
Galectin-1 Inhibition as a Strategy for Malignant Peripheral Nerve Sheath Tumor Treatment

 and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Viability and Apoptosis Assay
2.3. Immunoblotting Analysis
2.4. Protein–Protein Docking Simulation
2.5. Plasmid Construction and Protein Production and Purification
2.6. Pull-Down Assay
2.7. Co-Immunoprecipitation Assay
2.8. In Vivo Animal Assays
2.9. Ras Activation Assay
2.10. Transcriptome Sequencing and Enrichment Analysis
2.11. Gene Set Enrichment Analysis (GSEA)
2.12. Statistics Analysis
3. Results
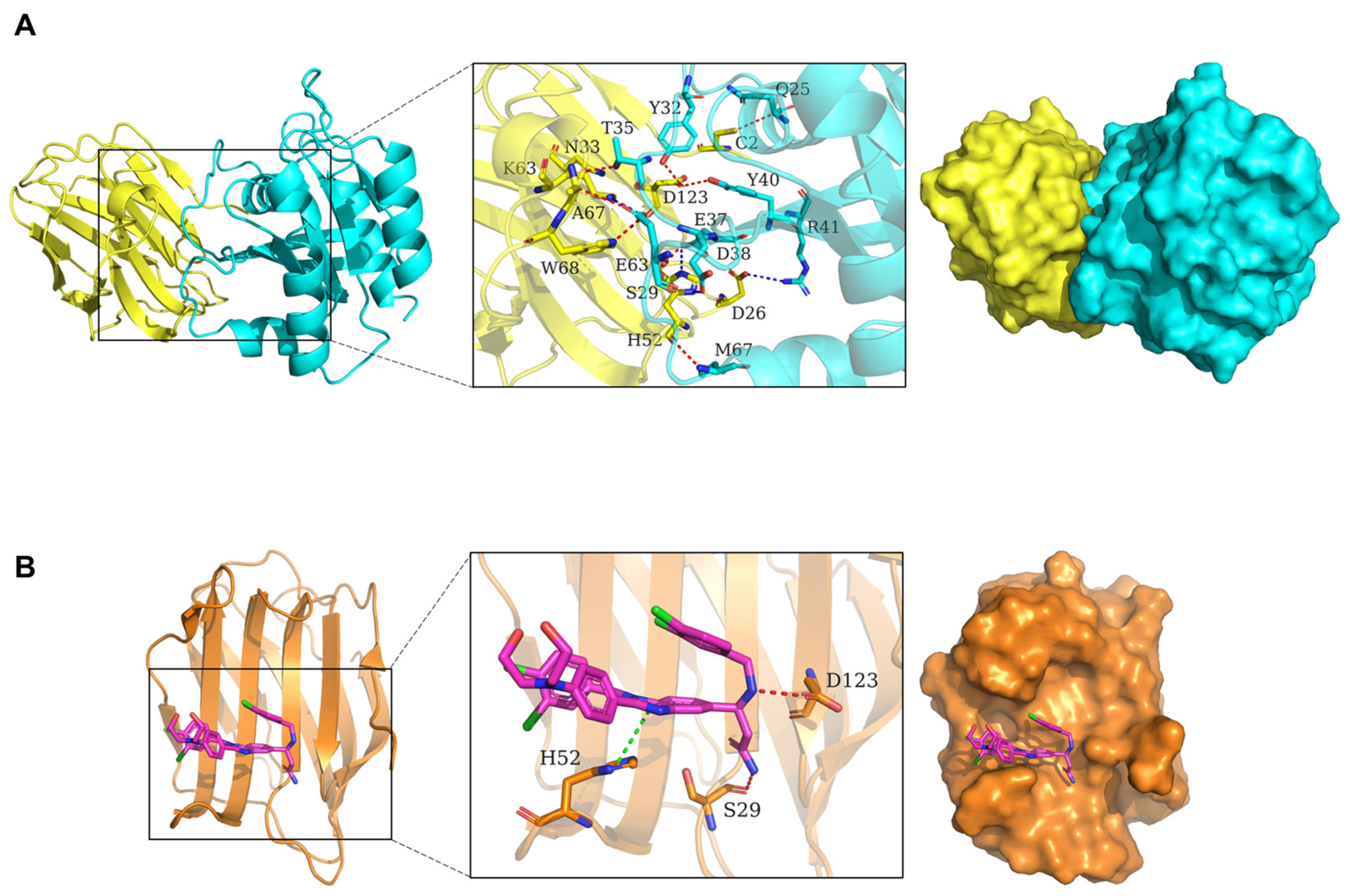
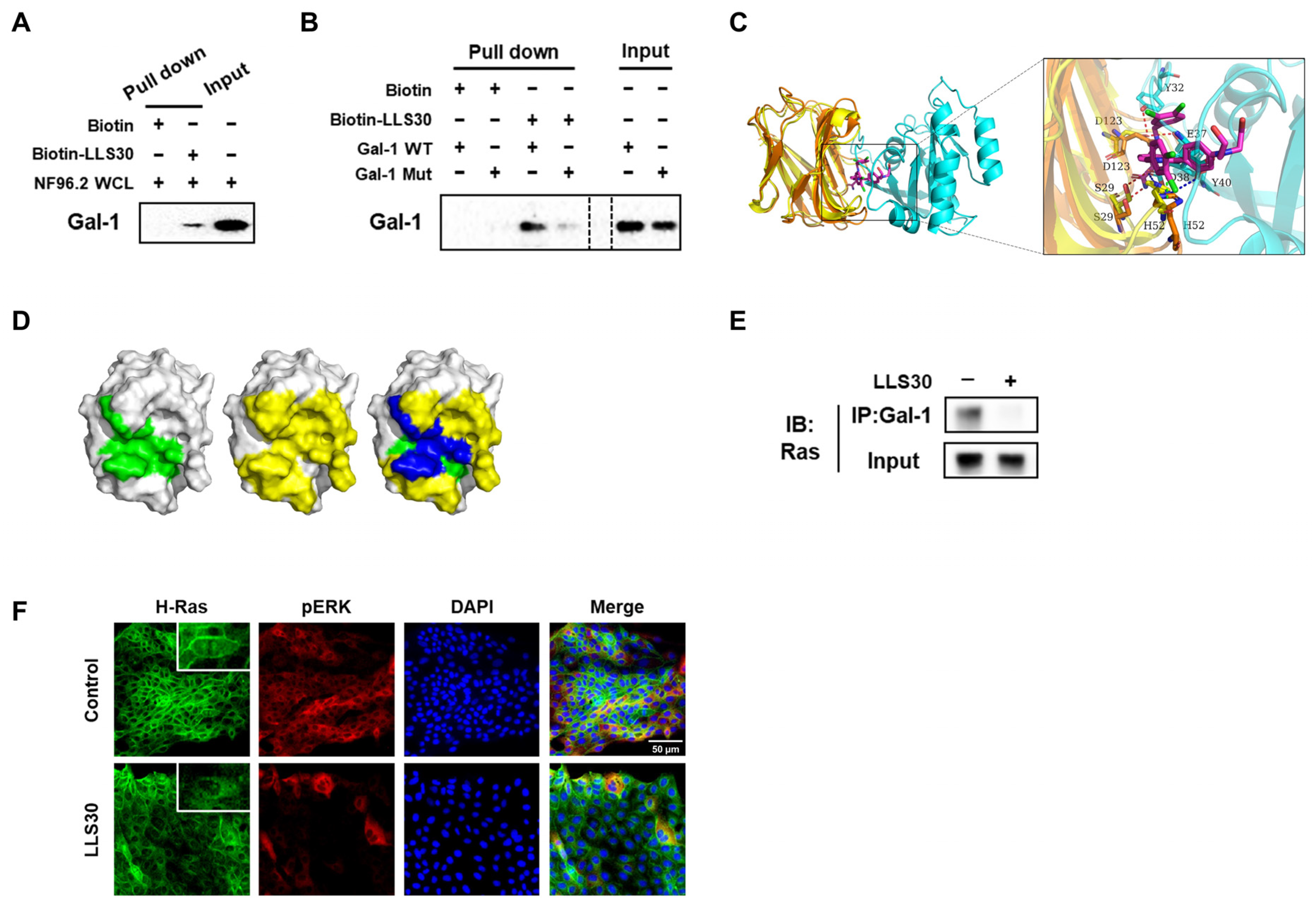
3.1. Molecular Dynamics Simulation of Gal-1, Ras, and LLS30 Interactions
3.2. LLS30 Disrupts Gal-1/Ras Interactions and Resulting Delocalization of RAS
3.3. Anti-Cancer Activity of LLS30 Against MPNST Cells In Vitro
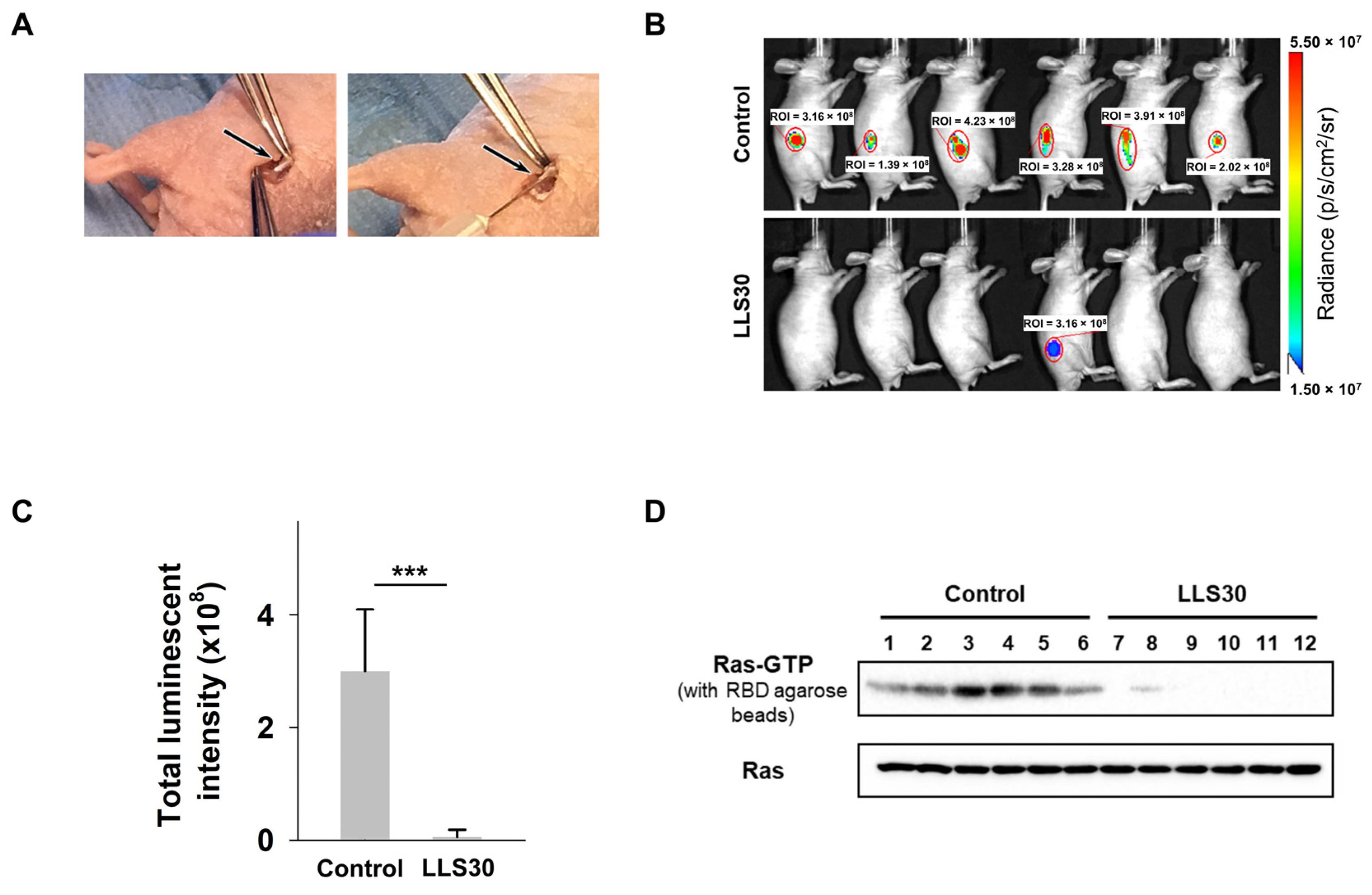
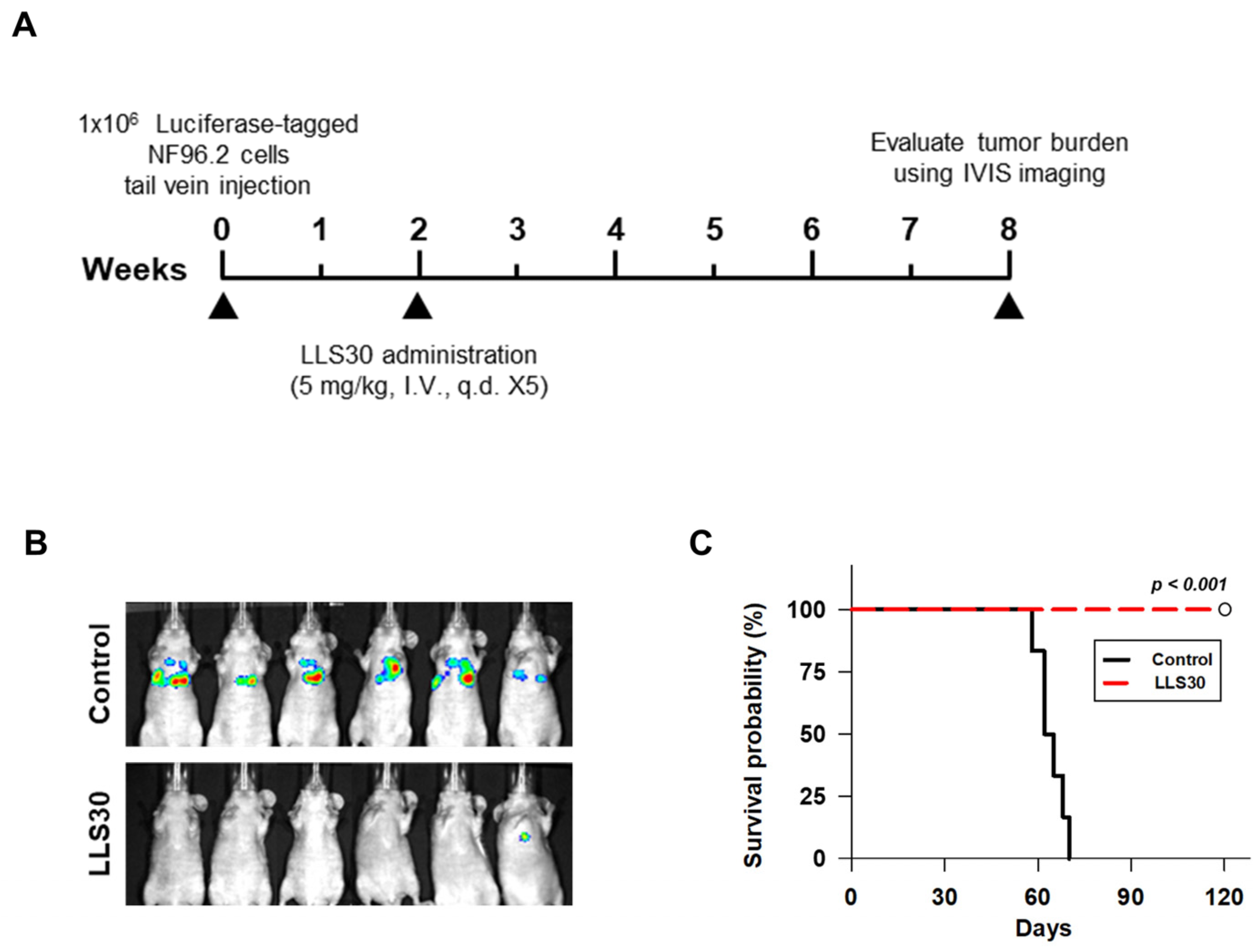
3.4. LLS30 Has Anti-Tumor Activity in MPNST Orthotopic Xenograft Mouse Models
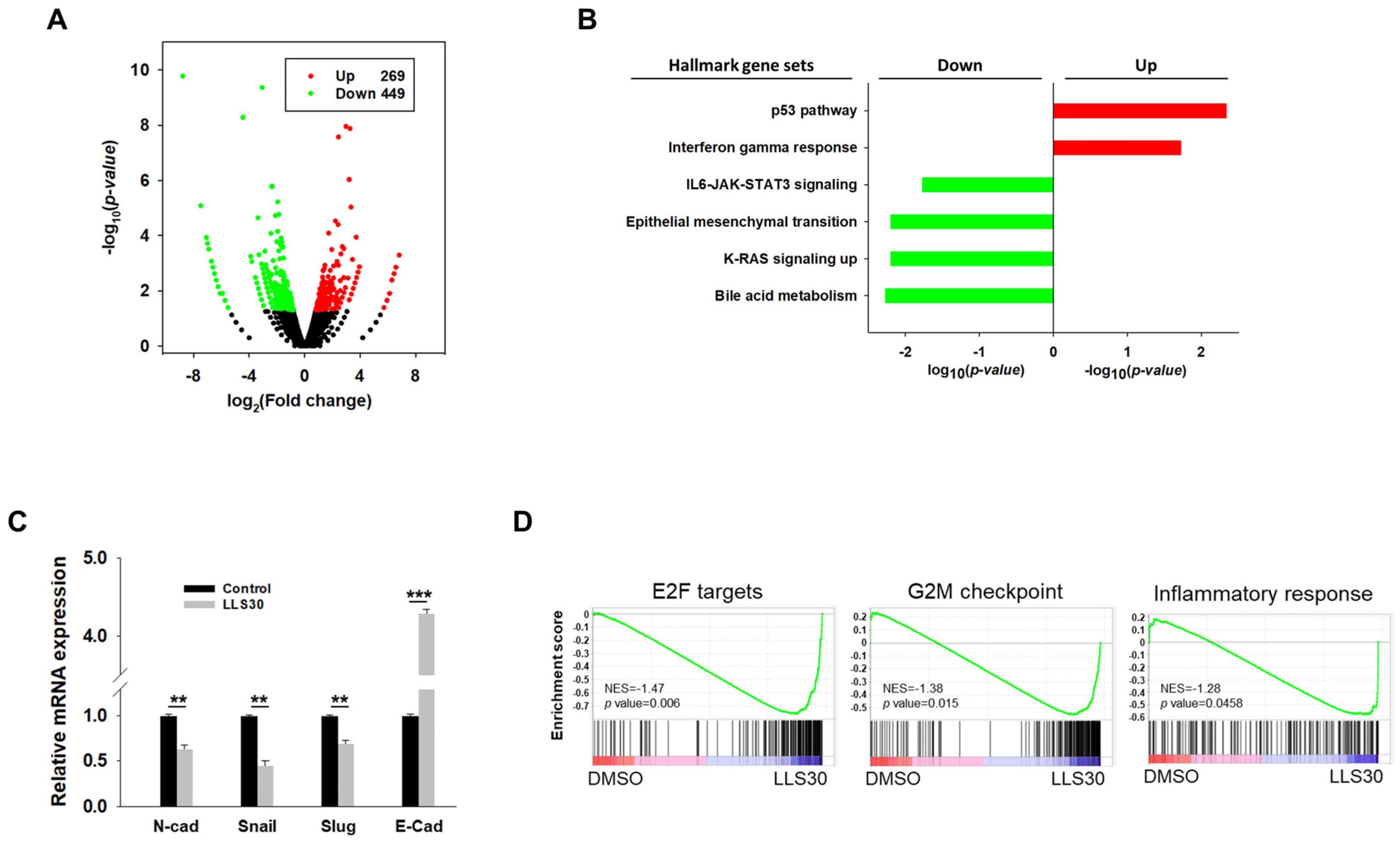
3.5. RNA-seq Analysis Reveals the Implication of LLS30 in Various Cancer Pathways
3.6. LLS30 Inhibits Formation and Growth of Experimental MPNST Metastasis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. Part A 2010, 152a, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Jouhilahti, E.-M.; Peltonen, S.; Callens, T.; Jokinen, E.; Heape, A.M.; Messiaen, L.; Peltonen, J. The development of cutaneous neurofibromas. Am. J. Pathol. 2011, 178, 500–505. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Bates, J.E.; Peterson, C.R.; Dhakal, S.; Giampoli, E.J.; Constine, L.S. Malignant peripheral nerve sheath tumors (MPNST): A SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr. Blood Cancer 2014, 61, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Gutmann, D.H. The management of neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors: Challenges, progress, and future prospects. Expert Opin. Orphan Drugs 2017, 5, 623–631. [Google Scholar] [CrossRef]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef]
- Carli, M.; Ferrari, A.; Mattke, A.; Zanetti, I.; Casanova, M.; Bisogno, G.; Cecchetto, G.; Alaggio, R.; De Sio, L.; Koscielniak, E.; et al. Pediatric malignant peripheral nerve sheath tumor: The Italian and German soft tissue sarcoma cooperative group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8422–8430. [Google Scholar] [CrossRef]
- Stucky, C.C.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Ann. Surg. Oncol. 2012, 19, 878–885. [Google Scholar] [CrossRef]
- Natalie Wu, L.M.; Lu, Q.R. Therapeutic Targets for Malignant Peripheral nerve Sheath Tumors. Future Neurol. 2019, 14, FNL7. [Google Scholar] [CrossRef]
- Ferner, R.E. Neurofibromatosis 1. Eur. J. Hum. Genet. 2006, 15, 131–138. [Google Scholar] [CrossRef]
- Valentin, T.; Le Cesne, A.; Ray-Coquard, I.; Italiano, A.; Decanter, G.; Bompas, E.; Isambert, N.; Thariat, J.; Linassier, C.; Bertucci, F.; et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur. J. Cancer 2016, 56, 77–84. [Google Scholar] [CrossRef]
- DeClue, J.E.; Cohen, B.D.; Lowy, D.R. Identification and characterization of the neurofibromatosis type 1 protein product. Proc. Natl. Acad. Sci. USA 1991, 88, 9914–9918. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.A.; Hunter, J.L.; Eccleston, J.F.; Webb, M.R. The Mechanism of Ras GTPase Activation by Neurofibromin. Biochemistry 2003, 42, 3956–3965. [Google Scholar] [CrossRef]
- Sherman, L.S.; Atit, R.; Rosenbaum, T.; Cox, A.D.; Ratner, N. Single Cell Ras-GTP Analysis Reveals Altered Ras Activity in a Subpopulation of Neurofibroma Schwann Cells but Not Fibroblasts. J. Biol. Chem. 2000, 275, 30740–30745. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef]
- Casey, P.J.; Seabra, M.C. Protein Prenyltransferases. J. Biol. Chem. 1996, 271, 5289–5292. [Google Scholar] [CrossRef]
- Azoulay-Alfaguter, I.; Strazza, M.; Mor, A. Chaperone-mediated specificity in Ras and Rap signaling. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 194–202. [Google Scholar] [CrossRef]
- Rorive, S.; Belot, N.; Decaestecker, C.; Lefranc, F.; Gordower, L.; Micik, S.; Maurage, C.A.; Kaltner, H.; Ruchoux, M.M.; Danguy, A.; et al. Galectin-1 is highly expressed in human gliomas with relevance for modulation of invasion of tumor astrocytes into the brain parenchyma. Glia 2001, 33, 241–255. [Google Scholar] [CrossRef]
- van den Brûle, F.A.; Waltregny, D.; Castronovo, V. Increased expression of galectin-1 in carcinoma-associated stroma predicts poor outcome in prostate carcinoma patients. J. Pathol. 2001, 193, 80–87. [Google Scholar] [CrossRef]
- Ingrassia, L.; Camby, I.; Lefranc, F.; Mathieu, V.; Nshimyumukiza, P.; Darro, F.; Kiss, R. Anti-galectin compounds as potential anti-cancer drugs. Curr. Med. Chem. 2006, 13, 3513–3527. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Heusschen, R.; Caers, J.; Griffioen, A.W. Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim. Biophys. Acta 2015, 1855, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Alonso, M.; Bruger, A.M.; van der Bruggen, P. Extracellular galectins as controllers of cytokines in hematological cancer. Blood 2018, 132, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.-C.; Fan, Y.; Kiss, S.; Li, X.; Deng, X.N.; Liu, R.; Chen, X.-J.; Carney, R.; Chen, A.; Ghosh, P.M.; et al. Galectin-1 inhibition induces cell apoptosis through dual suppression of CXCR4 and Ras pathways in human malignant peripheral nerve sheath tumors. Neuro Oncol. 2019, 21, 1389–1400. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef]
- Brockman, Q.R.; Scherer, A.; McGivney, G.R.; Gutierrez, W.R.; Voigt, A.P.; Isaacson, A.L.; Laverty, E.A.; Roughton, G.; Knepper-Adrian, V.; Darbro, B.; et al. PRC2 loss drives MPNST metastasis and matrix remodeling. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Stannard, K.A.; Collins, P.M.; Ito, K.; Sullivan, E.M.; Scott, S.A.; Gabutero, E.; Darren Grice, I.; Low, P.; Nilsson, U.J.; Leffler, H.; et al. Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett. 2010, 299, 95–110. [Google Scholar] [CrossRef]
- Blanchard, H.; Bum-Erdene, K.; Bohari, M.H.; Yu, X. Galectin-1 inhibitors and their potential therapeutic applications: A patent review. Expert Opin. Ther. Pat. 2016, 26, 537–554. [Google Scholar] [CrossRef]
- Sethi, A.; Sanam, S.; Alvala, R.; Alvala, M. An updated patent review of galectin-1 and galectin-3 inhibitors and their potential therapeutic applications (2016-present). Expert Opin. Ther. Pat. 2021, 31, 709–721. [Google Scholar] [CrossRef]
- Laaf, D.; Bojarová, P.; Elling, L.; Křen, V. Galectin-Carbohydrate Interactions in Biomedicine and Biotechnology. Trends Biotechnol. 2019, 37, 402–415. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Rabinovich, G.A.; Bieche, I.; Vidaud, M.; de Gramont, A.; Martinet, M.; Cvitkovic, E.; et al. OTX008, a selective small-molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur. J. Cancer 2014, 50, 2463–2477. [Google Scholar] [CrossRef]
- Koonce, N.A.; Griffin, R.J.; Dings, R.P.M. Galectin-1 Inhibitor OTX008 Induces Tumor Vessel Normalization and Tumor Growth Inhibition in Human Head and Neck Squamous Cell Carcinoma Models. Int. J. Mol. Sci. 2017, 18, 2671. [Google Scholar] [CrossRef] [PubMed]
- Leung, Z.; Ko, F.C.F.; Tey, S.K.; Kwong, E.M.L.; Mao, X.; Liu, B.H.M.; Ma, A.P.Y.; Fung, Y.M.E.; Che, C.M.; Wong, D.K.H.; et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J. Exp. Clin. Cancer Res. CR 2019, 38, 423. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Spranger, S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Shurell, E.; Singh, A.S.; Crompton, J.G.; Jensen, S.; Li, Y.; Dry, S.; Nelson, S.; Chmielowski, B.; Bernthal, N.; Federman, N.; et al. Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes. Oncotarget 2016, 7, 64300–64308. [Google Scholar] [CrossRef] [PubMed]
- Sobczuk, P.; Teterycz, P.; Czarnecka, A.M.; Świtaj, T.; Koseła-Paterczyk, H.; Kozak, K.; Falkowski, S.; Rutkowski, P. Systemic Treatment for Advanced and Metastatic Malignant Peripheral Nerve Sheath Tumors-A Sarcoma Reference Center Experience. J. Clin. Med. 2020, 9, 3157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.Y.; Gu, Y.H.; Cui, X.W.; Long, M.M.; Wang, W.; Wei, C.J.; Gu, B.; Zhang, H.B.; Li, Q.F.; Wang, Z.C. Protein Tyrosine Phosphatase Receptor S Acts as a Metastatic Suppressor in Malignant Peripheral Nerve Sheath Tumor via Profilin 1-Induced Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2020, 8, 582220. [Google Scholar] [CrossRef]
- Bacigalupo, M.L.; Manzi, M.; Espelt, M.V.; Gentilini, L.D.; Compagno, D.; Laderach, D.J.; Wolfenstein-Todel, C.; Rabinovich, G.A.; Troncoso, M.F. Galectin-1 triggers epithelial-mesenchymal transition in human hepatocellular carcinoma cells. J. Cell. Physiol. 2015, 230, 1298–1309. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Y.; Zhang, H.; Liu, Y.; Sun, H.; Zhang, P. Galectin-1 induces metastasis and epithelial-mesenchymal transition (EMT) in human ovarian cancer cells via activation of the MAPK JNK/p38 signalling pathway. Am. J. Transl. Res. 2019, 11, 3862–3878. [Google Scholar]
- Tang, Z.; Liu, X.; Li, Z.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SpaRx: Elucidate single-cell spatial heterogeneity of drug responses for personalized treatment. Brief. Bioinform. 2023, 24, bbad338. [Google Scholar] [CrossRef]
- Chen, M.; Jia, S.; Xue, M.; Huang, H.; Xu, Z.; Yang, D.; Zhu, W.; Song, Q. Dual-Stream Subspace Clustering Network for revealing gene targets in Alzheimer’s disease. Comput. Biol. Med. 2022, 151, 106305. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chain 1 | Residue | Chain 2 | Residue | Interaction Type |
|---|---|---|---|---|
| H-Ras(G12V) | Gln25.CA | Gal-1 | Cys2.SG | Hydrogen bond interaction |
| H-Ras(G12V) | Tyr32.OH | Gal-1 | Asp123.OD1 | Hydrogen bond interaction |
| H-Ras(G12V) | Thr35.OG1 | Gal-1 | Asn33.OD1 | Hydrogen bond interaction |
| H-Ras(G12V) | Glu37.OE1 | Gal-1 | His52.ND1 | Salt bridge |
| H-Ras(G12V) | Asp38.OD1 | Gal-1 | Ser29.OG | Hydrogen bond interaction |
| H-Ras(G12V) | Asp38.OD1 | Gal-1 | His52.NE2 | Salt bridge |
| H-Ras(G12V) | Tyr40.OH | Gal-1 | Asp123.OD1 | Hydrogen bond interaction |
| H-Ras(G12V) | Arg41.NH2 | Gal-1 | Asp26.OD2 | Salt bridge |
| H-Ras(G12V) | Glu63.OE1/OE2 | Gal-1 | Lys63.NZ | Salt bridge |
| H-Ras(G12V) | Glu63.OE1 | Gal-1 | Ala67.CA | Hydrogen bond interaction |
| H-Ras(G12V) | Glu63.OE2 | Gal-1 | Trp68.NE1 | Hydrogen bond interaction |
| H-Ras(G12V) | Met67.N | Gal-1 | His52.O | Hydrogen bond interaction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-C.; Torres, K.E.; Xia, R.; Malogolowkin, M.H.; Hsu, S.-W.; Chen, C.-H.; Shih, T.-C. Galectin-1 Inhibition as a Strategy for Malignant Peripheral Nerve Sheath Tumor Treatment. Cells 2025, 14, 515. https://doi.org/10.3390/cells14070515
Wang H-C, Torres KE, Xia R, Malogolowkin MH, Hsu S-W, Chen C-H, Shih T-C. Galectin-1 Inhibition as a Strategy for Malignant Peripheral Nerve Sheath Tumor Treatment. Cells. 2025; 14(7):515. https://doi.org/10.3390/cells14070515
Chicago/Turabian StyleWang, Hsiao-Chi, Keila E. Torres, Roger Xia, Marcio H. Malogolowkin, Ssu-Wei Hsu, Ching-Hsien Chen, and Tsung-Chieh Shih. 2025. "Galectin-1 Inhibition as a Strategy for Malignant Peripheral Nerve Sheath Tumor Treatment" Cells 14, no. 7: 515. https://doi.org/10.3390/cells14070515
APA StyleWang, H.-C., Torres, K. E., Xia, R., Malogolowkin, M. H., Hsu, S.-W., Chen, C.-H., & Shih, T.-C. (2025). Galectin-1 Inhibition as a Strategy for Malignant Peripheral Nerve Sheath Tumor Treatment. Cells, 14(7), 515. https://doi.org/10.3390/cells14070515