
mTORopathies in Epilepsy and Neurodevelopmental Disorders: The Future of Therapeutics and the Role of Gene Editing

 , , , , , and
, , , , , and
Abstract
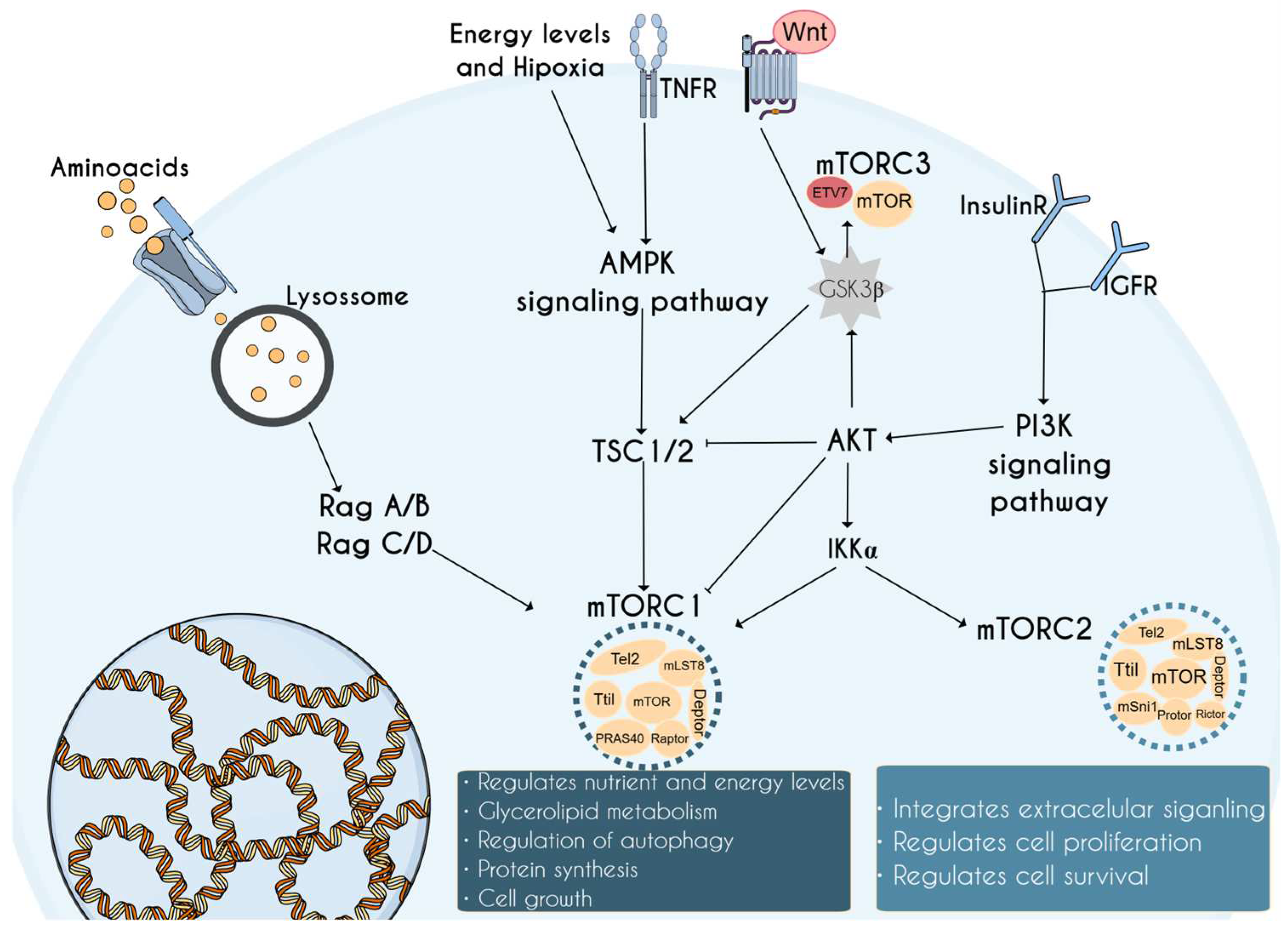
1. Introduction to the mTOR Complex and Its Relevance in Neurodevelopment
2. mTORopathies: mTOR-Related Disorders
3. Gene Editing in the Treatment of mTORopathies

{kind=link}
{kind=link}
| Organism/Cell Type | Target | Editing Method | Year | Reference |
|---|---|---|---|---|
| Mice | STRADA | shRNA | 2013 | [78] |
| Mice | Depdc5 | TALEN | 2016 | [79] |
| Mice | PTEN | Cre-Lox recombination | 2016 | [80] |
| Mice | Depdc5 | Cre-Lox recombination | 2018 | [63] |
| Rat | Depdc5 | CRISPR | 2018 | [87] |
| Mice | Depdc5 | Cre-Lox recombination | 2020 | [88] |
| Mice | STRADA | shRNA | 2010 | [54] |
| Mice | TSC1 | Rous sarcoma virus-based retroviral vectors | 2001 | [89] |
| Mice | TSC1 | Cre-Lox recombination | 2007 | [90] |
| Mice | TSC1 | Cre-Lox recombination | 2012 | [91] |
| Mice | TSC1 | Cre-Lox recombination | 2013 | [92] |
| Mice | Rictor | Cre-Lox recombination | 2020 | [32] |
| Mice | TSC2 | Cre-Lox recombination | 2013 | [93] |
| Mice | TSC1 | Cre-Lox recombination | 2011 | [94] |
| Mice | RHEB | Plasmid construct | 2019 | [95] |
| hiPSCs | TSC2 | CRISPR | 2018 | [96] |
| hiPSCs | TSC2 | ZFN | 2016 | [97] |
| hESCs | PTEN | CRISPR | 2019 | [98] |
| N2a | TSC2/Depdc5 | CRISPR | 2021 | [99] |
| Mice | TSC2 | Cre-Lox recombination | 2021 | [84] |
| Mice | TSC1 | Cre-Lox recombination | 2019 | [83] |
| Mice | TSC1 | Cre-Lox recombination | 2016 | [82] |
4. Available Types of Gene Editing
5. Current Advances in mTOR Gene Therapies
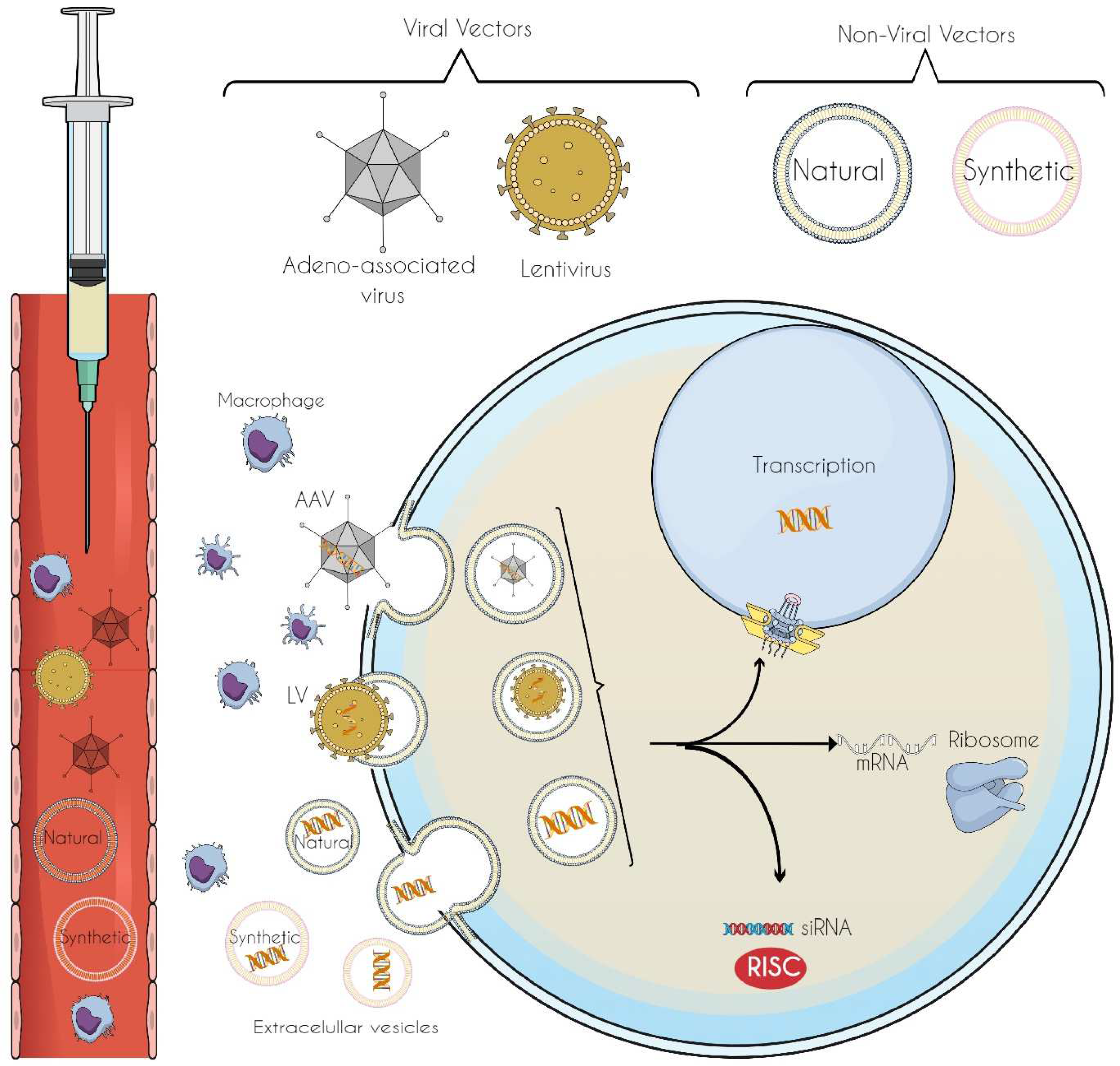
6. Vectors for Gene-Editing Tool Delivery: Innovations in Precision and Targeted Therapeutics
6.1. Viral Vectors
6.2. Non-Viral Vectors
7. The Future of Gene Editing for mTORopathies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wipperman, M.F.; Montrose, D.C.; Gotto, A.M., Jr.; Hajjar, D.P. Mammalian Target of Rapamycin: A Metabolic Rheostat for Regulating Adipose Tissue Function and Cardiovascular Health. Am. J. Pathol. 2019, 189, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Committee, H.G.N. MTOR (Mechanistic Target Of Rapamycin Kinase). Available online: https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:3942 (accessed on 19 January 2025).
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- el Hage, A.; Dormond, O. Combining mTOR Inhibitors and T Cell-Based Immunotherapies in Cancer Treatment. Cancers 2021, 13, 1359. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygiel, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Driedger, J.H.; Schroter, J.; Group, P.R.-S.; Syrbe, S.; Saffari, A. Long-term neuropsychologic outcome of pre-emptive mTOR inhibitor treatment in children with tuberous sclerosis complex (TSC) under 4 months of age (PROTECT), a two-arm, randomized, observer-blind, controlled phase IIb national multicentre clinical trial: Study protocol. Orphanet J. Rare Dis. 2025, 20, 2. [Google Scholar] [CrossRef]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef]
- Koike-Kumagai, M.; Fujimoto, M.; Wataya-Kaneda, M. Sirolimus relieves seizures and neuropsychiatric symptoms via changes of microglial polarity in tuberous sclerosis complex model mice. Neuropharmacology 2022, 218, 109203. [Google Scholar] [CrossRef]
- Cui, F.; Gu, S.; Gu, Y.; Yin, J.; Fang, C.; Liu, L. Alteration in the mRNA expression profile of the autophagy-related mTOR pathway in schizophrenia patients treated with olanzapine. BMC Psychiatry 2021, 21, 388. [Google Scholar] [CrossRef] [PubMed]
- Nawwar, D.A.; Zaki, H.F.; Sayed, R.H. Role of the NRG1/ErbB4 and PI3K/AKT/mTOR signaling pathways in the anti-psychotic effects of aripiprazole and sertindole in ketamine-induced schizophrenia-like behaviors in rats. Inflammopharmacology 2022, 30, 1891–1907. [Google Scholar] [CrossRef] [PubMed]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.J.; Reis, G.; Kang, H.; Gingras, A.C.; Sonenberg, N.; Schuman, E.M. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, L.; Wu, Y.; Singh, K.; Su, B.; Zhang, N.; Liu, X.; Shen, Y.; Huang, S. Rapamycin inhibits mSin1 phosphorylation independently of mTORC1 and mTORC2. Oncotarget 2015, 6, 4286. [Google Scholar] [CrossRef] [PubMed]
- Harwood, F.C.; Klein Geltink, R.I.; O’Hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Harwood, F.; Have, S.T.; Lamond, A.; Phillips, A.H.; Kriwacki, R.W.; Halder, P.; Cardone, M.; Grosveld, G.C. Assembly of mTORC3 Involves Binding of ETV7 to Two Separate Sequences in the mTOR Kinase Domain. Int. J. Mol. Sci. 2024, 25, 42. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Inamura, N.; Kawamura, M.; Namba, H.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 2004, 24, 9760–9769. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Avruch, J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J. Biol. Chem. 2005, 280, 38121–38124. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol 2010, 20, 427–434. [Google Scholar] [CrossRef]
- Zhao, W.; Xie, C.; Zhang, X.; Liu, J.; Liu, J.; Xia, Z. Advances in the mTOR signaling pathway and its inhibitor rapamycin in epilepsy. Brain Behav. 2023, 13, e2995. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Y.; Sun, X.; Cui, Q.; Bai, X.; Dong, G.; Gao, Z.; Wang, Y.; Gao, C.; Sun, S.; et al. The PI3K/AKT/mTOR signaling pathway is aberrantly activated in primary central nervous system lymphoma and correlated with a poor prognosis. BMC Cancer 2022, 22, 190. [Google Scholar] [CrossRef]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Chen, C.J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 2019, 25, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Chen, G.; Li, Y.; Guo, Z.; Zhang, X. The molecular genetics of PI3K/PTEN/AKT/mTOR pathway in the malformations of cortical development. Genes Dis. 2024, 11, 101021. [Google Scholar] [CrossRef]
- Girodengo, M.; Ultanir, S.K.; Bateman, J.M. Mechanistic target of rapamycin signaling in human nervous system development and disease. Front. Mol. Neurosci. 2022, 15, 1005631. [Google Scholar] [CrossRef]
- Luo, C.; Ye, W.R.; Shi, W.; Yin, P.; Chen, C.; He, Y.B.; Chen, M.F.; Zu, X.B.; Cai, Y. Perfect match: mTOR inhibitors and tuberous sclerosis complex. Orphanet J. Rare Dis. 2022, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, A.; Baldassari, S.; Baulac, S. mTOR pathway: Insights into an established pathway for brain mosaicism in epilepsy. Neurobiol. Dis. 2023, 182, 106144. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Bordey, A. Convergent and Divergent Mechanisms of Epileptogenesis in mTORopathies. Front. Neuroanat. 2021, 15, 664695. [Google Scholar] [CrossRef]
- Liu, A.C.; Shen, Y.; Serbinski, C.R.; He, H.; Roman, D.; Endale, M.; Aschbacher-Smith, L.; King, K.A.; Granadillo, J.L.; Lopez, I.; et al. Clinical and functional studies of MTOR variants in Smith-Kingsmore syndrome reveal deficits of circadian rhythm and sleep-wake behavior. HGG Adv. 2024, 5, 100333. [Google Scholar] [CrossRef] [PubMed]
- Besterman, A.D.; Althoff, T.; Elfferich, P.; Gutierrez-Mejia, I.; Sadik, J.; Bernstein, J.A.; van Ierland, Y.; Kattentidt-Mouravieva, A.A.; Nellist, M.; Abramson, J.; et al. Functional and structural analyses of novel Smith-Kingsmore Syndrome-Associated MTOR variants reveal potential new mechanisms and predictors of pathogenicity. PLoS Genet. 2021, 17, e1009651. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.L.; Curry, P.D.K.; Marcinkute, R.; Brewer, C.; Coman, D.; Hobson, E.; Johnson, D.; Lynch, S.A.; Saggar, A.; Searle, C.; et al. Delineating the Smith-Kingsmore syndrome phenotype: Investigation of 16 patients with the MTOR c.5395G > A p.(Glu1799Lys) missense variant. Am. J. Med. Genet. A 2021, 185, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Gordo, G.; Tenorio, J.; Arias, P.; Santos-Simarro, F.; Garcia-Minaur, S.; Moreno, J.C.; Nevado, J.; Vallespin, E.; Rodriguez-Laguna, L.; de Mena, R.; et al. mTOR mutations in Smith-Kingsmore syndrome: Four additional patients and a review. Clin. Genet. 2018, 93, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Baldassari, S.; Ribierre, T.; Marsan, E.; Adle-Biassette, H.; Ferrand-Sorbets, S.; Bulteau, C.; Dorison, N.; Fohlen, M.; Polivka, M.; Weckhuysen, S.; et al. Dissecting the genetic basis of focal cortical dysplasia: A large cohort study. Acta Neuropathol. 2019, 138, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Baulac, S. mTOR-therapy and targeted treatment opportunities in mTOR-related epilepsies associated with cortical malformations. Rev. Neurol. 2023, 179, 337–344. [Google Scholar] [CrossRef]
- Lee, W.S.; Baldassari, S.; Stephenson, S.E.M.; Lockhart, P.J.; Baulac, S.; Leventer, R.J. Cortical Dysplasia and the mTOR Pathway: How the Study of Human Brain Tissue Has Led to Insights into Epileptogenesis. Int. J. Mol. Sci. 2022, 23, 1344. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, C.; Battaglia, D.; Pietrini, D.; Piastra, M.; Massimi, L. Hemimegalencephaly: Clinical implications and surgical treatment. Childs Nerv. Syst. 2006, 22, 852–866. [Google Scholar] [CrossRef]
- Garcia, C.A.B.; Carvalho, S.C.S.; Yang, X.; Ball, L.L.; George, R.D.; James, K.N.; Stanley, V.; Breuss, M.W.; Thome, U.; Santos, M.V.; et al. mTOR pathway somatic variants and the molecular pathogenesis of hemimegalencephaly. Epilepsia Open 2020, 5, 97–106. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Poduri, A. Megalencephaly and hemimegalencephaly: Breakthroughs in molecular etiology. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166C, 156–172. [Google Scholar] [CrossRef]
- Sidira, C.; Vargiami, E.; Dragoumi, P.; Zafeiriou, D.I. Hemimegalencephaly and tuberous sclerosis complex: A rare yet challenging association. Eur. J. Paediatr. Neurol. 2021, 30, 58–65. [Google Scholar] [CrossRef]
- Venot, Q.; Canaud, G. [PIK3CA-related overgrowth syndrome (PROS)]. Nephrol. Ther. 2017, 13 (Suppl. 1), S155–S156. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Rios, J.J.; Parker, V.E.; Semple, R.K.; Lindhurst, M.J.; Sapp, J.C.; Alomari, A.; Ezaki, M.; Dobyns, W.; Biesecker, L.G. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am. J. Med. Genet. A 2015, 167A, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.T.; Vaid, S.; Lin, G.; Swaminathan, P.; Safran, J.; Loughman, A.; Lee, M.; Glenn, T.; Majolo, F.; Crino, P.B.; et al. STRADA-mutant human cortical organoids model megalencephaly and exhibit delayed neuronal differentiation. Dev. Neurobiol. 2021, 81, 696–709. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Gorter, J.A.; Cepeda, C. Finding a better drug for epilepsy: The mTOR pathway as an antiepileptogenic target. Epilepsia 2012, 53, 1119–1130. [Google Scholar] [CrossRef]
- Puffenberger, E.G.; Strauss, K.A.; Ramsey, K.E.; Craig, D.W.; Stephan, D.A.; Robinson, D.L.; Hendrickson, C.L.; Gottlieb, S.; Ramsay, D.A.; Siu, V.M.; et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 2007, 130, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Orlova, K.A.; Parker, W.E.; Heuer, G.G.; Tsai, V.; Yoon, J.; Baybis, M.; Fenning, R.S.; Strauss, K.; Crino, P.B. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J. Clin. Investig. 2010, 120, 1591–1602. [Google Scholar] [CrossRef]
- Haijes, H.A.; Koster, M.J.E.; Rehmann, H.; Li, D.; Hakonarson, H.; Cappuccio, G.; Hancarova, M.; Lehalle, D.; Reardon, W.; Schaefer, G.B.; et al. De Novo Heterozygous POLR2A Variants Cause a Neurodevelopmental Syndrome with Profound Infantile-Onset Hypotonia. Am. J. Hum. Genet. 2019, 105, 283–301. [Google Scholar] [CrossRef]
- Giacomini, T.; Scala, M.; Nobile, G.; Severino, M.; Tortora, D.; Nobili, L.; Accogli, A.; Torella, A.; Capra, V.; Mancardi, M.M.; et al. De novo POLR2A p.(Ile457Thr) variant associated with early-onset encephalopathy and cerebellar atrophy: Expanding the phenotypic spectrum. Brain Dev. 2022, 44, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Zeb, A.; Cheng, L.F. Exploring the molecular mechanism of hepatitis virus inducing hepatocellular carcinoma by microarray data and immune infiltrates analysis. Front. Immunol. 2022, 13, 1032819. [Google Scholar] [CrossRef] [PubMed]
- Kreis, P.; Leondaritis, G.; Lieberam, I.; Eickholt, B.J. Subcellular targeting and dynamic regulation of PTEN: Implications for neuronal cells and neurological disorders. Front. Mol. Neurosci. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Cullen, E.R.; Safari, M.; Mittelstadt, I.; Weston, M.C. Hyperactivity of mTORC1- and mTORC2-dependent signaling mediates epilepsy downstream of somatic PTEN loss. Elife 2024, 12, RP91323. [Google Scholar] [CrossRef] [PubMed]
- Tariq, K.; Cullen, E.; Getz, S.A.; Conching, A.K.S.; Goyette, A.R.; Prina, M.L.; Wang, W.; Li, M.; Weston, M.C.; Luikart, B.W. Disruption of mTORC1 rescues neuronal overgrowth and synapse function dysregulated by Pten loss. Cell Rep. 2022, 41, 111574. [Google Scholar] [CrossRef] [PubMed]
- Baulac, S.; Baldassari, S. DEPDC5-Related Epilepsy. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Samanta, D. DEPDC5-related epilepsy: A comprehensive review. Epilepsy Behav. 2022, 130, 108678. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Jones, B.M.; Wolfson, R.L.; Super, C.E.; Dhamne, S.C.; Rotenberg, A.; Sabatini, D.M.; Sahin, M.; Poduri, A. A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 2018, 111, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Adashi, E.Y.; Gruppuso, P.A.; Cohen, I.G. CRISPR Therapy of Sickle Cell Disease: The Dawning of the Gene Editing Era. Am. J. Med. 2024, 137, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Pauling, L.; Itano, H.A.; Singer, S.J.; Wells, I.C. Sickle cell anemia a molecular disease. Science 1949, 110, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type Iii. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Clin. Orthop. Relat. Res. 2007, 462, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Lowenthal, A.; Terheggen, H.G.; Columbo, J.P. Induction of arginase activity with the Shope papilloma virus in tissue culture cells from an argininemic patient. J. Exp. Med. 1973, 137, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Terheggen, H.G.; Lowenthal, A.; Lavinha, F.; Colombo, J.P.; Rogers, S. Unsuccessful trial of gene replacement in arginase deficiency. Z. Kinderheilkd 1975, 119, 1–3. [Google Scholar] [CrossRef]
- Beutler, E. The Cline affair. Mol. Ther. 2001, 4, 396–397. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Anderson, W.F.; Blaese, M.; Hwu, P.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Ettinghausen, S.E.; et al. The development of gene therapy for the treatment of cancer. Ann. Surg. 1993, 218, 455–463, discussion 463–454. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Yla-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Advanced Therapy Medicinal Products: Overview. Available online: https://www.ema.europa.eu/en/human-regulatory-overview/advanced-therapy-medicinal-products-overview (accessed on 19 January 2025).
- Available online: https://www.fda.gov/ (accessed on 19 January 2025).
- Blair, J.D.; Bateup, H.S. New frontiers in modeling tuberous sclerosis with human stem cell-derived neurons and brain organoids. Dev. Dyn. 2020, 249, 46–55. [Google Scholar] [CrossRef]
- Parker, W.E.; Orlova, K.A.; Parker, W.H.; Birnbaum, J.F.; Krymskaya, V.P.; Goncharov, D.A.; Baybis, M.; Helfferich, J.; Okochi, K.; Strauss, K.A.; et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci. Transl. Med. 2013, 5, 182ra153. [Google Scholar] [CrossRef] [PubMed]
- Marsan, E.; Ishida, S.; Schramm, A.; Weckhuysen, S.; Muraca, G.; Lecas, S.; Liang, N.; Treins, C.; Pende, M.; Roussel, D.; et al. Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol. Dis. 2016, 89, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Cupolillo, D.; Hoxha, E.; Faralli, A.; De Luca, A.; Rossi, F.; Tempia, F.; Carulli, D. Autistic-Like Traits and Cerebellar Dysfunction in Purkinje Cell PTEN Knock-Out Mice. Neuropsychopharmacology 2016, 41, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Siciliano, B.; Wen, Z. Modeling tuberous sclerosis complex with human induced pluripotent stem cells. World J. Pediatr. 2024, 20, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.; Zhang, X.; Goto, J.; Han, S.; Lai, C.; Bronson, R.; Sena-Esteves, M.; Ramesh, V.; Stemmer-Rachamimov, A.; Kwiatkowski, D.J.; et al. Survival benefit and phenotypic improvement by hamartin gene therapy in a tuberous sclerosis mouse brain model. Neurobiol. Dis. 2015, 82, 22–31. [Google Scholar] [CrossRef]
- Prabhakar, S.; Cheah, P.S.; Zhang, X.; Zinter, M.; Gianatasio, M.; Hudry, E.; Bronson, R.T.; Kwiatkowski, D.J.; Stemmer-Rachamimov, A.; Maguire, C.A.; et al. Long-Term Therapeutic Efficacy of Intravenous AAV-Mediated Hamartin Replacement in Mouse Model of Tuberous Sclerosis Type 1. Mol. Ther. Methods Clin. Dev. 2019, 15, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.S.; Prabhakar, S.; Yellen, D.; Beauchamp, R.L.; Zhang, X.; Kasamatsu, S.; Bronson, R.T.; Thiele, E.A.; Kwiatkowski, D.J.; Stemmer-Rachamimov, A.; et al. Gene therapy for tuberous sclerosis complex type 2 in a mouse model by delivery of AAV9 encoding a condensed form of tuberin. Sci. Adv. 2021, 7, eabb1703. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Leandro, K.; Rufino-Ramos, D.; Breyne, K.; Di Ianni, E.; Lopes, S.M.; Jorge Nobre, R.; Kleinstiver, B.P.; Perdigao, P.R.L.; Breakefield, X.O.; Pereira de Almeida, L. Exploring the potential of cell-derived vesicles for transient delivery of gene editing payloads. Adv. Drug Deliv. Rev. 2024, 211, 115346. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Knowlton, R.C.; Watson, B.O.; Glanowska, K.M.; Murphy, G.G.; Parent, J.M.; Wang, Y. Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol. 2018, 84, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Klofas, L.K.; Short, B.P.; Zhou, C.; Carson, R.P. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum. Mol. Genet. 2020, 29, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767. [Google Scholar] [CrossRef] [PubMed]
- Meikle, L.; Talos, D.M.; Onda, H.; Pollizzi, K.; Rotenberg, A.; Sahin, M.; Jensen, F.E.; Kwiatkowski, D.J. A mouse model of tuberous sclerosis: Neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 2007, 27, 5546–5558. [Google Scholar] [CrossRef]
- Carson, R.P.; Van Nielen, D.L.; Winzenburger, P.A.; Ess, K.C. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 2012, 45, 369–380. [Google Scholar] [CrossRef]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Reith, R.M.; McKenna, J.; Wu, H.; Hashmi, S.S.; Cho, S.H.; Dash, P.K.; Gambello, M.J. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 2013, 51, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, D.M.; Su, T.; Lopez, J.; Platel, J.C.; Bordey, A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Investig. 2011, 121, 1596–1607. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Mahadeo, T.; Bordey, A. mTOR Hyperactivity Levels Influence the Severity of Epilepsy and Associated Neuropathology in an Experimental Model of Tuberous Sclerosis Complex and Focal Cortical Dysplasia. J. Neurosci. 2019, 39, 2762–2773. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Aigner, S.; Vukcevic, M.; Sauter, E.; Behr, K.; Ebeling, M.; Dunkley, T.; Friedlein, A.; Zoffmann, S.; Meyer, C.A.; et al. mTORC1 Inhibition Corrects Neurodevelopmental and Synaptic Alterations in a Human Stem Cell Model of Tuberous Sclerosis. Cell Rep. 2016, 15, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef] [PubMed]
- Iffland, P.H., 2nd; Barnes, A.E.; Baybis, M.; Crino, P.B. Dynamic analysis of 4E-BP1 phosphorylation in neurons with Tsc2 or Depdc5 knockout. Exp. Neurol. 2020, 334, 113432. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Madsen, R.R.; Vanhaesebroeck, B. Cracking the context-specific PI3K signaling code. Sci. Signal. 2020, 13, eaay2940. [Google Scholar] [CrossRef] [PubMed]
- Redenbaugh, V.; Coulter, T. Disorders Related to PI3Kdelta Hyperactivation: Characterizing the Clinical and Immunological Features of Activated PI3-Kinase Delta Syndromes. Front. Pediatr. 2021, 9, 702872. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Azam, Z.; Guo, J.; To, S.S.T. Oncogenic potential of PIK3CD in glioblastoma is exerted through cytoskeletal proteins PAK3 and PLEK2. Lab. Investig. 2022, 102, 1314–1322. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhou, X.; Guan, K.L.; Zhang, J. Rheb regulates nuclear mTORC1 activity independent of farnesylation. Cell Chem. Biol. 2022, 29, 1037–1045.e4. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Liu, D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023, 24, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5, 2078. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Francis, D.; Krycer, J.R.; Larance, M.; Zhang, Z.; Novotny, C.J.; Diaz-Vegas, A.; Shokat, K.M.; James, D.E. Dissecting the biology of mTORC1 beyond rapamycin. Sci. Signal 2021, 14, eabe0161. [Google Scholar] [CrossRef] [PubMed]
- Almacellas Barbanoj, A.; Graham, R.T.; Maffei, B.; Carpenter, J.C.; Leite, M.; Hoke, J.; Hardjo, F.; Scott-Solache, J.; Chimonides, C.; Schorge, S.; et al. Anti-seizure gene therapy for focal cortical dysplasia. Brain 2024, 147, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.S.; Lee, J.Y.; Choi, J.S.; Kim, H.J.; Kim, J.; Cha, S.; Lee, K.J.; Woo, H.N.; Park, K.; Lee, H. mTOR inhibition as a novel gene therapeutic strategy for diabetic retinopathy. PLoS ONE 2022, 17, e0269951. [Google Scholar] [CrossRef]
- Cha, S.; Seo, W.I.; Woo, H.N.; Kim, H.J.; Lee, S.H.S.; Kim, J.; Choi, J.S.; Park, K.; Lee, J.Y.; Lee, B.J.; et al. AAV expressing an mTOR-inhibiting siRNA exhibits therapeutic potential in retinal vascular disorders by preserving endothelial integrity. FEBS Open Bio 2022, 12, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A. Adeno-associated virus: From defective virus to effective vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Tanenhaus, A.; Stowe, T.; Young, A.; McLaughlin, J.; Aeran, R.; Lin, I.W.; Li, J.; Hosur, R.; Chen, M.; Leedy, J.; et al. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum. Gene Ther. 2022, 33, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, N.P.; Vink, C.A. The impact of lentiviral vector genome size and producer cell genomic to gag-pol mRNA ratios on packaging efficiency and titre. Mol. Ther. Methods Clin. Dev. 2021, 21, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Parr-Brownlie, L.C.; Bosch-Bouju, C.; Schoderboeck, L.; Sizemore, R.J.; Abraham, W.C.; Hughes, S.M. Lentiviral vectors as tools to understand central nervous system biology in mammalian model organisms. Front. Mol. Neurosci. 2015, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Kotin, R.M. Large-scale recombinant adeno-associated virus production. Hum. Mol. Genet. 2011, 20, R2–R6. [Google Scholar] [CrossRef]
- Melin, E.; Andersson, M.; Gotzsche, C.R.; Wickham, J.; Huang, Y.; Szczygiel, J.A.; Boender, A.; Christiansen, S.H.; Pinborg, L.; Woldbye, D.P.D.; et al. Combinatorial gene therapy for epilepsy: Gene sequence positioning and AAV serotype influence expression and inhibitory effect on seizures. Gene Ther. 2023, 30, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Di Berardino, C.; Massimino, L.; Ungaro, F.; Colasante, G. Gene therapy for Dravet syndrome: Promises and impact on disease trigger and secondary modifications. Rare Dis. Orphan Drugs J. 2024, 3, 21. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Tian, H.; Wang, L.; Guo, B.; Wang, Y.; Li, W.; Wang, F.; Sun, T. SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 2021, 12, 743726. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, M.; Luo, B.; Jing, H.; Yu, Y.; Wang, S.; Luo, S. Lrp4 in hippocampal astrocytes serves as a negative feedback factor in seizures. Cell Biosci. 2020, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Barral, S.; De La Fuente Barrigon, C.; Lignani, G.; Erdem, F.A.; Wallings, R.; Privolizzi, R.; Rossignoli, G.; Alrashidi, H.; Heasman, S.; et al. Gene therapy restores dopamine transporter expression and ameliorates pathology in iPSC and mouse models of infantile parkinsonism. Sci. Transl. Med. 2021, 13, eaaw1564. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. mTOR Signaling in Metabolism and Cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef]
- Pakola, S.A.; Peltola, K.J.; Clubb, J.H.A.; Jirovec, E.; Haybout, L.; Kudling, T.V.; Alanko, T.; Korpisaari, R.; Juteau, S.; Jaakkola, M.; et al. Safety, Efficacy, and Biological Data of T-Cell-Enabling Oncolytic Adenovirus TILT-123 in Advanced Solid Cancers from the TUNIMO Monotherapy Phase I Trial. Clin. Cancer Res. 2024, 30, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, X.; Li, X.; Liu, H.; Liu, M.; Tao, J.; Shan, Y.; Raza, H.K.; Liu, Y.; Zhong, W.; et al. Preliminary efficacy and safety of YSCH-01 in patients with advanced solid tumors: An investigator-initiated trial. J. Immunother. Cancer 2024, 12, e008999. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Janson, C.G.; Bilianuk, L.; Wang, Z.; Sorgi, F.; Huang, L.; Matalon, R.; Kaul, R.; Zeng, Z.; Freese, A.; et al. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann. Neurol. 2000, 48, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; Sondhi, D.; Hackett, N.R.; Kaminsky, S.M.; Worgall, S.; Stieg, P.; Souweidane, M.; Hosain, S.; Heier, L.; Ballon, D.; et al. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum. Gene Ther. 2004, 15, 1131–1154. [Google Scholar] [CrossRef]
- McPhee, S.W.; Janson, C.G.; Li, C.; Samulski, R.J.; Camp, A.S.; Francis, J.; Shera, D.; Lioutermann, L.; Feely, M.; Freese, A.; et al. Immune responses to AAV in a phase I study for Canavan disease. J. Gene Med. 2006, 8, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Sondhi, D.; Hackett, N.R.; Kosofsky, B.; Kekatpure, M.V.; Neyzi, N.; Dyke, J.P.; Ballon, D.; Heier, L.; Greenwald, B.M.; et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther. 2008, 19, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Buch, S.; Yao, H.; Wu, C.; Tong, H.I.; Wang, Y.; Lu, Y. Monocytes-derived macrophages mediated stable expression of human brain-derived neurotrophic factor, a novel therapeutic strategy for neuroAIDS. PLoS ONE 2014, 9, e82030. [Google Scholar] [CrossRef]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wan, J.; Wu, Y.; Tian, Y.; Yao, Y.; Yao, S.; Ji, X.; Wang, S.; Su, Z.; Xu, H. Challenges in adeno-associated virus-based treatment of central nervous system diseases through systemic injection. Life Sci. 2021, 270, 119142. [Google Scholar] [CrossRef] [PubMed]
- Puhl, D.L.; D’Amato, A.R.; Gilbert, R.J. Challenges of gene delivery to the central nervous system and the growing use of biomaterial vectors. Brain Res. Bull. 2019, 150, 216–230. [Google Scholar] [CrossRef]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Spunde, K.; Korotkaja, K.; Zajakina, A. Recombinant Viral Vectors for Therapeutic Programming of Tumour Microenvironment: Advantages and Limitations. Biomedicines 2022, 10, 2142. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Han, Y.; Liu, Y.; Zhou, Y.; Yu, J.; von Brunn, A.; Lei, J. Viral vector-based cancer treatment and current clinical applications. MedComm Oncol. 2023, 2, e55. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, M.; Li, G.; Zhang, S.; Zhang, J.; Fu, X.; Sun, F. Applications and Research Advances in the Delivery of CRISPR/Cas9 Systems for the Treatment of Inherited Diseases. Int. J. Mol. Sci. 2023, 24, 3202. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, C.A.; Wasko, K.M.; Hamilton, J.R.; Doudna, J.A. Targeted nonviral delivery of genome editors in vivo. Proc. Natl. Acad. Sci. USA 2024, 121, e2307796121. [Google Scholar] [CrossRef] [PubMed]
- Aljabali, A.A.A.; El-Tanani, M.; Tambuwala, M.M. Principles of CRISPR-Cas9 technology: Advancements in genome editing and emerging trends in drug delivery. J. Drug Deliv. Sci. Technol. 2024, 92, 105338. [Google Scholar] [CrossRef]
- Dubey, S.; Chen, Z.; Jiang, Y.J.; Talis, A.; Molotkov, A.; Ali, A.; Mintz, A.; Momen-Heravi, F. Small extracellular vesicles (sEVs)-based gene delivery platform for cell-specific CRISPR/Cas9 genome editing. Theranostics 2024, 14, 2777–2793. [Google Scholar] [CrossRef]
- Malik, S.; Muhammad, K.; Waheed, Y. Emerging Applications of Nanotechnology in Healthcare and Medicine. Molecules 2023, 28, 6624. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Muhammad, K.; Waheed, Y. Nanotechnology: A Revolution in Modern Industry. Molecules 2023, 28, 661. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Quarto, R.; Tasso, R. Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems. Pharmaceutics 2019, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, H.; Liu, J.; Chen, J.; Cui, Y.; Wang, S.; Zhang, X.; Yang, Z. Extracellular Vesicles: A New Star for Gene Drug Delivery. Int. J. Nanomed. 2024, 19, 2241–2264. [Google Scholar] [CrossRef]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target Ther. 2024, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Zanirati, G.; Dos Santos, P.G.; Alcara, A.M.; Bruzzo, F.; Ghilardi, I.M.; Wietholter, V.; Xavier, F.A.C.; Goncalves, J.I.B.; Marinowic, D.; Shetty, A.K.; et al. Extracellular Vesicles: The Next Generation of Biomarkers and Treatment for Central Nervous System Diseases. Int. J. Mol. Sci. 2024, 25, 7371. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.; Almotairy, A.R.Z.; Henidi, H.; Alshehri, O.Y.; Aldughaim, M.S. Nanoparticles as Drug Delivery Systems: A Review of the Implication of Nanoparticles’ Physicochemical Properties on Responses in Biological Systems. Polymers 2023, 15, 1596. [Google Scholar] [CrossRef]
- Wang, H.; Qin, L.; Zhang, X.; Guan, J.; Mao, S. Mechanisms and challenges of nanocarriers as non-viral vectors of therapeutic genes for enhanced pulmonary delivery. J. Control. Release 2022, 352, 970–993. [Google Scholar] [CrossRef] [PubMed]
- Witika, B.A.; Makoni, P.A.; Matafwali, S.K.; Chabalenge, B.; Mwila, C.; Kalungia, A.C.; Nkanga, C.I.; Bapolisi, A.M.; Walker, R.B. Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches. Nanomaterials 2020, 10, 1649. [Google Scholar] [CrossRef]
- Kunzmann, A.; Andersson, B.; Thurnherr, T.; Krug, H.; Scheynius, A.; Fadeel, B. Toxicology of engineered nanomaterials: Focus on biocompatibility, biodistribution and biodegradation. Biochim. Biophys. Acta 2011, 1810, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jiang, Y.; He, Y.; Zhang, W.; Zou, J.; Magar, K.T.; Boucetta, H.; Teng, C.; He, W. Approved Nanomedicine against Diseases. Pharmaceutics 2023, 15, 774. [Google Scholar] [CrossRef] [PubMed]
- Akombaetwa, N.; Ilangala, A.B.; Thom, L.; Memvanga, P.B.; Witika, B.A.; Buya, A.B. Current Advances in Lipid Nanosystems Intended for Topical and Transdermal Drug Delivery Applications. Pharmaceutics 2023, 15, 656. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Huang, X.; He, T.; Liang, X.; Xiang, Z.; Liu, C.; Zhou, S.; Luo, R.; Bai, L.; Kou, X.; Li, X.; et al. Advances and applications of nanoparticles in cancer therapy. MedComm Oncol. 2024, 3, e67. [Google Scholar] [CrossRef]
- Wang, B.; Hu, S.; Teng, Y.; Chen, J.; Wang, H.; Xu, Y.; Wang, K.; Xu, J.; Cheng, Y.; Gao, X. Current advance of nanotechnology in diagnosis and treatment for malignant tumors. Signal Transduct. Target Ther. 2024, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Gavas, S.; Quazi, S.; Karpinski, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Fathizadeh, H.; Afshar, S.; Masoudi, M.R.; Gholizadeh, P.; Asgharzadeh, M.; Ganbarov, K.; Kose, S.; Yousefi, M.; Kafil, H.S. SARS-CoV-2 (Covid-19) vaccines structure, mechanisms and effectiveness: A review. Int. J. Biol. Macromol. 2021, 188, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Rutkowski, P.L.; Thariat, J.; Carrere, S.; Ducassou, A.; Sunyach, M.P.; Agoston, P.; Hong, A.; Mervoyer, A.; Rastrelli, M.; et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): A multicentre, phase 2-3, randomised, controlled trial. Lancet Oncol 2019, 20, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Swanson, D.; Swett, M.C.; Patel, A.; Berardino, K.; Amgalan, A.; Berger, A.A.; Kassem, H.; Kaye, A.D.; Viswanath, O. Correction to: A Review of Patisiran (ONPATTRO(R)) for the Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2021, 10, 407. [Google Scholar] [CrossRef]
- Preda, A.; Shapiro, B.B. A safety evaluation of aripiprazole in the treatment of schizophrenia. Expert Opin. Drug Saf. 2020, 19, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef]
- Vergote, I.; Bergfeldt, K.; Franquet, A.; Lisyanskaya, A.S.; Bjermo, H.; Heldring, N.; Buyse, M.; Brize, A. A randomized phase III trial in patients with recurrent platinum sensitive ovarian cancer comparing efficacy and safety of paclitaxel micellar and Cremophor EL-paclitaxel. Gynecol. Oncol. 2020, 156, 293–300. [Google Scholar] [CrossRef]
- Zong, Y.; Wu, J.; Shen, K. Nanoparticle albumin-bound paclitaxel as neoadjuvant chemotherapy of breast cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 17360–17372. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.D.; Armstead-Williams, C.; Hyatali, F.; Cox, K.S.; Kaye, R.J.; Eng, L.K.; Farooq Anwar, M.A.; Patel, P.V.; Patil, S.; Cornett, E.M. Exparel for Postoperative Pain Management: A Comprehensive Review. Curr. Pain Headache Rep. 2020, 24, 73. [Google Scholar] [CrossRef]
- Du, Y.; Liu, Y.; Hu, J.; Peng, X.; Liu, Z. CRISPR/Cas9 systems: Delivery technologies and biomedical applications. Asian J. Pharm. Sci. 2023, 18, 100854. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target Ther. 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Razi Soofiyani, S.; Baradaran, B.; Lotfipour, F.; Kazemi, T.; Mohammadnejad, L. Gene therapy, early promises, subsequent problems, and recent breakthroughs. Adv. Pharm. Bull. 2013, 3, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, C.; Yong, H.; Wang, F.; Bo, T.; Zhao, Y.; Ma, B.; He, W.; Li, M. Emerging non-viral vectors for gene delivery. J. Nanobiotechnol. 2023, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Wallen, M.; Aqil, F.; Spencer, W.; Gupta, R.C. Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy. Pharmaceutics 2023, 15, 1832. [Google Scholar] [CrossRef] [PubMed]
- Butt, M.H.; Zaman, M.; Ahmad, A.; Khan, R.; Mallhi, T.H.; Hasan, M.M.; Khan, Y.H.; Hafeez, S.; Massoud, E.E.S.; Rahman, M.H.; et al. Appraisal for the Potential of Viral and Nonviral Vectors in Gene Therapy: A Review. Genes 2022, 13, 1370. [Google Scholar] [CrossRef] [PubMed]
- Shintaro, F.; Shigeru, K.; Mitsuru, H.; Koyo, N. Chapter 1: Targeted Gene Delivery: Importance of Administration Routes. In Novel Gene Therapy Approaches; Ming, W., David, G., Eds.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Gessler, D.J.; Tai, P.W.L.; Li, J.; Gao, G. Intravenous Infusion of AAV for Widespread Gene Delivery to the Nervous System. Methods Mol. Biol. 2019, 1950, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Rheima, A.M.; Kadhim, M.M.; Ahmed, N.N.; Mohammed, S.H.; Abbas, F.H.; Abed, Z.T.; Mahdi, Z.M.; Abbas, Z.S.; Hachim, S.K.; et al. An overview of nanoparticles in drug delivery: Properties and applications. S. Afr. J. Chem. Eng. 2023, 46, 233–270. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.S.A.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N.; et al. Nanoparticles in Drug Delivery: From History to Therapeutic Applications. Nanomaterials 2022, 12, 4494. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Q.; Xia, G.; Adilijiang, N.; Li, Y.; Hou, Z.; Fan, Z.; Li, J. Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy. Pharmaceutics 2023, 15, 2233. [Google Scholar] [CrossRef] [PubMed]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Moloney, P.B.; Cavalleri, G.L.; Delanty, N. Epilepsy in the mTORopathies: Opportunities for precision medicine. Brain Commun. 2021, 3, fcab222. [Google Scholar] [CrossRef] [PubMed]
- Dentel, B.; Escamilla, C.O.; Tsai, P.T. Therapeutic Targeting of mTORC2 in mTORopathies. Neuron 2019, 104, 1032–1033. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Orozco, J.M.; Adelmann, C.H.; Spinelli, J.B.; van der Helm, P.W.; Roberts, J.M.; Kunchok, T.; Sabatini, D.M. Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2021, 118, e2022120118. [Google Scholar] [CrossRef]
- Imanaga, H.; Semba, Y.; Sasaki, K.; Setoguchi, K.; Maniriho, H.; Yamauchi, T.; Terasaki, T.; Hirabayashi, S.; Nakao, F.; Nogami, J.; et al. Central role of the mTORC1 pathway in glucocorticoid activity against B-ALL cells. Blood Neoplasia 2024, 1, 100015. [Google Scholar] [CrossRef]
- Coleman, M.; Pinares-Garcia, P.; Stephenson, S.E.; Lee, W.S.; Kooshavar, D.; McLean, C.A.; Howell, K.B.; Leventer, R.J.; Reid, C.A.; Lockhart, P.J. Ectopic HCN4 Provides a Target Biomarker for the Genetic Spectrum of mTORopathies. Neurol. Genet. 2024, 10, e200135. [Google Scholar] [CrossRef]
- Drago, D.; Foss-Campbell, B.; Wonnacott, K.; Barrett, D.; Ndu, A. Global regulatory progress in delivering on the promise of gene therapies for unmet medical needs. Mol. Ther. Methods Clin. Dev. 2021, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Munawar, N.; Khan, Z.; Qusmani, A.T.; Khan, S.H.; Jamil, A.; Ashraf, S.; Ghouri, M.Z.; Aslam, S.; Mubarik, M.S.; et al. An Outlook on Global Regulatory Landscape for Genome-Edited Crops. Int. J. Mol. Sci. 2021, 22, 1753. [Google Scholar] [CrossRef] [PubMed]
| Approval Year | Virus/Serotype | Clinical Application | Drug/Cargo Type | Reference |
|---|---|---|---|---|
| 2000 | pAAV | Canavan disease | Human aspartoacylase cDNA | [135] |
| 2004 | AAV2 | Late infantile neuronal ceroid lipofuscinosis | human CLN2 cDNA | [136] |
| 2006 | AAV2 | Canavan disease | Human aspartoacylase cDNA | [137] |
| 2008 | AAV2 | Late infantile neuronal ceroid lipofuscinosis | Human aspartoacylase cDNA | [138] |
| 2014 | Lentivirus | neuroAIDS | human (h)BDNF | [139] |
| 2016 | Lentivirus | Metachromatic leukodystrophy | Human ARSA cDNA | [140] |
| Approval Year | Nanoparticle Type | Clinical Application | Drug/Cargo Type | Reference |
|---|---|---|---|---|
| 2021 | Liposome | COVID-19 | mRNA-1273 Nucleoside-modified RNA (modRNA) | [168,169] |
| 2019 | Metal nanoparticle | Advanced soft-tissue sarcoma | Hafnium oxide | [170] |
| 2018 | Liposome | Polyneuropathy caused by hereditary ATTR amyloidosis | siRNA | [171] |
| 2018 | Nanocrystals | Schizophrenia | Aripiprazole lauroxil | [172] |
| 2017 | Liposome | Acute myeloid leukemia | Daunorubicin and cytarabine | [173] |
| 2015 | Liposome | Metastatic pancreatic cancer | Irinotecan chemotherapy | [174] |
| 2015 | Polymeric nanoparticle | Ovarian cancer | Paclitaxel chemotherapy | [175] |
| 2014 | Polymeric nanoparticle | Multiple sclerosis | Polymer–protein conjugate (PEGylated IFN beta-1a) | [163] |
| 2013 | Polymeric nanoparticle | Crohn’s disease; Rheumatoid arthritis; Psoriatic arthritis; Ankylosing spondylitis | PEGylated antibody fragment (Certolizumab) | [163] |
| 2013 | Protein nanoparticles | Breast cancer; Non-small-cell lung cancer; Pancreatic cancer | Albumin-bound paclitaxel chemotherapy | [176] |
| 2011 | Liposome | Bupivacaine | Pain treatment | [177] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boff, M.O.; Xavier, F.A.C.; Diz, F.M.; Gonçalves, J.B.; Ferreira, L.M.; Zambeli, J.; Pazzin, D.B.; Previato, T.T.R.; Erwig, H.S.; Gonçalves, J.I.B.; et al. mTORopathies in Epilepsy and Neurodevelopmental Disorders: The Future of Therapeutics and the Role of Gene Editing. Cells 2025, 14, 662. https://doi.org/10.3390/cells14090662
Boff MO, Xavier FAC, Diz FM, Gonçalves JB, Ferreira LM, Zambeli J, Pazzin DB, Previato TTR, Erwig HS, Gonçalves JIB, et al. mTORopathies in Epilepsy and Neurodevelopmental Disorders: The Future of Therapeutics and the Role of Gene Editing. Cells. 2025; 14(9):662. https://doi.org/10.3390/cells14090662
Chicago/Turabian StyleBoff, Marina Ottmann, Fernando Antônio Costa Xavier, Fernando Mendonça Diz, Júlia Budelon Gonçalves, Laura Meireles Ferreira, Jean Zambeli, Douglas Bottega Pazzin, Thales Thor Ramos Previato, Helena Scartassini Erwig, João Ismael Budelon Gonçalves, and et al. 2025. "mTORopathies in Epilepsy and Neurodevelopmental Disorders: The Future of Therapeutics and the Role of Gene Editing" Cells 14, no. 9: 662. https://doi.org/10.3390/cells14090662
APA StyleBoff, M. O., Xavier, F. A. C., Diz, F. M., Gonçalves, J. B., Ferreira, L. M., Zambeli, J., Pazzin, D. B., Previato, T. T. R., Erwig, H. S., Gonçalves, J. I. B., Bruzzo, F. T. K., Marinowic, D., da Costa, J. C., & Zanirati, G. (2025). mTORopathies in Epilepsy and Neurodevelopmental Disorders: The Future of Therapeutics and the Role of Gene Editing. Cells, 14(9), 662. https://doi.org/10.3390/cells14090662