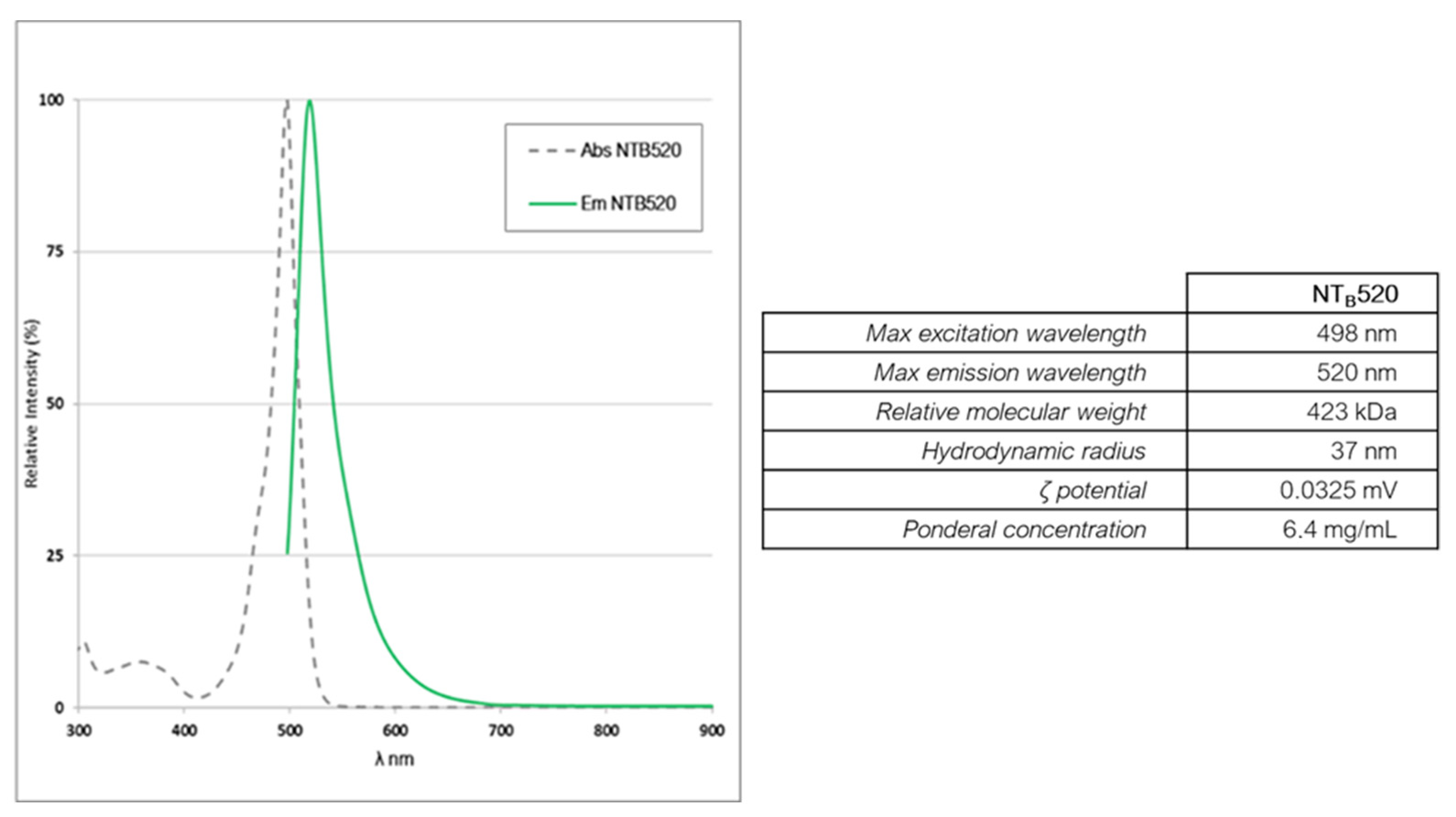
3.1. Characterization of Fluorescent Silica Nanoparticles
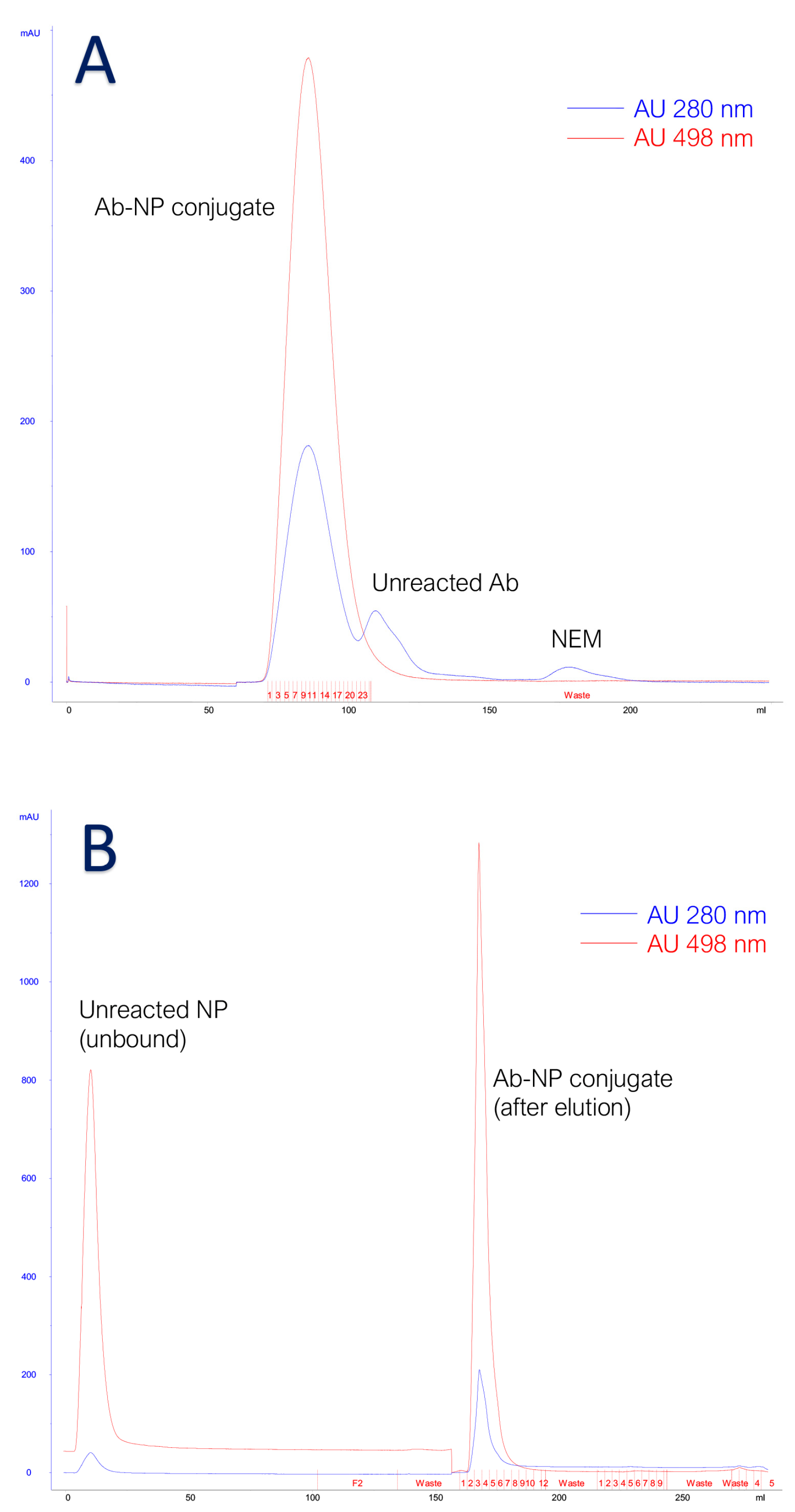
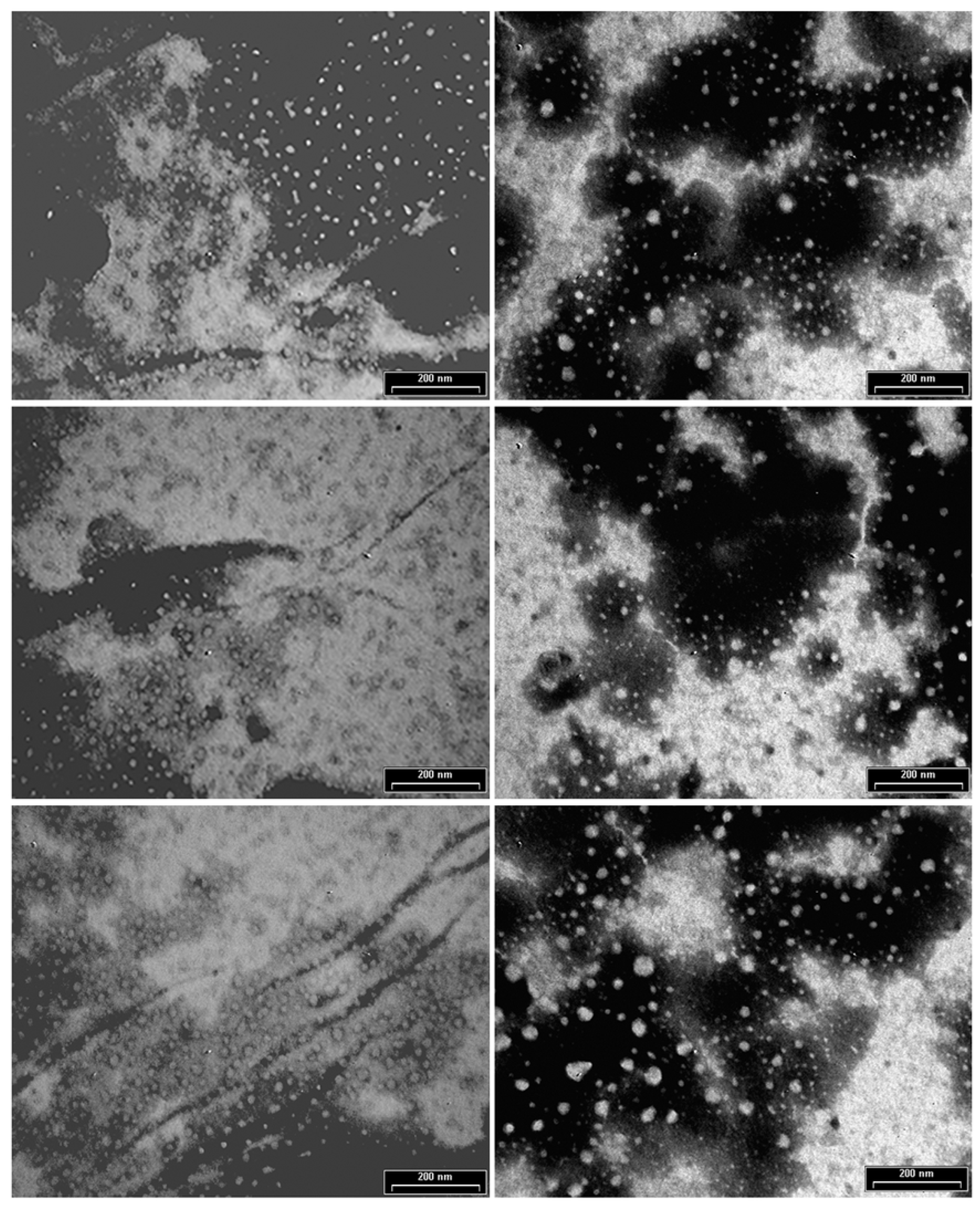
3.2. Characterization of Antibody Conjugated to Fluorescent Silica Nanoparticles
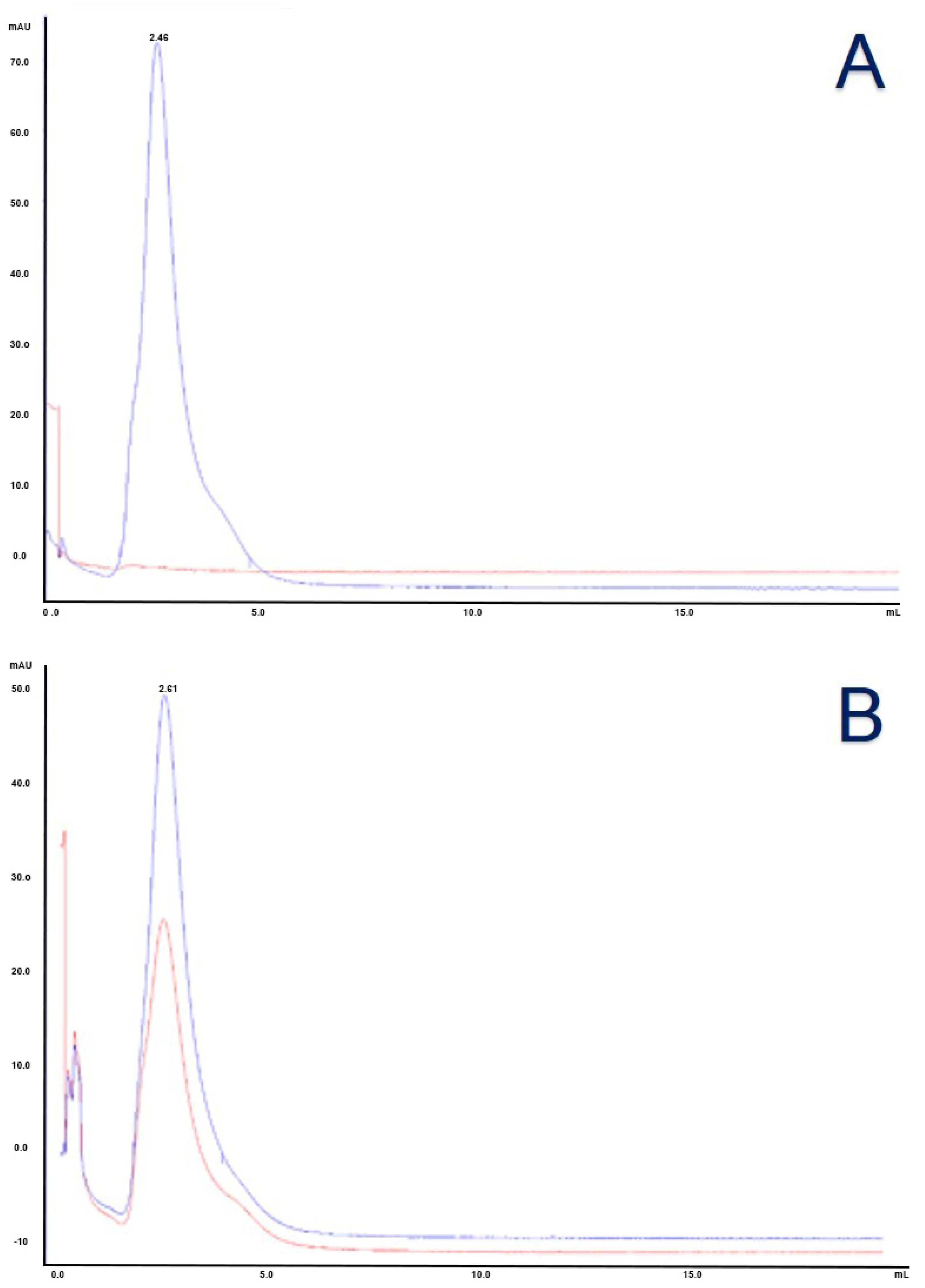
3.3. Characterization of Antibody Conjugated to Monomethyl Auristatin E
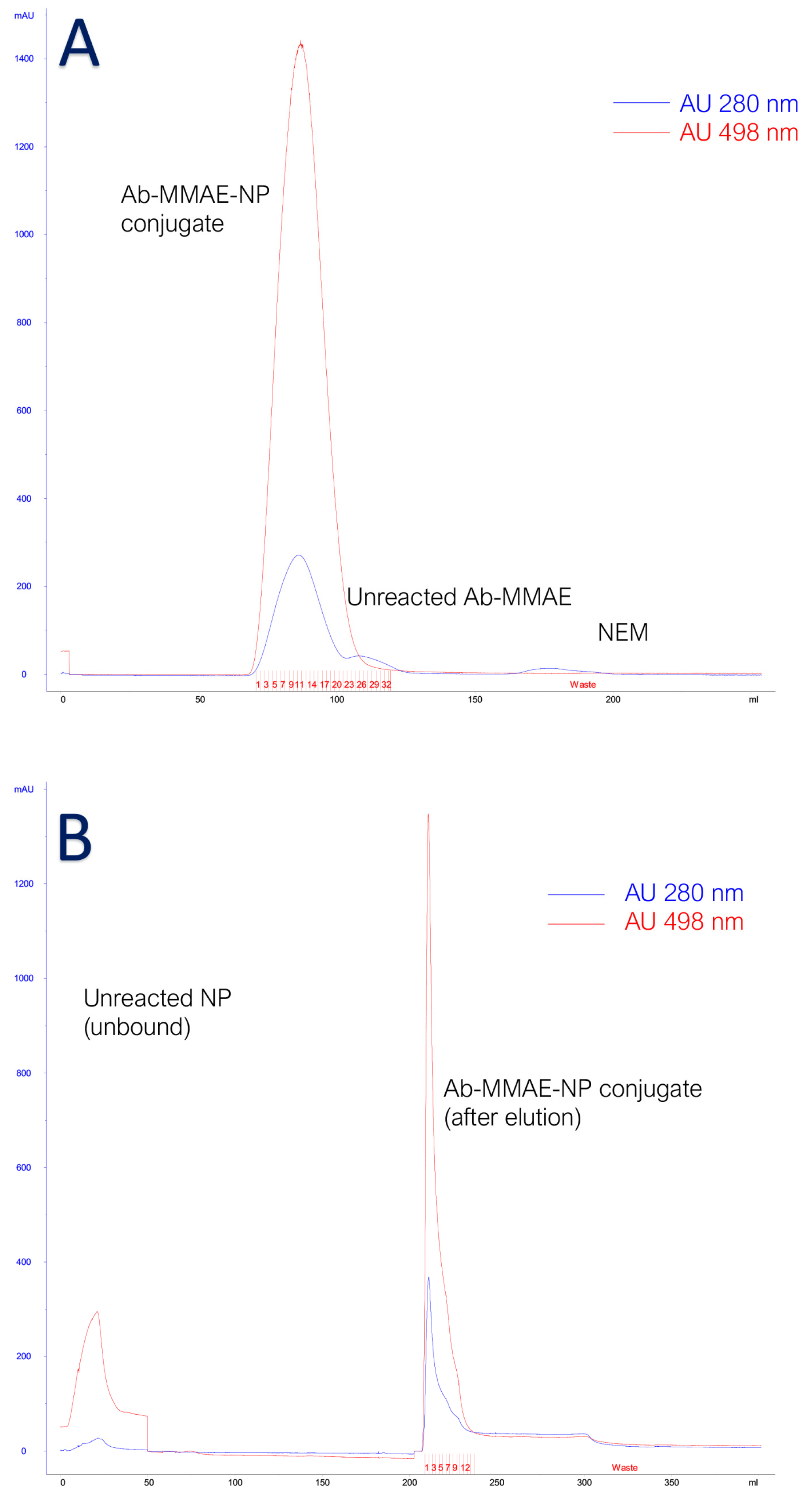
3.4. Characterization of Antibody Conjugated to Fluorescent Silica Nanoparticles and Monomethyl Auristatin E
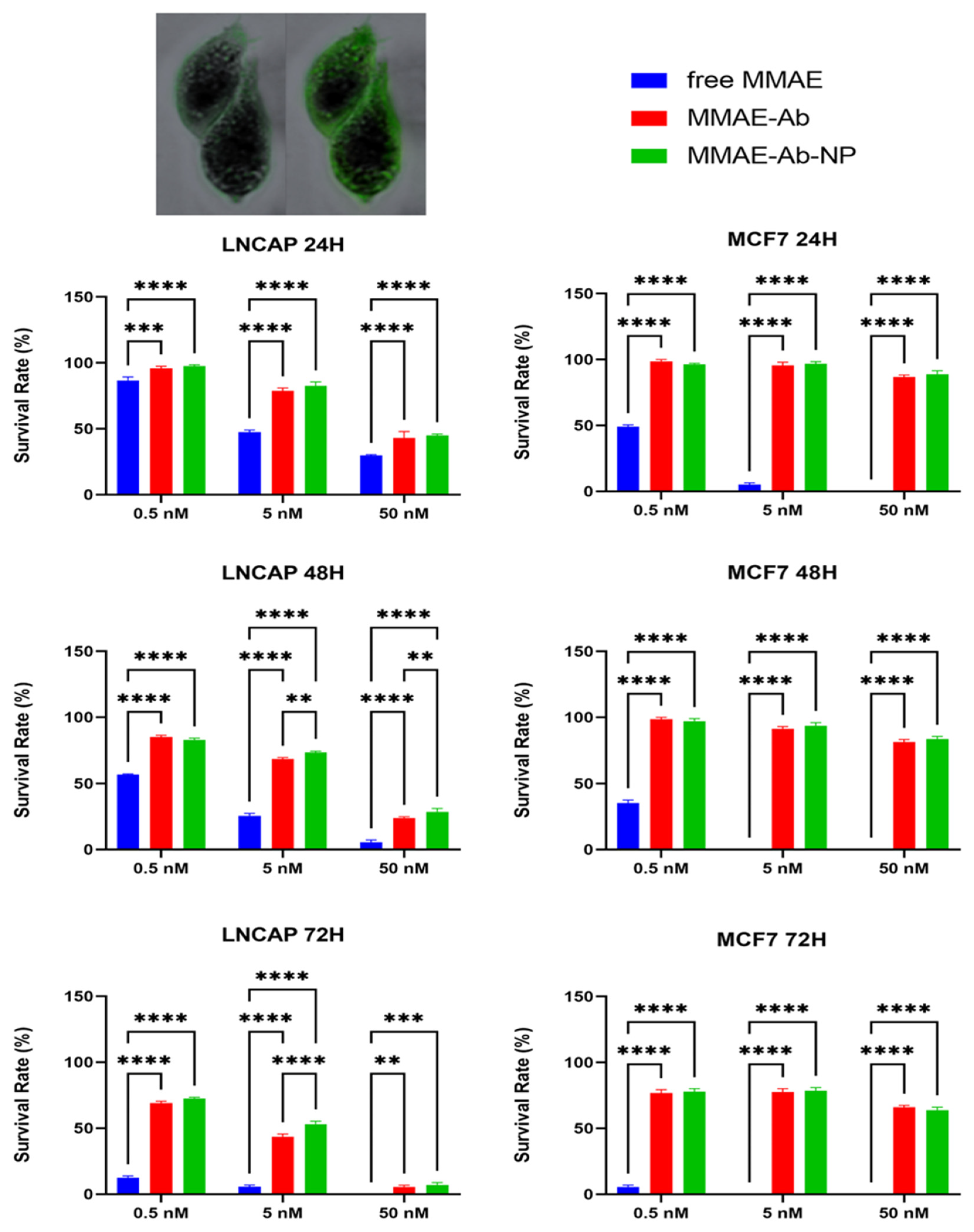
3.5. Comparison of Different Conjugates Toxicity Effects on Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody drug conjugate |
| ADCNP | Antibody drug conjugated nanoparticle |
| DAR | Drug antibody ratio |
| DLS | Dynamic light scattering |
| DMSO | Dimethyl sulfoxide |
| DTNB | 5,5′-dithiobis(2-nitrobenzoic acid) |
| DTT | 1,4-dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| EPR | Enhanced permeability and retention |
| FBS | Fetal bovine serum |
| FTIR | Fourier transform infrared spectroscopy |
| IC50 | Half-maximal inhibitory concentration |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| MMAE | Monomethyl auristatin E |
| MoAb | Monoclonal antibody |
| NEM | N-ethylmaleimide |
| NHS | N-hydroxysuccinimide |
| NP | Nanoparticle |
| PEG | Polyethylene glycol |
| PES | Polyether sulfone |
| PSMA | Prostate specific membrane antigen |
| PVDF | Polyvinylidene fluoride |
| SEC | Size exclusion chromatography |
| SMCC | Succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate |
| TCEP | Tris(2-carboxyethyl)phosphine |
| TEM | Transmission electron microscopy |
| TNB | 5-thio-2-nitrobenzoic acid |
References
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.D.; Chan, W.C.W. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem. Soc. Rev. 2012, 41, 2780–2799. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Zhang, Y.; Chen, T.; Xiao, S.; Liang, H. Morphological and mechanical determinants of cellular uptake of deformable nanoparticles. Nanoscale 2018, 10, 11969–11979. [Google Scholar] [CrossRef]
- Gunasekaran, T.; Haile, T.; Nigusse, T.; Dhanaraju, M.D. Nanotechnology: An effective tool for enhancing bioavailability and bioactivity of phytomedicine. Asian Pac. J. Trop. Biomed. 2014, 4, S1–S7. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef]
- Kumari, M.; Acharya, A.; Krishnamurthy, P.T. Antibody-conjugated nanoparticles for target-specific drug delivery of chemotherapeutics. Beilstein J. Nanotechnol. 2023, 14, 912–926. [Google Scholar] [CrossRef]
- Hui, L.; Chen, W.; Najlah, M. The challenges to develop antibody-conjugated nanomedicine products. Nano TransMed 2023, 2, 100018. [Google Scholar] [CrossRef]
- Cho, H.; Shim, M.K.; Moon, Y.; Song, S.; Kim, J.; Choi, J.; Kim, J.; Lee, Y.; Park, J.Y.; Kim, Y.; et al. Tumor-Specific Monomethyl Auristatin E (MMAE) Prodrug Nanoparticles for Safe and Effective Chemotherapy. Pharmaceutics 2022, 14, 2131. [Google Scholar] [CrossRef]
- Xie, C.; Wang, Y.; Cai, Z.; Du, J.; Chen, Z.; Wang, J.; Peng, X. MMAE-loaded PLGA nanomedicine with improved biosafety to achieve efficient antitumor treatment. J. Innov. Opt. Health Sci. 2024, 17, 2350024. [Google Scholar] [CrossRef]
- Pellegrino, C.; Volpe, A.; Juris, R.; Menna, M.; Calabrese, V.; Sola, F.; Barattini, C.; Ventola, A. Multiple Dye Doped Core-Shell Silica Nanoparticles: Outstanding Stability and Signal Intensity Exploiting FRET Phenomenon for Biomedical Applications. J. Nanomater. Mol. Nanotechnol. 2018, s6, 2. [Google Scholar] [CrossRef]
- Baù, L.; Tecilla, P.; Mancin, F. Sensing with fluorescent nanoparticles. Nanoscale 2011, 3, 121–133. [Google Scholar] [CrossRef]
- Caponetti, V.; Trzcinski, J.W.; Cantelli, A.; Tavano, R.; Papini, E.; Mancin, F.; Montalti, M. Self-assembled biocompatible fluorescent nanoparticles for bioimaging. Front. Chem. 2019, 7, 168. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Newcombe, A.R.; Cresswell, C.; Davies, S.; Watson, K.; Harris, G.; Odonovan, K.; Francis, R. Optimised affinity purification of polyclonal antibodies from hyper immunised ovine serum using a synthetic Protein A adsorbent, MAbsorbent® A2P. J. Chromatogr. B 2005, 814, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction−alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjugate Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Kawinski, E.; Murphy, G.P. Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients. Anticancer Res. 1987, 7, 927–935. [Google Scholar] [CrossRef]
- Gorges, T.M.; Riethdorf, S.; von Ahsen, O.; Nastały, P.; Röck, K.; Boede, M.; Peine, S.; Kuske, A.; Schmid, E.; Kneip, C.; et al. Heterogeneous PSMA expression on circulating tumor cells—A potential basis for stratification and monitoring of PSMA-directed therapies in prostate cancer. Oncotarget 2016, 7, 34930–34941. [Google Scholar] [CrossRef]
- Sutherland, R.; Hall, R.; Taylor, I. Cell Proliferation Kinetics of MCF-7 Human Mammary Carcinoma Cells in Culture and Effects of Tamoxifen on Exponentially Growing and Plateau-Phase Cells. Cancer Res. 1983, 43, 3998–4006. [Google Scholar] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Nunes, J.P.M.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.B.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody–drug conjugate (ADC). Chem. Commun. 2015, 51, 10624–10627. [Google Scholar] [CrossRef]
- Pal, A.; Albusairi, W.; Liu, F.; Tuhin, T.H.; Miller, M.; Liang, D.; Joo, H.; Amin, T.U.; Wilson, E.A.; Faridi, J.S.; et al. Hydrophilic Small Molecules That Harness Transthyretin To Enhance the Safety and Efficacy of Targeted Chemotherapeutic Agents. Mol. Pharm. 2019, 16, 3237–3252. [Google Scholar] [CrossRef]
- Cunningham, D.; Parajuli, K.R.; Zhang, C.; Wang, G.; Mei, J.; Zhang, Q.; Liu, S.; You, Z. Monomethyl Auristatin E Phosphate Inhibits Human Prostate Cancer Growth. Prostate 2016, 76, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Lahnif, H.; Grus, T.; Salvanou, E.-A.; Deligianni, E.; Stellas, D.; Bouziotis, P.; Rösch, F. Old Drug, New Delivery Strategy: MMAE Repackaged. Int. J. Mol. Sci. 2023, 24, 8543. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, K.; Tian, R.; Lu, C. Substrate-assisted visualization of surfactant micelles via transmission electron microscopy. Front. Chem. 2019, 7, 242. [Google Scholar] [CrossRef]
- Richert, L.E.; Servid, A.E.; Harmsen, A.L.; Rynda-Apple, A.; Han, S.; Wiley, J.A.; Douglas, T.; Harmsen, A.G. A virus-like particle vaccine platform elicits heightened and hastened local lung mucosal antibody production after a single dose. Vaccine 2012, 30, 3653–3665. [Google Scholar] [CrossRef]
- Luangtana-Anan, M.; Limmatvapirat, S.; Nunthanid, J.; Chalongsuk, R.; Yamamoto, K. Polyethylene glycol on stability of chitosan microparticulate carrier for protein. AAPS PharmSciTech 2010, 11, 1376–1382. [Google Scholar] [CrossRef]
- Eid, M.M. Characterization of Nanoparticles by FTIR and FTIR-Microscopy. In Handbook of Consumer Nanoproducts; Springer: Singapore, 2021. [Google Scholar]
- Machulkin, A.E.; Uspenskaya, A.A.; Nimenko, E.A.; Ber, A.P.; Petrov, S.A.; Polshakov, V.I.; Shafikov, R.R.; Skvortsov, D.A.; Plotnikova, E.A.; Pankratov, A.A.; et al. Synthesis, Characterization, and Preclinical Evaluation of a Small-Molecule Prostate-Specific Membrane Antigen-Targeted Monomethyl Auristatin E Conjugate. J. Med. Chem. 2021, 64, 17123–17145. [Google Scholar] [CrossRef]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjugate Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef]
- Cruz, E.; Kayser, V. Synthesis and enhanced cellular uptake in vitro of anti-HER2 multifunctional gold nanoparticles. Cancers 2019, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Drug-to-antibody ratio (DAR) by UV/Vis spectroscopy. In Antibody-Drug Conjugates; Humana Press: Totowa, NJ, USA, 2013; Volume 1045. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Complete lysosomal disruption: A route to necrosis, not to the inflammasome. Cell Cycle 2013, 12, 1995. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular basis of valine-citrulline-PABC linker instability in site-specific ADCs and its mitigation by linker design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Yu, H.; Yang, Z.; Li, F.; Xu, L.; Sun, Y. Cell-mediated targeting drugs delivery systems. Drug Deliv. 2020, 27, 1425–1437. [Google Scholar] [CrossRef]
- Szijj, P.A.; Bahou, C.; Chudasama, V. Minireview: Addressing the retro-Michael instability of maleimide bioconjugates. Drug Discov. Today Technol. 2018, 30, 27–34. [Google Scholar] [CrossRef]
- Lu, D.; Jin, J.Y.; Girish, S.; Agarwal, P.; Li, D.; Prabhu, S.; Dere, R.C.; Saad, O.M.; Nazzal, D.; Koppada, N.; et al. Semi-mechanistic multiple-analyte pharmacokinetic model for an antibody-drug-conjugate in cynomolgus monkeys. Pharm. Res. 2014, 32, 1907–1919. [Google Scholar] [CrossRef]
- Lahnsteiner, M.; Kastner, A.; Mayr, J.; Roller, A.; Keppler, B.K.; Kowol, C.R. Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting. Chem.—A Eur. J. 2020, 26, 15867–15870. [Google Scholar] [CrossRef]
- Fontaine, S.D.; Reid, R.; Robinson, L.; Ashley, G.W.; Santi, D.V. Long-term stabilization of maleimide–thiol conjugates. Bioconjugate Chem. 2014, 26, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G. Preparation of well-defined antibody–drug conjugates through glycan remodeling and strain-promoted azide–alkyne cycloadditions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Shi, W.; Huang, W. Homogeneous Antibody–Drug Conjugates via Glycoengineering. In Bioconjugation: Methods and Protocols; Humana: New York, NY, USA, 2019; Volume 2033. [Google Scholar] [CrossRef]
- Akilesh, S.; Huber, T.B.; Wu, H.; Wang, G.; Hartleben, B.; Kopp, J.B.; Miner, J.H.; Roopenian, D.C.; Unanue, E.R.; Shaw, A.S. Podocytes use FcRn to clear IgG from the glomerular basement membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 967–972. [Google Scholar] [CrossRef]
- Elias, D.R.; Poloukhtine, A.; Popik, V.; Tsourkas, A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 194–201. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef]
- Georgiev, N.I.; Bakov, V.V.; Anichina, K.K.; Bojinov, V.B. Fluorescent Probes as a Tool in Diagnostic and Drug Delivery Systems. Pharmaceuticals 2023, 16, 381. [Google Scholar] [CrossRef]
- Etrych, T.; Janoušková, O.; Chytil, P. Fluorescence imaging as a tool in preclinical evaluation of polymer-based nano-dds systems intended for cancer treatment. Pharmaceutics 2019, 11, 471. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barattini, C.; Volpe, A.; Gori, D.; Lopez, D.; Ventola, A.; Papa, S.; Montanari, M.; Canonico, B. Early Development of an Innovative Nanoparticle-Based Multimodal Tool for Targeted Drug Delivery: A Step-by-Step Approach. Cells 2025, 14, 670. https://doi.org/10.3390/cells14090670
Barattini C, Volpe A, Gori D, Lopez D, Ventola A, Papa S, Montanari M, Canonico B. Early Development of an Innovative Nanoparticle-Based Multimodal Tool for Targeted Drug Delivery: A Step-by-Step Approach. Cells. 2025; 14(9):670. https://doi.org/10.3390/cells14090670
Chicago/Turabian StyleBarattini, Chiara, Angela Volpe, Daniele Gori, Daniele Lopez, Alfredo Ventola, Stefano Papa, Mariele Montanari, and Barbara Canonico. 2025. "Early Development of an Innovative Nanoparticle-Based Multimodal Tool for Targeted Drug Delivery: A Step-by-Step Approach" Cells 14, no. 9: 670. https://doi.org/10.3390/cells14090670
APA StyleBarattini, C., Volpe, A., Gori, D., Lopez, D., Ventola, A., Papa, S., Montanari, M., & Canonico, B. (2025). Early Development of an Innovative Nanoparticle-Based Multimodal Tool for Targeted Drug Delivery: A Step-by-Step Approach. Cells, 14(9), 670. https://doi.org/10.3390/cells14090670