
Bilateral Germ Cell Tumor of the Testis: Biological and Clinical Implications for a Stem Versus Genetic Origin of Cancers

 , , , ,
, , , , 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor DNA Isolation
2.2. Whole-Exome Sequencing (WES)
2.3. DNA Methylation Analysis
3. Results
3.1. Clinical and Pathological Features of Total Cohort
3.2. Clinical and Pathological Features of Nine Patients
3.3. Genomic Profiles
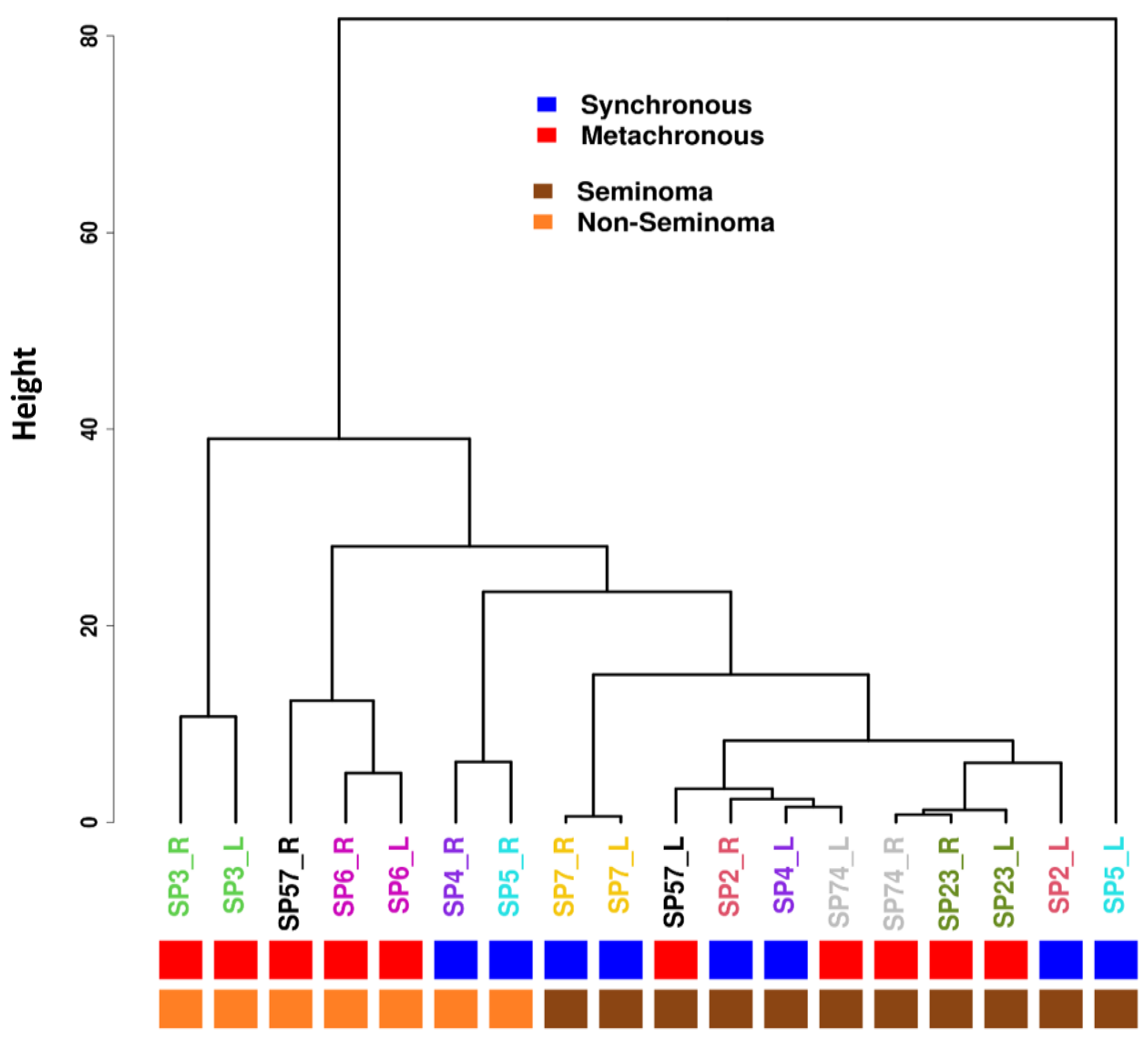
3.4. Methylation Profiles
3.5. Differential Methylation Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GCT | Germ cell tumor of the testis |
| WES | Whole-exome sequencing |
| RRBS | Reduced representation bisulfite sequencing |
| PGC | Primordial germ cells |
| FFPE | Formalin-fixed paraffin-embedded |
References
- Castrillon, D.H.; Quade, B.J.; Wang, T.Y.; Quigley, C.; Crum, C.P. The human VASA gene is specifically expressed in the germ cell lineage. Proc. Natl. Acad. Sci. USA 2000, 97, 9585–9590. [Google Scholar] [CrossRef]
- Ohinata, Y.; Payer, B.; O’Carroll, D.; Ancelin, K.; Ono, Y.; Sano, M.; Barton, S.C.; Obukhanych, T.; Nussenzweig, M.; Tarakhovsky, A.; et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 2005, 436, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Barton, S.C.; Surani, M.A. A molecular programme for the specification of germ cell fate in mice. Nature 2002, 418, 293–300. [Google Scholar] [CrossRef]
- Tilgner, K.; Atkinson, S.P.; Golebiewska, A.; Stojkovic, M.; Lako, M.; Armstrong, L. Isolation of primordial germ cells from differentiating human embryonic stem cells. Stem Cells 2008, 26, 3075–3085. [Google Scholar] [CrossRef]
- Theodore, C.; Terrier-Lacombe, M.J.; Laplanche, A.; Benoit, G.; Fizazi, K.; Stamerra, O.; Wibault, P. Bilateral germ-cell tumours: 22-year experience at the Institut Gustave Roussy. Br. J. Cancer 2004, 90, 55–59. [Google Scholar] [CrossRef]
- Zequi Sde, C.; da Costa, W.H.; Santana, T.B.; Favaretto, R.L.; Sacomani, C.A.; Guimaraes, G.C. Bilateral testicular germ cell tumours: A systematic review. BJU Int. 2012, 110, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; Labreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef]
- Cutcutache, I.; Suzuki, Y.; Tan, I.B.; Ramgopal, S.; Zhang, S.; Ramnarayanan, K.; Gan, A.; Lee, H.H.; Tay, S.T.; Ooi, A.; et al. Exome-wide Sequencing Shows Low Mutation Rates and Identifies Novel Mutated Genes in Seminomas. Eur. Urol. 2015, 68, 77–83. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Summersgill, B.; Grygalewicz, B.; Gillis, A.J.; Stoop, J.; van Gurp, R.J.; Dennis, N.; Fisher, C.; Huddart, R.; Cooper, C.; et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 2005, 65, 8085–8089. [Google Scholar] [CrossRef]
- Kemmer, K.; Corless, C.L.; Fletcher, J.A.; McGreevey, L.; Haley, A.; Griffith, D.; Cummings, O.W.; Wait, C.; Town, A.; Heinrich, M.C. KIT mutations are common in testicular seminomas. Am. J. Pathol. 2004, 164, 305–313. [Google Scholar] [CrossRef]
- Coffey, J.; Linger, R.; Pugh, J.; Dudakia, D.; Sokal, M.; Easton, D.F.; Timothy Bishop, D.; Stratton, M.; Huddart, R.; Rapley, E.A. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: Report of 220 tumors and review of literature. Genes Chromosomes Cancer 2008, 47, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Meric-Bernstam, F.; Zhao, H.; Zhang, Q.; Ezzeddine, N.; Tang, L.Y.; Qi, Y.; Mao, Y.; Chen, T.; Chong, Z.; et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin. Chem. 2015, 61, 544–553. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Consortium, G.R. Genome Assembly GRCh37. 27 February 2009. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/#:~:text=GRCh37%20Genome%20Reference%20Consortium%20Human,GCA_000001405.1%20(replaced)%20RefSeq (accessed on 4 February 2016).
- Pozzi, E.; Ventimiglia, E.; Corsini, C.; Negri, F.; Rusconi, F.; Hernandez, G.; Ramasamy, R.; Montorsi, F.; Salonia, A. EPIDEMIOLOGICAL PATTERNS, OUTCOMES, AND VARIATIONS IN SYNCHRONOUS AND METACHRONOUS SEMINOMATOUS TESTICULAR GERM CELL CANCER—FINDINGS FROM A LARGE NETWORK DATABASE. J. Urol. 2024, 211, e3. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef]
- Jaszczak, R.G.; Zussman, J.W.; Wagner, D.E.; Laird, D.J. Comprehensive profiling of migratory primordial germ cells reveals niche-specific differences in non-canonical Wnt and Nodal-Lefty signaling in anterior vs posterior migrants. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Skotheim, R.I.; Abeler, V.M.; Nesland, J.M.; Fossa, S.D.; Holm, R.; Wagner, U.; Florenes, V.A.; Aass, N.; Kallioniemi, O.P.; Lothe, R.A. Candidate genes for testicular cancer evaluated by in situ protein expression analyses on tissue microarrays. Neoplasia 2003, 5, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Aubry, F.; Satie, A.P.; Rioux-Leclercq, N.; Rajpert-De Meyts, E.; Spagnoli, G.C.; Chomez, P.; De Backer, O.; Jegou, B.; Samson, M. MAGE-A4, a germ cell specific marker, is expressed differentially in testicular tumors. Cancer 2001, 92, 2778–2785. [Google Scholar] [CrossRef]
- Grassetti, D.; Giannandrea, F.; Paoli, D.; Masciandaro, P.; Figura, V.; Carlini, T.; Rizzo, F.; Lombardo, F.; Lenzi, A.; Gandini, L. Androgen receptor polymorphisms and testicular cancer risk. Andrology 2015, 3, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, E.C.; Siddiqui, B.A.; Hwang, M.J.; Joon, A.Y.; Maity, T.; Westerman, M.E.; Merriman, K.W.; Alhasson, H.; Uthup, J.; Guo, T.; et al. Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor. Cancers 2020, 12, 3755. [Google Scholar] [CrossRef]
- Oliver, R.T. HLA phenotype and clinicopathological behaviour of germ cell tumours: Possible evidence for clonal evolution from seminomas to nonseminomas. Int. J. Androl. 1987, 10, 85–93. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Berthelsen, J.G.; Giwercman, A.; Muller, J. Carcinoma-in-situ of the testis: Possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int. J. Androl. 1987, 10, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Lin, S.H.; Logothetis, C.J. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002, 3, 508–513. [Google Scholar] [CrossRef]
- Tu, S.M.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.F.; Wood, C.G.; et al. Intratumoral heterogeneity: Role of differentiation in a potentially lethal phenotype of testicular cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef]
- Looijenga, L.H. Testicular germ cell tumors. Pediatr. Endocrinol. Rev. 2014, 11 (Suppl. S2), 251–262. [Google Scholar]
- Kernek, K.M.; Ulbright, T.M.; Zhang, S.; Billings, S.D.; Cummings, O.W.; Henley, J.D.; Michael, H.; Brunelli, M.; Martignoni, G.; Foster, R.S.; et al. Identical allelic losses in mature teratoma and other histologic components of malignant mixed germ cell tumors of the testis. Am. J. Pathol. 2003, 163, 2477–2484. [Google Scholar] [CrossRef]
- Jones, T.D.; Wang, M.; Sung, M.T.; Zhang, S.; Ulbright, T.M.; Eble, J.N.; Beck, S.D.; Foster, R.S.; Anagnostou, J.J., Jr.; Conner, C.; et al. Clonal origin of metastatic testicular teratomas. Clin. Cancer Res. 2006, 12, 5377–5383. [Google Scholar] [CrossRef] [PubMed]
- Kum, J.B.; Ulbright, T.M.; Williamson, S.R.; Wang, M.; Zhang, S.; Foster, R.S.; Grignon, D.J.; Eble, J.N.; Beck, S.D.; Cheng, L. Molecular genetic evidence supporting the origin of somatic-type malignancy and teratoma from the same progenitor cell. Am. J. Surg. Pathol. 2012, 36, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.C. Origin of testicular teratomas from primordial germ cells in mice. J. Natl. Cancer Inst. 1967, 38, 549–552. [Google Scholar]
- Stevens, L.C. The biology of teratomas including evidence indicating their origin form primordial germ cells. Annee Biol. 1962, 1, 585–610. [Google Scholar]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sorlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Borresen-Dale, A.L.; Pollack, J.R. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef]
- De Felici, M.; Klinger, F.G.; Campolo, F.; Balistreri, C.R.; Barchi, M.; Dolci, S. To Be or Not to Be a Germ Cell: The Extragonadal Germ Cell Tumor Paradigm. Int. J. Mol. Sci. 2021, 22, 5982. [Google Scholar] [CrossRef]
- Park, T.W.; Felix, J.C.; Wright, T.C., Jr. X chromosome inactivation and microsatellite instability in early and advanced bilateral ovarian carcinomas. Cancer Res. 1995, 55, 4793–4796. [Google Scholar]
- Yin, X.; Jing, Y.; Cai, M.C.; Ma, P.; Zhang, Y.; Xu, C.; Zhang, M.; Di, W.; Zhuang, G. Clonality, Heterogeneity, and Evolution of Synchronous Bilateral Ovarian Cancer. Cancer Res. 2017, 77, 6551–6561. [Google Scholar] [CrossRef]
- Li, C.; Bonazzoli, E.; Bellone, S.; Choi, J.; Dong, W.; Menderes, G.; Altwerger, G.; Han, C.; Manzano, A.; Bianchi, A.; et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc. Natl. Acad. Sci. USA 2019, 116, 619–624. [Google Scholar] [CrossRef]
- Abeln, E.C.; Kuipers-Dijkshoorn, N.J.; Berns, E.M.; Henzen-Logmans, S.C.; Fleuren, G.J.; Cornelisse, C.J. Molecular genetic evidence for unifocal origin of advanced epithelial ovarian cancer and for minor clonal divergence. Br. J. Cancer 1995, 72, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, D.; Ryan, A.; Ayhan, A.; McCluggage, W.G.; Feakins, R.; Santibanez-Koref, M.F.; Mein, C.A.; Gayther, S.A.; Jacobs, I.J. A molecular genetic and statistical approach for the diagnosis of dual-site cancers. J. Natl. Cancer Inst. 2004, 96, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.; Czene, K.; Reilly, M.; Adolfsson, J.; Bergh, J.; Adami, H.O.; Dickman, P.W.; Hall, P. Incidence and prognosis of synchronous and metachronous bilateral breast cancer. J. Clin. Oncol. 2007, 25, 4210–4216. [Google Scholar] [CrossRef] [PubMed]
- Giannakeas, V.; Lim, D.W.; Narod, S.A. Bilateral Mastectomy and Breast Cancer Mortality. JAMA Oncol. 2024, 10, 1228–1236. [Google Scholar] [CrossRef]
- Noor, D.A.M.; Jeyapalan, J.N.; Alhazmi, S.; Carr, M.; Squibb, B.; Wallace, C.; Tan, C.; Cusack, M.; Hughes, J.; Reader, T.; et al. Genome-wide methylation analysis identifies genes silenced in non-seminoma cell lines. NPJ Genom. Med. 2016, 1, 15009. [Google Scholar] [CrossRef]
- Amatruda, J.F.; Ross, J.A.; Christensen, B.; Fustino, N.J.; Chen, K.S.; Hooten, A.J.; Nelson, H.; Kuriger, J.K.; Rakheja, D.; Frazier, A.L.; et al. DNA methylation analysis reveals distinct methylation signatures in pediatric germ cell tumors. BMC Cancer 2013, 13, 313. [Google Scholar] [CrossRef]
- Orosz, F. The Role of Tubulin Polymerization-Promoting Protein2 (TPPP2) in Spermatogenesis: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 7017. [Google Scholar] [CrossRef]
- Milardi, D.; Grande, G.; Vincenzoni, F.; Pierconti, F.; Pontecorvi, A. Proteomics for the Identification of Biomarkers in Testicular Cancer-Review. Front. Endocrinol. 2019, 10, 462. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Q.; An, Y.; Chen, X.; Liu, Z.; Li, Z.; Gao, J.; Aung, L.H.H.; Li, P. Role of apoptosis repressor with caspase recruitment domain (ARC) in cancer. Oncol. Lett. 2019, 18, 5691–5698. [Google Scholar] [CrossRef]
- Pangon, L.; Ng, I.; Giry-Laterriere, M.; Currey, N.; Morgan, A.; Benthani, F.; Tran, P.N.; Al-Sohaily, S.; Segelov, E.; Parker, B.L.; et al. JRK is a positive regulator of beta-catenin transcriptional activity commonly overexpressed in colon, breast and ovarian cancer. Oncogene 2016, 35, 2834–2841. [Google Scholar] [CrossRef]
- Chueh, A.C.; Advani, G.; Foroutan, M.; Smith, J.; Ng, N.; Nandurkar, H.; Lio, D.S.; Zhu, H.J.; Chong, Y.P.; Verkade, H.; et al. CSK-homologous kinase (CHK/MATK) is a potential colorectal cancer tumour suppressor gene epigenetically silenced by promoter methylation. Oncogene 2021, 40, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, T.; Fu, G.; Xu, Y.; Zhang, N.; Han, L.; Yang, M. The allelic regulation of tumor suppressor ADARB2 in papillary thyroid carcinoma. Endocr. Relat. Cancer 2023, 30, e220189. [Google Scholar] [CrossRef] [PubMed]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Kaprara, A.; Pazaitou-Panayiotou, K.; Kortsaris, A.; Chatzaki, E. The corticotropin releasing factor system in cancer: Expression and pathophysiological implications. Cell Mol. Life Sci. 2010, 67, 1293–1306. [Google Scholar] [CrossRef]
- Zhong, S.; Jeong, J.H.; Huang, C.; Chen, X.; Dickinson, S.I.; Dhillon, J.; Yang, L.; Luo, J.L. Targeting INMT and interrupting its methylation pathway for the treatment of castration resistant prostate cancer. J. Exp. Clin. Cancer Res. 2021, 40, 307. [Google Scholar] [CrossRef]
- Jianfeng, W.; Yutao, W.; Jianbin, B. Indolethylamine-N-Methyltransferase Inhibits Proliferation and Promotes Apoptosis of Human Prostate Cancer Cells: A Mechanistic Exploration. Front. Cell Dev. Biol. 2022, 10, 805402. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, L.; Jing, D.; Xu, G.; Zhang, J.; Lin, L.; Zhao, J.; Yao, Z.; Lin, H. Galectin-9 Expression Predicts Favorable Clinical Outcome in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Physiol. 2018, 9, 452. [Google Scholar] [CrossRef]
- Urinary and Male Genital Tumors; World Health Organization: Geneva, Switzerland, 2022; Volume 5.
- Garolla, A.; Ferlin, A.; Vinanzi, C.; Roverato, A.; Sotti, G.; Artibani, W.; Foresta, C. Molecular analysis of the androgen receptor gene in testicular cancer. Endocr. Relat. Cancer 2005, 12, 645–655. [Google Scholar] [CrossRef]
- Pluta, J.; Pyle, L.C.; Nead, K.T.; Wilf, R.; Li, M.; Mitra, N.; Weathers, B.; D’Andrea, K.; Almstrup, K.; Anson-Cartwright, L.; et al. Identification of 22 susceptibility loci associated with testicular germ cell tumors. Nat. Commun. 2021, 12, 4487. [Google Scholar] [CrossRef]
- Hofer, M.D.; Browne, T.J.; He, L.; Skotheim, R.I.; Lothe, R.A.; Rubin, M.A. Identification of two molecular groups of seminomas by using expression and tissue microarrays. Clin. Cancer Res. 2005, 11, 5722–5729. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Total (n = 38) | Synchronous (n = 7) | Metachronous (n = 31) | p-Value | |
|---|---|---|---|---|
| Race/Ethnicity (%) | ||||
| Caucasian/White | 27 (71.1) | 5 (71.4) | 22 (71.0) | >0.99 |
| African American/Black | 10 (26.3) | 2 (28.6) | 8 (25.8) | |
| Hispanic | 1 (2.6) | 0 (0) | 1 (3.2) | |
| Median Follow-Up (Months; IQR) | 134.2 (69.5–222.1) | 50.3 (22.8–99.7) | 157.3 (91.8–252.0) | 0.004 |
| Median Age at 1st Orchiectomy (IQR) | 27. 5 (20–32) | 26 (20–34) | 27.5 (20–31) | 0.77 |
| Median Age at 2nd Orchiectomy (IQR) | 33 (26–37) | 26 (20–34) | 33 (29–37) | 0.11 |
| Stage | I—28 (73.7) | I—6 (85.7) | I—22 (71.0) | 0.65 |
| II—9 (23.7) | II—1 (14.3) | II—8 (25.8) | ||
| III—1 (2.6) | III—1 (3.2) | |||
| Pathologic Subtype (%) | ||||
| Bilateral Seminoma | 13 (34.2) | 3 (42.9) | 10 (32.3) | 0.66 |
| Bilateral Nonseminoma | 12 (31.6) | 1 (14.3) | 11 (35.5) | |
| Seminoma/Nonseminoma | 13 (34.2) | 3 (42.9) | 10 (32.3) | |
| Disease Progression (%) | ||||
| Yes | 7 (18.4) | 0 (0) | 7 (22.6) | 0.31 |
| No | 31 (81.6) | 7 (100) | 24 (77.4) | |
| Survival (%) | ||||
| Alive | 36 (94.7) | 7 (100) | 29 (93.5) | >0.99 |
| Dead | 2 (5.3) | 0 (0) | 2 (6.5) |
| Sample ID | Laterality | Size (cm) | Interval (Months) | Stage | Treatment | Pathology |
|---|---|---|---|---|---|---|
| SP2_L_S2 | Left | 7.8 | S—0 | I | XRT * | Seminoma |
| SP2_R_S1 | Right | 5.6 | S—0 | I | Seminoma | |
| SP3_L_S4 | Left | 1.2 | M—21.2 | I | BEP (X2) | Nonseminoma (20% embryonal carcinoma, 20% yolk sac tumor, 60% teratoma) |
| SP3_R_S3 | Right | N/a | M—0 | I | Nonseminoma (20% yolk sac tumor, 80% teratoma) | |
| SP4_L_S6 | Left | 2.6 | S—0 | I | BEP (x3) | Seminoma |
| SP4_R_S5 | Right | 5 | S—0 | I | Nonseminoma (10% embryonal carcinoma, 90% seminoma) | |
| SP5_L_S8 | Left | 3.5 | S—0 | I | BEP (x3) | Seminoma |
| SP5_R_S7 | Right | 9.2 | S—0 | I | Nonseminoma (85% yolk sac tumor, 10% immature teratoma, 5% embryonal carcinoma) | |
| SP6_L_S10 | Left | 2.7 | M—66.2 | I | VIP (x3) | Nonseminoma (90% embryonal carcinoma, 5% seminoma, 4% teratoma, 1% yolk sac tumor) |
| SP6_R_S9 | Right | 2.8 | M—0 | II | BEP (x3) | Nonseminoma (90% embryonal carcinoma, 10% yolk sac tumor) |
| SP7_L_S12 | Left | 2.9 | S—0 | I | None | Seminoma |
| SP7_R_S11 | Right | 2.2 | S—0 | I | Seminoma | |
| SP23_L_S14 | Left | 2.7 | M—0 | I | XRT | Seminoma |
| SP23_R_S13 | Right | 3.0 | M—10 | I | Seminoma | |
| SP57_L_S16 | Left | 2.9 | M—158.3 | I | None | Seminoma |
| SP57_R_S15 | Right | 3.1 | M—0 | I | Nonseminoma (80% embryonal carcinoma, 10% teratoma, 5% choriocarcinoma, 4% seminoma, 1% yolk sac tumor) | |
| SP74_L_S18 | Left | 1.5 | M—18.8 | I | XRT | Seminoma |
| SP74_R_S17 | Right | 0.9 | M—0 | I | Seminoma |
| Chromosome | Position | Mean Nonseminoms | Mean Seminomas | Mean Difference | p-Value | Gene |
|---|---|---|---|---|---|---|
| 8 | 144000000 | 79.46 | 34.56 | 44.90 | <0.0001 | ARC, JRK |
| 19 | 3778280 | 67.81 | 20.41 | 47.40 | <0.0001 | MATK |
| 10 | 1701540 | 73.76 | 28.79 | 44.97 | <0.0001 | ADARB2 |
| 5 | 677993 | 80.97 | 35.25 | 45.72 | 0.0001 | TPPP1 |
| 5 | 678007 | 87.82 | 42.96 | 44.87 | 0.0003 | TPPP1 |
| 7 | 56620283 | 74.92 | 28.81 | 46.12 | 0.0003 | LOC101928401, LOC401357 |
| 7 | 30765560 | 76.14 | 28.98 | 47.16 | 0.0003 | CRHR2, INMT |
| 17 | 2699778 | 52.22 | 6.27 | 45.95 | 0.0004 | RAP1GAP2 |
| 12 | 133000000 | 83.65 | 37.61 | 46.04 | 0.0004 | GALNT9 |
| 17 | 7569655 | 76.20 | 29.12 | 47.08 | 0.0005 | LOC100507351, LOC100132174 |
| Chromosome | Position | Mean Nonseminomas | Mean Seminomas | Mean Difference | p-Value | Genes |
|---|---|---|---|---|---|---|
| X | 151081290 | 82.31 | 35.96 | 46.34 | 0.001 | MAGE-A4 |
| X | 151081284 | 81.97 | 34.80 | 47.17 | 0.003 | MAGE-A4 |
| X | 151081291 | 85.49 | 35.97 | 49.52 | 0.008 | MAGE-A4 |
| X | 151081297 | 1.74 | 0 | 1.74 | 0.018 | MAGE-A4 |
| X | 151081296 | 82.31 | 35.96 | 47.95 | 0.031 | MAGE-A4 |
| 17 | 39942776 | 81.97 | 34.80 | 35.47 | 0.013 | JUP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, J.C.; Sanchez, D.; Joon, A.Y.; Estecio, M.R.; Johns, A.C.; Shah, A.Y.; Campbell, M.; Ward, J.F.; Pisters, L.L.; Guo, C.C.; et al. Bilateral Germ Cell Tumor of the Testis: Biological and Clinical Implications for a Stem Versus Genetic Origin of Cancers. Cells 2025, 14, 658. https://doi.org/10.3390/cells14090658
Jackson JC, Sanchez D, Joon AY, Estecio MR, Johns AC, Shah AY, Campbell M, Ward JF, Pisters LL, Guo CC, et al. Bilateral Germ Cell Tumor of the Testis: Biological and Clinical Implications for a Stem Versus Genetic Origin of Cancers. Cells. 2025; 14(9):658. https://doi.org/10.3390/cells14090658
Chicago/Turabian StyleJackson, Jamaal C., Darren Sanchez, Aron Y. Joon, Marcos R. Estecio, Andrew C. Johns, Amishi Y. Shah, Matthew Campbell, John F. Ward, Louis L. Pisters, Charles C. Guo, and et al. 2025. "Bilateral Germ Cell Tumor of the Testis: Biological and Clinical Implications for a Stem Versus Genetic Origin of Cancers" Cells 14, no. 9: 658. https://doi.org/10.3390/cells14090658
APA StyleJackson, J. C., Sanchez, D., Joon, A. Y., Estecio, M. R., Johns, A. C., Shah, A. Y., Campbell, M., Ward, J. F., Pisters, L. L., Guo, C. C., Zhang, M., Zacharias, N. M., & Tu, S.-M. (2025). Bilateral Germ Cell Tumor of the Testis: Biological and Clinical Implications for a Stem Versus Genetic Origin of Cancers. Cells, 14(9), 658. https://doi.org/10.3390/cells14090658