
The Efficacy of Targeted Monoclonal IgA Antibodies Against Pancreatic Ductal Adenocarcinoma

 , , , , ,
, , , , , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Lines
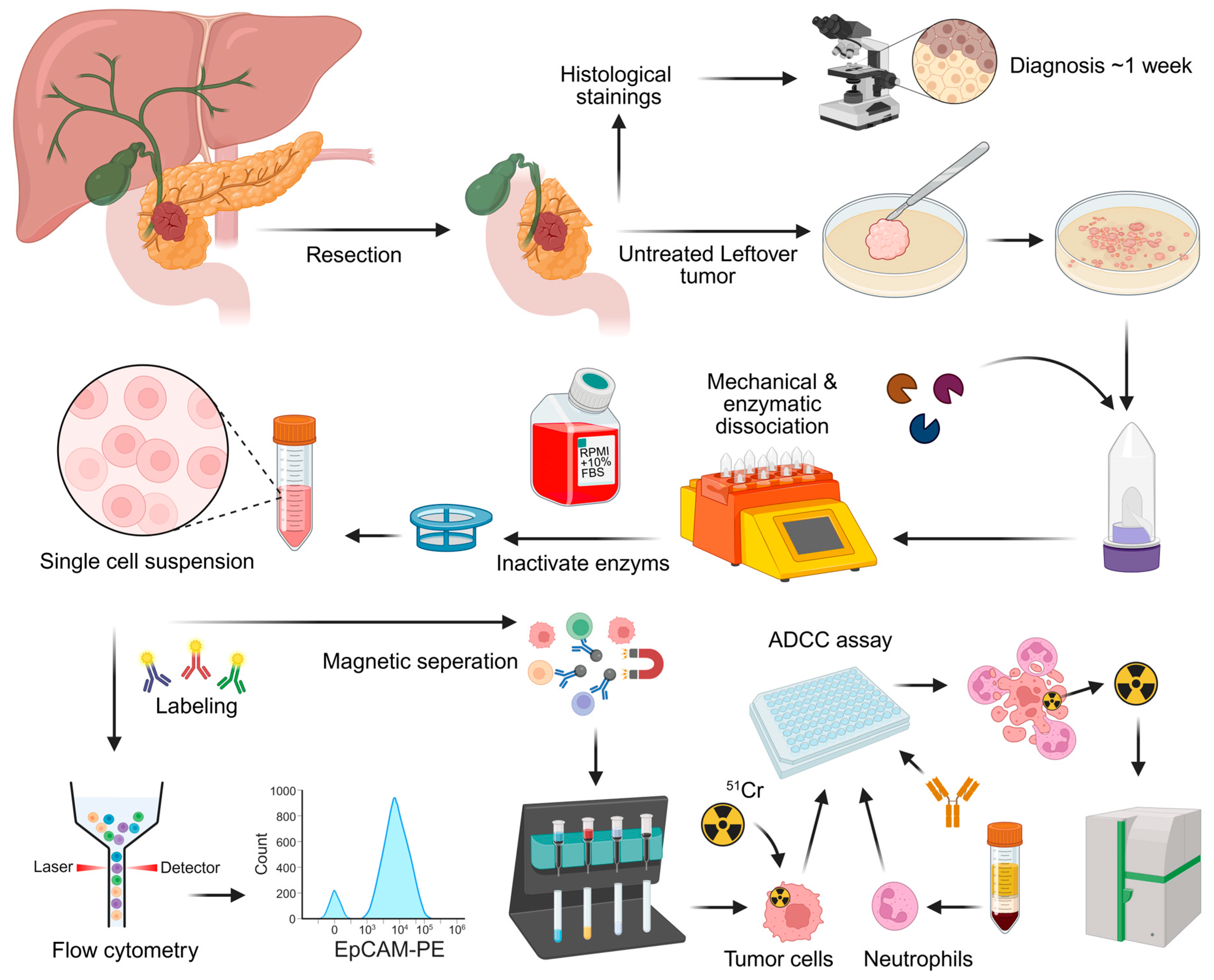
2.2. Patient Samples and Processing
2.3. Antibody Production
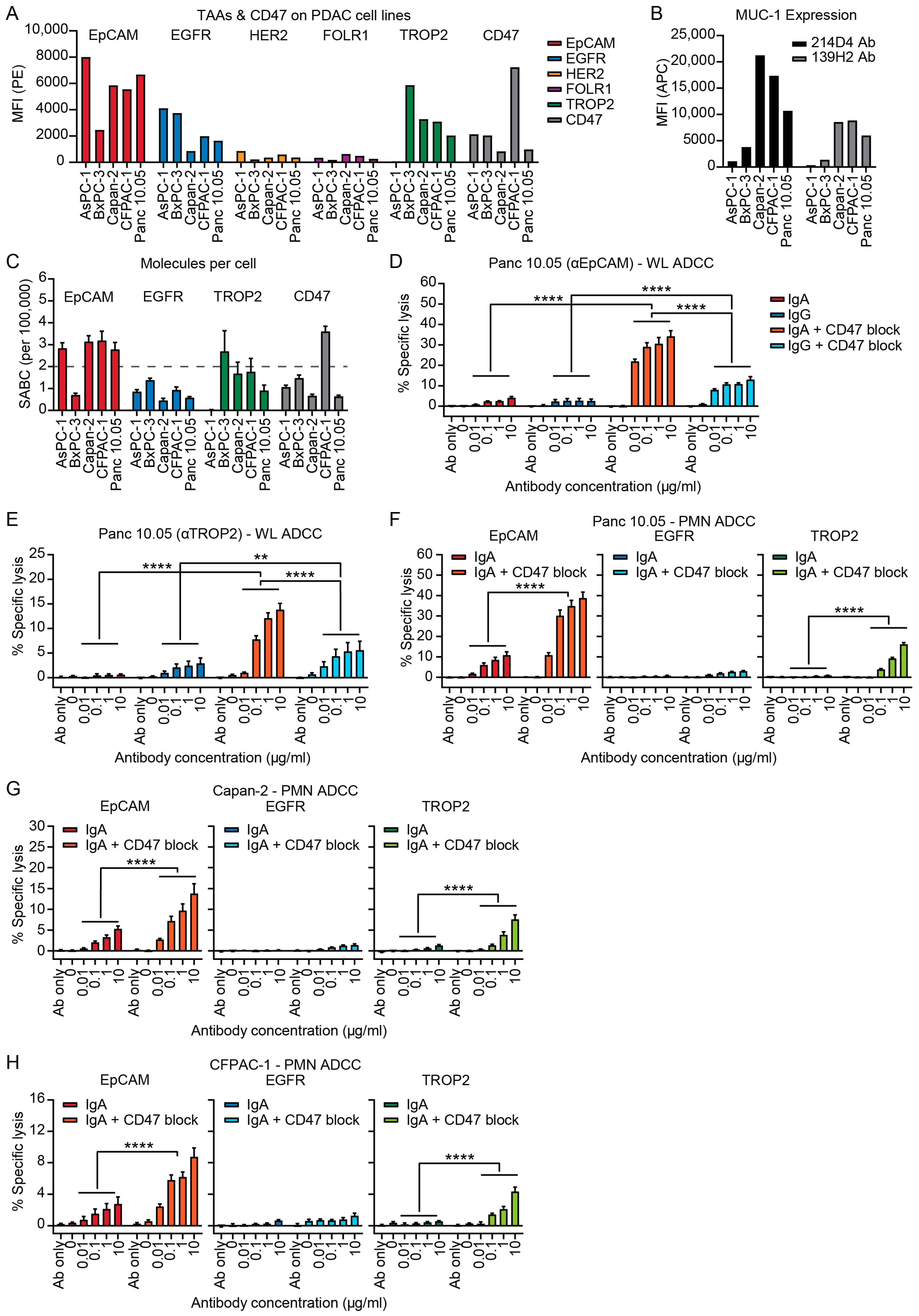
2.4. TAA Detection
2.5. Quantification of TAA Expression
2.6. Analysis of Immune Cell Subsets
2.7. ADCC Assay
2.8. Data Processing and Statistical Analyses
3. Results
3.1. IgA Antibodies Against Tumor-Associated Antigens (TAAs) Can Activate Neutrophils to Induce ADCC of PDAC Cell Lines
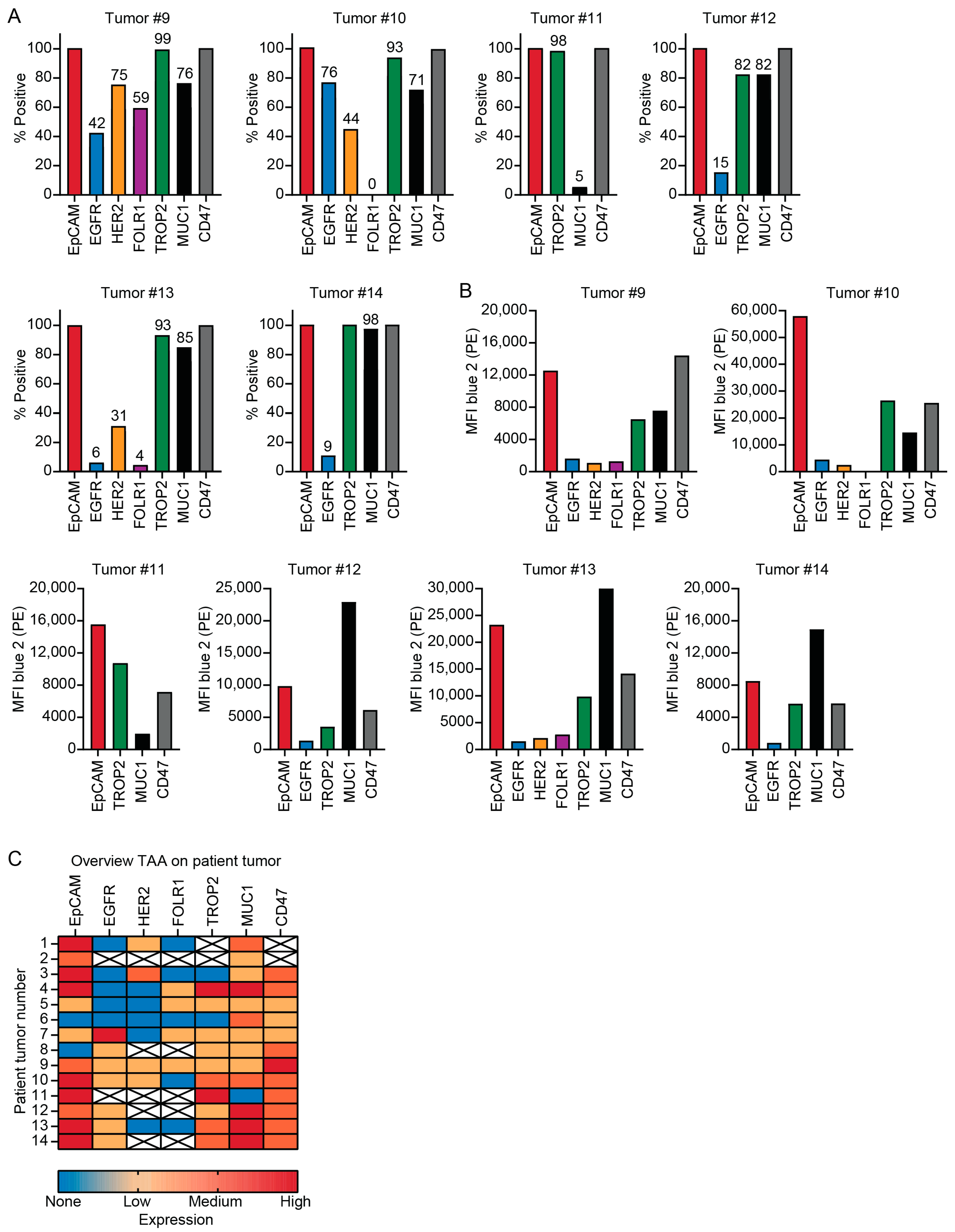
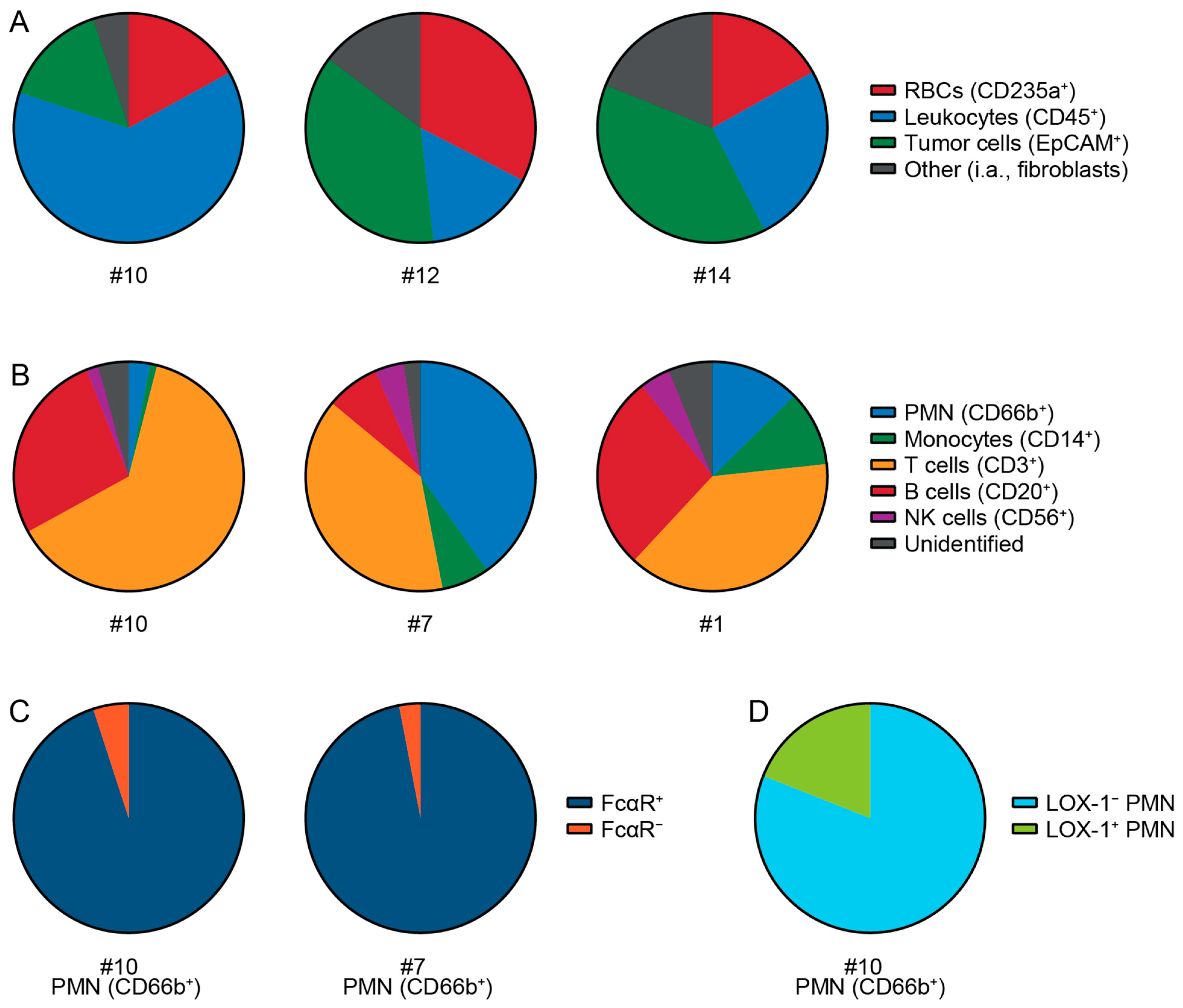
3.2. Patient Tumors Express Similar TAAs as PDAC Cell Lines
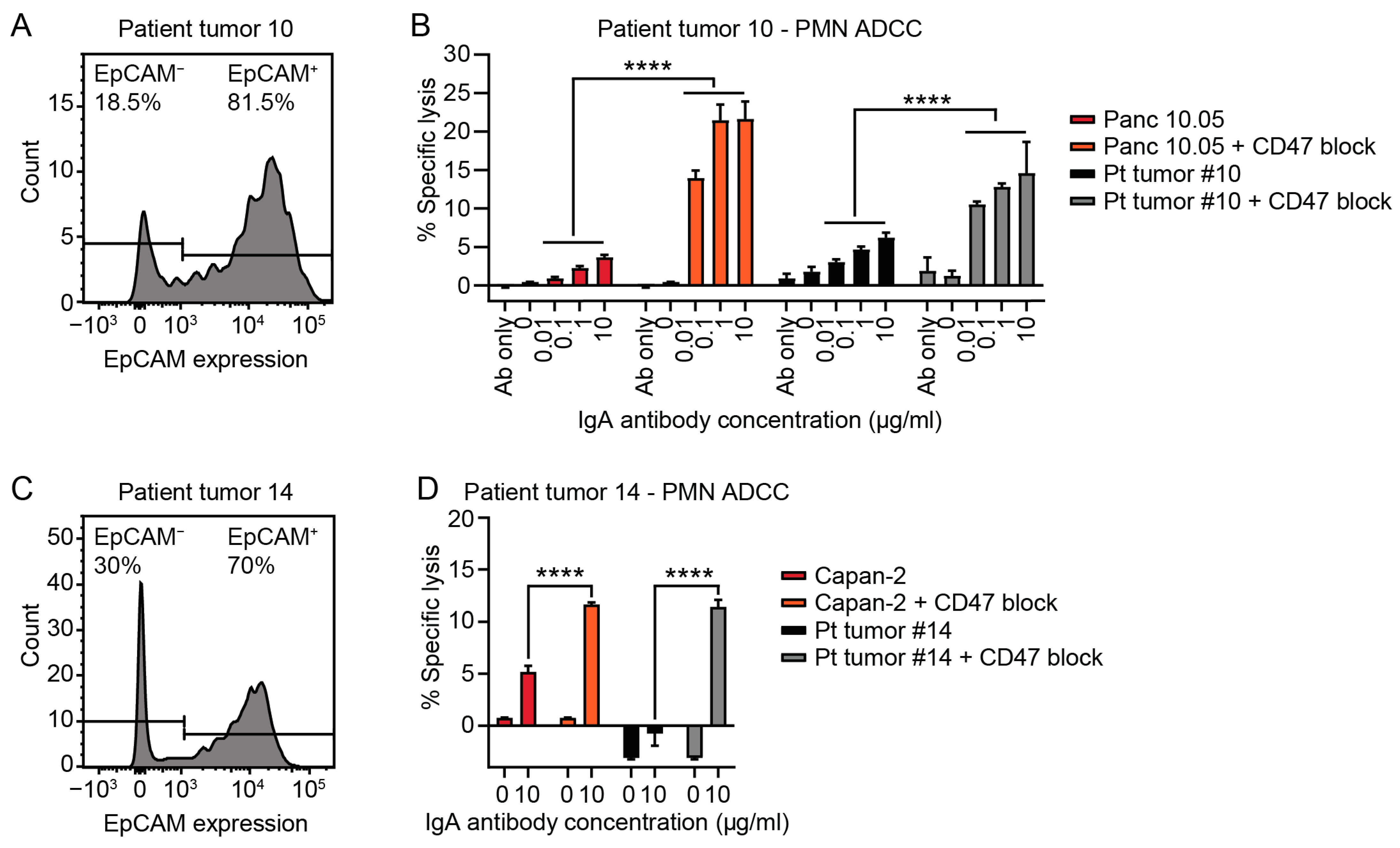
3.3. Tumor Cells Isolated from Patient PDAC Can Be Killed by Neutrophils with IgA mAbs
3.4. Cell Types and Immune Cells in Patient Tumor Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | pancreatic ductal adenocarcinoma |
| TME | tumor microenvironment |
| mAb(s) | monoclonal antibody (antibodies) |
| TAA(s) | tumor-associated antigen(s) |
| EpCAM | epithelial cell adhesion molecule |
| TROP2 (TACSTD2) | trophoblast cell surface antigen 2 |
| MUC(1) | mucin(−1) |
| ADCC(s) | antibody-dependent cellular cytotoxicity assay(s) |
| EGFR | epidermal growth factor receptor |
| SIRPα | signal regulatory protein alpha |
| ATCC | American Type Culture Collection |
| Pen/Step | penicillin–streptomycin |
| FCS | fetal calf serum |
| PBS | phosphate buffered saline |
| RAKU | Regional Academic Cancer Center Utrecht |
| RBC(s) | red blood cell(s) |
| PE | phycoerythrin |
| Cy | cyanine |
| HER2 | human epidermal growth factor receptor 2 |
| FOLR1 | folate receptor 1 |
| 7-AAD | 7-amino-actinomycin D |
| BV | brilliant violet |
| AF | Alexa Fluor |
| 51Cr | chromium-51 |
| FITC | fluorescein isothiocyanate |
| PB | pacific blue |
| H7 | hilite 7 |
| PerCp | peridinin–chlorophyll–protein |
| PMN(s) | polymorphonuclear leukocyte(s) |
| WL | whole leukocytes |
| CPM | counts per minute |
| SEM | standard error of the mean |
| TA | tumor-associated |
| FcαR | Fc alpha receptor |
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Latenstein, A.E.J.; van der Geest, L.G.M.; Bonsing, B.A.; Groot Koerkamp, B.; Haj Mohammad, N.; de Hingh, I.; de Meijer, V.E.; Molenaar, I.Q.; van Santvoort, H.C.; van Tienhoven, G.; et al. Nationwide trends in incidence, treatment and survival of pancreatic ductal adenocarcinoma. Eur. J. Cancer 2020, 125, 83–93. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Ferreira, M.; Reckamp, K.L. Editorial: Impact of immunotherapy in lung cancer. Front. Oncol. 2022, 12, 1083524. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef]
- Katz, M.H.G.; Petroni, G.R.; Bauer, T.; Reilley, M.J.; Wolpin, B.M.; Stucky, C.C.; Bekaii-Saab, T.S.; Elias, R.; Merchant, N.; Dias Costa, A.; et al. Multicenter randomized controlled trial of neoadjuvant chemoradiotherapy alone or in combination with pembrolizumab in patients with resectable or borderline resectable pancreatic adenocarcinoma. J. Immunother. Cancer 2023, 11, e007586. [Google Scholar] [CrossRef] [PubMed]
- Neoadjuvant Triple Treatment with FOLFIRINOX Plus Pembrolizumab and SABR in Patients with Borderline Resectable Pancreatic Cancer (PREOPANC-5): A Multicenter Single Arm Phase I/II Trial of the Dutch Pancreatic Cancer Group. 2024. Available online: https://adisinsight.springer.com/trials/700374043 (accessed on 27 November 2024).
- Chouari, T.; La Costa, F.S.; Merali, N.; Jessel, M.D.; Sivakumar, S.; Annels, N.; Frampton, A.E. Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 4265. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pinilla, G.A.; Modjtahedi, H. Therapeutic Application of Monoclonal Antibodies in Pancreatic Cancer: Advances, Challenges and Future Opportunities. Cancers 2021, 13, 1781. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Fumagalli, M.R.; D’Anna, R.; Sollai, M.; Bozzarelli, S.; Nappo, G.; Zanini, D.; Parente, R.; Garlanda, C.; Rimassa, L.; et al. Depicting the cellular complexity of pancreatic adenocarcinoma by Imaging Mass Cytometry: Focus on cancer-associated fibroblasts. Front. Immunol. 2024, 15, 1472433. [Google Scholar] [CrossRef]
- Vayrynen, S.A.; Zhang, J.; Yuan, C.; Vayrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef]
- Sherman, M.H.; Beatty, G.L. Tumor Microenvironment in Pancreatic Cancer Pathogenesis and Therapeutic Resistance. Annu. Rev. Pathol. 2023, 18, 123–148. [Google Scholar] [CrossRef]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, T.; Huang, L.; Wang, H.; Zhang, L.; Wang, Z.; Cui, Y. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem. Biophys. Res. Commun. 2018, 501, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, X.; Xiang, C.; Zhou, W. Neutrophils in pancreatic cancer: Potential therapeutic targets. Front. Oncol. 2022, 12, 1025805. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Jin, Y.; Christenson, E.S.; Zheng, L.; Li, K. Neutrophils in pancreatic ductal adenocarcinoma: Bridging preclinical insights to clinical prospects for improved therapeutic strategies. Expert Rev. Clin. Immunol. 2024, 20, 945–958. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Alshehri, M.S.A.; Miller, K.L.R.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers 2019, 11, 1627. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rosner, T.; Valerius, T.; Leusen, J.H.W.; Ten Broeke, T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef]
- Stip, M.C.; Evers, M.; Nederend, M.; Chan, C.; Reiding, K.R.; Damen, M.J.; Heck, A.J.R.; Koustoulidou, S.; Ramakers, R.; Krijger, G.C.; et al. IgA antibody immunotherapy targeting GD2 is effective in preclinical neuroblastoma models. J. Immunother. Cancer 2023, 11, e006948. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.; van Tetering, G.; Dechant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef]
- van Tetering, G.; Evers, M.; Chan, C.; Stip, M.; Leusen, J. Fc Engineering Strategies to Advance IgA Antibodies as Therapeutic Agents. Antibodies 2020, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Van Audenaerde, J.R.M.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E.L.J. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, 189, 31–44. [Google Scholar] [CrossRef]
- Chan, C.; Lustig, M.; Baumann, N.; Valerius, T.; van Tetering, G.; Leusen, J.H.W. Targeting Myeloid Checkpoint Molecules in Combination with Antibody Therapy: A Novel Anti-Cancer Strategy with IgA Antibodies? Front. Immunol. 2022, 13, 932155. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Chan, C.; Stip, M.; Nederend, M.; Jansen, M.; Passchier, E.; van den Ham, F.; Wienke, J.; van Tetering, G.; Leusen, J. Enhancing IgA-mediated neutrophil cytotoxicity against neuroblastoma by CD47 blockade. J. Immunother. Cancer 2024, 12, e008478. [Google Scholar] [CrossRef]
- Chan, C.; Jansen, J.H.M.; Hendriks, I.S.T.; van der Peet, I.C.; Verdonschot, M.E.L.; Passchier, E.M.; Tsioumpekou, M.; Nederend, M.; Klomp, S.A.; Peipp, M.; et al. Enhancing neutrophil cytotoxicity of a panel of clinical EGFR antibodies by Fc engineering to IgA3.0. Mol. Cancer Ther. 2024, 23, 1317–1331. [Google Scholar] [CrossRef]
- Chernyavska, M.; Hermans, C.K.J.C.; Chan, C.; Baumann, N.; Rösner, T.; Leusen, J.H.W.; Valerius, T.; Verdurmen, W.P.R. Evaluation of immunotherapies improving macrophage anti-tumor response using a microfluidic model. Organs-on-a-Chip 2022, 4, 100019. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Fan, J.Q.; Wang, M.F.; Chen, H.L.; Shang, D.; Das, J.K.; Song, J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer 2020, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Jung, Y.J.; Moon, S.H. Immunotherapy for pancreatic cancer. World J. Clin. Cases 2021, 9, 2969–2982. [Google Scholar] [CrossRef]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef]
- Dodson, L.F.; Hawkins, W.G.; Goedegebuure, P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy 2011, 3, 517–537. [Google Scholar] [CrossRef]
- Mas, L.; Cros, J.; Svrcek, M.; Van Laethem, J.L.; Emile, J.F.; Rebours, V.; Nicolle, R.; Bachet, J.B. Trop-2 is a ubiquitous and promising target in pancreatic adenocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102108. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, A.M.; Ten Broeke, T.; Nederend, M.; Meulenbroek, L.A.; van Tetering, G.; Meyer, S.; Jansen, J.H.; Beltran Buitrago, M.A.; Nagelkerke, S.Q.; Nemeth, I.; et al. Simultaneous Targeting of FcgammaRs and FcalphaRI Enhances Tumor Cell Killing. Cancer Immunol. Res. 2015, 3, 1316–1324. [Google Scholar] [CrossRef]
- Chan, C.; Cabanes, N.C.; Jansen, J.H.M.; Guillaume, J.; Nederend, M.; Passchier, E.M.; Gomez-Mellado, V.E.; Peipp, M.; Boes, M.; van Tetering, G.; et al. The relevance of tumor target expression levels on IgA-mediated cytotoxicity in cancer immunotherapy. Cancer Immunol. Immunother. 2024, 73, 238. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef]
- Macdonald, J.; Henri, J.; Roy, K.; Hays, E.; Bauer, M.; Veedu, R.N.; Pouliot, N.; Shigdar, S. EpCAM Immunotherapy versus Specific Targeted Delivery of Drugs. Cancers 2018, 10, 19. [Google Scholar] [CrossRef]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Schmidt, M.; Ruttinger, D.; Schuler, M.; Reinhardt, C.; et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef]
- de Bono, J.S.; Tolcher, A.W.; Forero, A.; Vanhove, G.F.; Takimoto, C.; Bauer, R.J.; Hammond, L.A.; Patnaik, A.; White, M.L.; Shen, S.; et al. ING-1, a monoclonal antibody targeting Ep-CAM in patients with advanced adenocarcinomas. Clin. Cancer Res. 2004, 10, 7555–7565. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Murr, A.; Kvesic, M.; Rau, D.; Mangold, S.; Pflanz, S.; Lumsden, J.; Volkland, J.; Fagerberg, J.; Riethmuller, G.; et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef]
- Fong, D.; Steurer, M.; Obrist, P.; Barbieri, V.; Margreiter, R.; Amberger, A.; Laimer, K.; Gastl, G.; Tzankov, A.; Spizzo, G. Ep-CAM expression in pancreatic and ampullary carcinomas: Frequency and prognostic relevance. J. Clin. Pathol. 2008, 61, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, G.; Fong, D.; Wurm, M.; Ensinger, C.; Obrist, P.; Hofer, C.; Mazzoleni, G.; Gastl, G.; Went, P. EpCAM expression in primary tumour tissues and metastases: An immunohistochemical analysis. J. Clin. Pathol. 2011, 64, 415–420. [Google Scholar] [CrossRef]
- Chan, C.; Lustig, M.; Jansen, J.H.M.; Garcia Villagrasa, L.; Raymakers, L.; Daamen, L.A.; Valerius, T.; van Tetering, G.; Leusen, J.H.W. Sialic Acids on Tumor Cells Modulate IgA Therapy by Neutrophils via Inhibitory Receptors Siglec-7 and Siglec-9. Cancers 2023, 15, 3405. [Google Scholar] [CrossRef]
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (mAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef]
- Caputo, R.; Buono, G.; Piezzo, M.; Martinelli, C.; Cianniello, D.; Rizzo, A.; Pantano, F.; Staropoli, N.; Cangiano, R.; Turano, S.; et al. Sacituzumab Govitecan for the treatment of advanced triple negative breast cancer patients: A multi-center real-world analysis. Front. Oncol. 2024, 14, 1362641. [Google Scholar] [CrossRef]
- Lombardi, P.; Filetti, M.; Falcone, R.; Altamura, V.; Paroni Sterbini, F.; Bria, E.; Fabi, A.; Giannarelli, D.; Scambia, G.; Daniele, G. Overview of Trop-2 in Cancer: From Pre-Clinical Studies to Future Directions in Clinical Settings. Cancers 2023, 15, 1744. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar] [PubMed]
- Seligson, J.M.; Patron, A.M.; Berger, M.J.; Harvey, R.D.; Seligson, N.D. Sacituzumab Govitecan-hziy: An Antibody-Drug Conjugate for the Treatment of Refractory, Metastatic, Triple-Negative Breast Cancer. Ann. Pharmacother. 2021, 55, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Effer, B.; Perez, I.; Ulloa, D.; Mayer, C.; Munoz, F.; Bustos, D.; Rojas, C.; Manterola, C.; Vergara-Gomez, L.; Dappolonnio, C.; et al. Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging. Biomedicines 2023, 11, 2086. [Google Scholar] [CrossRef]
- Guerra, E.; Trerotola, M.; Relli, V.; Lattanzio, R.; Tripaldi, R.; Ceci, M.; Boujnah, K.; Pantalone, L.; Sacchetti, A.; Havas, K.M.; et al. 3D-Informed Targeting of the Trop-2 Signal-Activation Site Drives Selective Cancer Vulnerability. Mol. Cancer Ther. 2023, 22, 790–804. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Z.; Zhang, S.; Zhu, P.; Ko, J.K.; Yung, K.K. MUC1: Structure, Function, and Clinic Application in Epithelial Cancers. Int. J. Mol. Sci. 2021, 22, 6567. [Google Scholar] [CrossRef]
- Striefler, J.K.; Riess, H.; Lohneis, P.; Bischoff, S.; Kurreck, A.; Modest, D.P.; Bahra, M.; Oettle, H.; Sinn, M.; Blaker, H.; et al. Mucin-1 Protein Is a Prognostic Marker for Pancreatic Ductal Adenocarcinoma: Results From the CONKO-001 Study. Front. Oncol. 2021, 11, 670396. [Google Scholar] [CrossRef]
- Bose, M.; Sanders, A.; De, C.; Zhou, R.; Lala, P.; Shwartz, S.; Mitra, B.; Brouwer, C.; Mukherjee, P. Targeting tumor-associated MUC1 overcomes anoikis-resistance in pancreatic cancer. Transl. Res. 2023, 253, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Lustig, M.; Chan, C.; Jansen, J.H.M.; Brautigam, M.; Kolling, M.A.; Gehlert, C.L.; Baumann, N.; Mester, S.; Foss, S.; Andersen, J.T.; et al. Disruption of the sialic acid/Siglec-9 axis improves antibody-mediated neutrophil cytotoxicity towards tumor cells. Front. Immunol. 2023, 14, 1178817. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Thompson, M.C.; Gardner, M.M.; Gendler, S.J. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J. Biol. Chem. 2001, 276, 13057–13064. [Google Scholar] [CrossRef]
- Bose, M.; Mukherjee, P. Potential of Anti-MUC1 Antibodies as a Targeted Therapy for Gastrointestinal Cancers. Vaccines 2020, 8, 659. [Google Scholar] [CrossRef]
- Vaughan, A.T.; Cragg, M.S.; Beers, S.A. Antibody modulation: Limiting the efficacy of therapeutic antibodies. Pharmacol. Res. 2015, 99, 269–275. [Google Scholar] [CrossRef]
- Dahal, L.N.; Huang, C.Y.; Stopforth, R.J.; Mead, A.; Chan, K.; Bowater, J.X.; Taylor, M.C.; Narang, P.; Chan, H.T.C.; Kim, J.H.; et al. Shaving Is an Epiphenomenon of Type I and II Anti-CD20-Mediated Phagocytosis, whereas Antigenic Modulation Limits Type I Monoclonal Antibody Efficacy. J. Immunol. 2018, 201, 1211–1221. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Bian, H.T.; Shen, Y.W.; Zhou, Y.D.; Nagle, D.G.; Guan, Y.Y.; Zhang, W.D.; Luan, X. CD47: Beyond an immune checkpoint in cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188771. [Google Scholar] [CrossRef]
- Song, X.; Lu, Z.; Xu, J. Targeting cluster of differentiation 47 improves the efficacy of anti-cytotoxic T-lymphocyte associated protein 4 treatment via antigen presentation enhancement in pancreatic ductal adenocarcinoma. Exp. Ther. Med. 2020, 20, 3301–3309. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, P.; Xu, Z.; Ye, H. Opportunities and challenges for anti-CD47 antibodies in hematological malignancies. Front. Immunol. 2024, 15, 1348852. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xun, Y.; You, H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark. Res. 2023, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Klaus, T.; Deshmukh, S. pH-responsive antibodies for therapeutic applications. J. Biomed. Sci. 2021, 28, 11. [Google Scholar] [CrossRef] [PubMed]
- Ibarlucea-Benitez, I.; Weitzenfeld, P.; Smith, P.; Ravetch, J.V. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2107424118. [Google Scholar] [CrossRef]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Eveline Li, R.J.; Kruijssen, L.; Bruijns, S.C.M.; van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; van der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun. 2021, 12, 1270. [Google Scholar] [CrossRef]
- Boelaars, K.; Rodriguez, E.; Huinen, Z.R.; Liu, C.; Wang, D.; Springer, B.O.; Olesek, K.; Goossens-Kruijssen, L.; van Ee, T.; Lindijer, D.; et al. Pancreatic cancer-associated fibroblasts modulate macrophage differentiation via sialic acid-Siglec interactions. Commun. Biol. 2024, 7, 430. [Google Scholar] [CrossRef]
- van der Haar Avila, I.; Marmol, P.; Kiessling, R.; Pico de Coana, Y. Evaluating Antibody-Dependent Cell-Mediated Cytotoxicity by Chromium Release Assay. Methods Mol. Biol. 2019, 1913, 167–179. [Google Scholar] [CrossRef]
- Tsioumpekou, M.; Krijgsman, D.; Leusen, J.H.W.; Olofsen, P.A. The Role of Cytokines in Neutrophil Development, Tissue Homing, Function and Plasticity in Health and Disease. Cells 2023, 12, 1981. [Google Scholar] [CrossRef]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Olajubutu, O.; Ogundipe, O.D.; Adebayo, A.; Adesina, S.K. Drug Delivery Strategies for the Treatment of Pancreatic Cancer. Pharmaceutics 2023, 15, 1318. [Google Scholar] [CrossRef]
- Muller, M.; Haghnejad, V.; Schaefer, M.; Gauchotte, G.; Caron, B.; Peyrin-Biroulet, L.; Bronowicki, J.P.; Neuzillet, C.; Lopez, A. The Immune Landscape of Human Pancreatic Ductal Carcinoma: Key Players, Clinical Implications, and Challenges. Cancers 2022, 14, 995. [Google Scholar] [CrossRef] [PubMed]
- Raymakers, L.; Demmers, T.J.; Meijer, G.J.; Molenaar, I.Q.; van Santvoort, H.C.; Intven, M.P.W.; Leusen, J.H.W.; Olofsen, P.A.; Daamen, L.A. The Effect of Radiation Treatment of Solid Tumors on Neutrophil Infiltration and Function: A Systematic Review. Int. J. Radiat. Oncol. Biol. Phys. 2024, 120, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Kuett, L.; Catena, R.; Ozcan, A.; Pluss, A.; Cancer Grand Challenges, I.C.; Schraml, P.; Moch, H.; de Souza, N.; Bodenmiller, B. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat. Cancer 2022, 3, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Fumagalli, M.R.; Zanini, D.; Candiello, E.; Tiberi, G.; Parente, R.; D’Anna, R.; Magrini, E.; Marchesi, F.; Cappello, P.; et al. Multiplexed Imaging Mass Cytometry Analysis in Preclinical Models of Pancreatic Cancer. Int. J. Mol. Sci. 2024, 25, 1389. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Tumor | Cells Extracted (×106) | TAA Staining | Leukocyte Composition Evaluation | ADCC Assay |
|---|---|---|---|---|
| 1 | 2.5 | + | + | |
| 2 | 0.75 | + | ||
| 3 | 2.2 | + | ||
| 4 | 15 | + | ||
| 5 | 3.0 | + | + | |
| 6 | 1.9 | + | ||
| 7 | 3.8 | + | + | |
| 8 | 0.80 | + | ||
| 9 | 0.30 | + | ||
| 10 | 4.9 | + | + | + |
| 11 | 0.80 | + | ||
| 12 | 0.50 | + | ||
| 13 | 0.42 | + | ||
| 14 | 1.2 | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raymakers, L.; Passchier, E.M.; Verdonschot, M.E.L.; Evers, M.; Chan, C.; Kuijpers, K.C.; Raicu, G.M.; Molenaar, I.Q.; van Santvoort, H.C.; Strijbis, K.; et al. The Efficacy of Targeted Monoclonal IgA Antibodies Against Pancreatic Ductal Adenocarcinoma. Cells 2025, 14, 632. https://doi.org/10.3390/cells14090632
Raymakers L, Passchier EM, Verdonschot MEL, Evers M, Chan C, Kuijpers KC, Raicu GM, Molenaar IQ, van Santvoort HC, Strijbis K, et al. The Efficacy of Targeted Monoclonal IgA Antibodies Against Pancreatic Ductal Adenocarcinoma. Cells. 2025; 14(9):632. https://doi.org/10.3390/cells14090632
Chicago/Turabian StyleRaymakers, Léon, Elsemieke M. Passchier, Meggy E. L. Verdonschot, Mitchell Evers, Chilam Chan, Karel C. Kuijpers, G. Mihaela Raicu, I. Quintus Molenaar, Hjalmar C. van Santvoort, Karin Strijbis, and et al. 2025. "The Efficacy of Targeted Monoclonal IgA Antibodies Against Pancreatic Ductal Adenocarcinoma" Cells 14, no. 9: 632. https://doi.org/10.3390/cells14090632
APA StyleRaymakers, L., Passchier, E. M., Verdonschot, M. E. L., Evers, M., Chan, C., Kuijpers, K. C., Raicu, G. M., Molenaar, I. Q., van Santvoort, H. C., Strijbis, K., Intven, M. P. W., Daamen, L. A., Leusen, J. H. W., & Olofsen, P. A. (2025). The Efficacy of Targeted Monoclonal IgA Antibodies Against Pancreatic Ductal Adenocarcinoma. Cells, 14(9), 632. https://doi.org/10.3390/cells14090632