

Platelet Reactive Oxygen Species, Oxidised Lipid Stress, Current Perspectives, and an Update on Future Directions
 , , , and
, , , and
Abstract
1. Introduction
2. Reactive Oxygen Species and Platelet Activation
2.1. ROS Generation in Platelets
2.1.1. NADPH Oxidases (NOX)
2.1.2. Cyclooxygenase-1 (COX1)
2.1.3. Xanthine Oxidase (XO)
2.1.4. Mitochondrial Respiratory Chain
2.2. Platelet Antioxidant Systems
2.2.1. Superoxide Dismutase (SOD)
2.2.2. Catalase
2.2.3. Glutathione Peroxidases (GPx)
2.3. The Role of ROS in Platelet Activation
2.4. ROS and Antioxidants in Disease and Ageing
3. Platelet Reactive Oxygen Species and Hyperlipidaemia
3.1. oxLDL and Platelet ROS
3.2. PCSK9 and Platelet ROS
3.3. Mitochondrial ROS and Hyperlipidaemia
4. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schwarz, U.R.; Walter, U.; Eigenthaler, M. Taming platelets with cyclic nucleotides. Biochem. Pharmacol. 2001, 62, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. An L-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc. Natl. Acad. Sci. USA 1990, 87, 5193–5197. [Google Scholar] [CrossRef] [PubMed]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–85. [Google Scholar] [CrossRef]
- Gibbins, J.M. Platelet adhesion signalling and the regulation of thrombus formation. J. Cell Sci. 2004, 117, 3415–3425. [Google Scholar] [CrossRef]
- André, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Hynes, R.O. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef]
- Heemskerk, J.W.M.; Mattheij, N.J.A.; Cosemans, J. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef]
- Marcus, A.J.; Silk, S.T.; Safier, L.B.; Ullman, H.L. Superoxide production and reducing activity in human platelets. J. Clin. Investig. 1977, 59, 149–158. [Google Scholar] [CrossRef]
- Iuliano, L.; Colavita, A.R.; Leo, R.; Praticò, D.; Violi, F. Oxygen free radicals and platelet activation. Free Radic. Biol. Med. 1997, 22, 999–1006. [Google Scholar] [CrossRef]
- Liao, R.; Wang, L.; Zeng, J.; Tang, X.; Huang, M.; Kantawong, F.; Huang, Q.; Mei, Q.; Huang, F.; Yang, Y.; et al. Reactive oxygen species: Orchestrating the delicate dance of platelet life and death. Redox Biol. 2025, 80, 103489. [Google Scholar] [CrossRef]
- Chlopicki, S.; Olszanecki, R.; Janiszewski, M.; Laurindo, F.R.; Panz, T.; Miedzobrodzki, J. Functional role of NADPH oxidase in activation of platelets. Antioxid. Redox Signal. 2004, 6, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wong, H.S. Are mitochondria the main contributor of reactive oxygen species in cells? J. Exp. Biol. 2021, 224 Pt 5, jeb221606. [Google Scholar] [CrossRef]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Diebold, B.A. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 2002, 100, 2692–2696. [Google Scholar] [CrossRef]
- Streeter, J.; Schickling, B.M.; Jiang, S.; Stanic, B.; Thiel, W.H.; Gakhar, L.; Houtman, J.C.; Miller, F.J., Jr. Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circ. Res. 2014, 115, 911–918. [Google Scholar] [CrossRef]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2020, 41, Atvbaha120315565. [Google Scholar] [CrossRef] [PubMed]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef]
- Carnevale, R.; Loffredo, L.; Nocella, C.; Bartimoccia, S.; Sanguigni, V.; Soresina, A.; Plebani, A.; Azzari, C.; Martire, B.; Pignata, C.; et al. Impaired platelet activation in patients with hereditary deficiency of p47(phox). Br. J. Haematol. 2018, 180, 454–456. [Google Scholar] [CrossRef]
- Gaspar, R.S.; Ferreira, P.M.; Mitchell, J.L.; Pula, G.; Gibbins, J.M. Platelet-derived extracellular vesicles express NADPH oxidase-1 (Nox-1), generate superoxide and modulate platelet function. Free Radic. Biol. Med. 2021, 165, 395–400. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J. 2020, 34, 13959–13977. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. 2013, 64, 409–421. [Google Scholar] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef]
- Morel, A.; Miller, E.; Bijak, M.; Saluk, J. The increased level of COX-dependent arachidonic acid metabolism in blood platelets from secondary progressive multiple sclerosis patients. Mol. Cell. Biochem. 2016, 420, 85–94. [Google Scholar] [CrossRef]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Kuwano, K.; Ikeda, H.; Oda, T.; Nakayama, H.; Koga, Y.; Toshima, H.; Imaizumi, T. Xanthine oxidase mediates cyclic flow variations in a canine model of coronary arterial thrombosis. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H1993–H1999. [Google Scholar] [CrossRef]
- Cecerska-Heryć, E.; Jesionowska, A.; Klaudyna, S.; Katarzyna, S.; Dominika, M.; Dominika, P.; Marta, U.; Dołęgowska, B. Xanthine oxidoreductase reference values in platelet-poor plasma and platelets in healthy volunteers. Oxid. Med. Cell. Longev. 2015, 2015, 341926. [Google Scholar] [CrossRef]
- Pandey, N.R.; Kaur, G.; Chandra, M.; Sanwal, G.G.; Misra, M.K. Enzymatic oxidant and antioxidants of human blood platelets in unstable angina and myocardial infarction. Int. J. Cardiol. 2000, 76, 33–38. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Vanden Hoek, T.L.; Becker, L.B.; Shao, Z.H.; Li, C.Q.; Schumacker, P.T. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 1998, 273, 18092–18098. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef]
- Lopez, J.J.; Salido, G.M.; Gómez-Arteta, E.; Rosado, J.A.; Pariente, J.A. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J. Thromb. Haemost. 2007, 5, 1283–1291. [Google Scholar] [CrossRef]
- Girish, K.S.; Paul, M.; Thushara, R.M.; Hemshekhar, M.; Shanmuga Sundaram, M.; Rangappa, K.S.; Kemparaju, K. Melatonin elevates apoptosis in human platelets via ROS mediated mitochondrial damage. Biochem. Biophys. Res. Commun. 2013, 438, 198–204. [Google Scholar] [CrossRef]
- Leytin, V.; Gyulkhandanyan, A.V.; Freedman, J. Role of mitochondrial membrane permeabilization and depolarization in platelet apoptosis. Br. J. Haematol. 2018, 181, 281–285. [Google Scholar] [CrossRef]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef]
- Yamagishi, S.; Edelstein, D.; Du, X.L.; Brownlee, M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Méndez, D.; Arauna, D.; Fuentes, F.; Araya-Maturana, R.; Palomo, I.; Alarcón, M.; Sebastián, D.; Zorzano, A.; Fuentes, E. Mitoquinone (MitoQ) Inhibits Platelet Activation Steps by Reducing ROS Levels. Int. J. Mol. Sci. 2020, 21, 6192. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef]
- Krötz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef]
- Dayal, S.; Gu, S.X.; Hutchins, R.D.; Wilson, K.M.; Wang, Y.; Fu, X.; Lentz, S.R. Deficiency of superoxide dismutase impairs protein C activation and enhances susceptibility to experimental thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1798–1804. [Google Scholar] [CrossRef]
- Fidler, T.P.; Rowley, J.W.; Araujo, C.; Boudreau, L.H.; Marti, A.; Souvenir, R.; Dale, K.; Boilard, E.; Weyrich, A.S.; Abel, E.D. Superoxide Dismutase 2 is dispensable for platelet function. Thromb. Haemost. 2017, 117, 1859–1867. [Google Scholar] [CrossRef]
- Sonkar, V.K.; Eustes, A.S.; Ahmed, A.; Jensen, M.; Solanki, M.V.; Swamy, J.; Kumar, R.; Fidler, T.P.; Houtman, J.C.D.; Allen, B.G.; et al. Endogenous SOD2 (Superoxide Dismutase) Regulates Platelet-Dependent Thrombin Generation and Thrombosis During Aging. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 79–91. [Google Scholar] [CrossRef]
- Manasa, K.; Vani, R. Influence of Oxidative Stress on Stored Platelets. Adv. Hematol. 2016, 2016, 4091461. [Google Scholar] [CrossRef]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Praticó, D.; Pasin, M.; Barry, O.P.; Ghiselli, A.; Sabatino, G.; Iuliano, L.; FitzGerald, G.A.; Violi, F. Iron-dependent human platelet activation and hydroxyl radical formation: Involvement of protein kinase C. Circulation 1999, 99, 3118–3124. [Google Scholar] [CrossRef]
- Bryant, R.W.; Simon, T.C.; Bailey, J.M. Role of glutathione peroxidase and hexose monophosphate shunt in the platelet lipoxygenase pathway. J. Biol. Chem. 1982, 257, 14937–14943. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Coulon, L.; Calzada, C.; Moulin, P.; Véricel, E.; Lagarde, M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic. Biol. Med. 2003, 35, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Calzada, C.; Véricel, E.; Mitel, B.; Coulon, L.; Lagarde, M. 12(S)-Hydroperoxy-eicosatetraenoic acid increases arachidonic acid availability in collagen-primed platelets. J. Lipid Res. 2001, 42, 1467–1473. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J.; Benoit, S.E.; Valeri, C.R.; Barnard, M.R.; Michelson, A.D. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J. Clin. Investig. 1996, 97, 979–987. [Google Scholar] [CrossRef]
- Jin, R.C.; Mahoney, C.E.; Coleman Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.Y.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Freedman, J.E.; Frei, B.; Welch, G.N.; Loscalzo, J. Glutathione peroxidase potentiates the inhibition of platelet function by S-nitrosothiols. J. Clin. Investig. 1995, 96, 394–400. [Google Scholar] [CrossRef]
- Becatti, M.; Fiorillo, C.; Gori, A.M.; Marcucci, R.; Paniccia, R.; Giusti, B.; Violi, F.; Pignatelli, P.; Gensini, G.F.; Abbate, R. Platelet and leukocyte ROS production and lipoperoxidation are associated with high platelet reactivity in Non-ST elevation myocardial infarction (NSTEMI) patients on dual antiplatelet treatment. Atherosclerosis 2013, 231, 392–400. [Google Scholar] [CrossRef]
- Marwali, M.R.; Hu, C.P.; Mohandas, B.; Dandapat, A.; Deonikar, P.; Chen, J.; Cawich, I.; Sawamura, T.; Kavdia, M.; Mehta, J.L. Modulation of ADP-induced platelet activation by aspirin and pravastatin: Role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J. Pharmacol. Exp. Ther. 2007, 322, 1324–1332. [Google Scholar] [CrossRef]
- Bakdash, N.; Williams, M.S. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic. Biol. Med. 2008, 45, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.F.; Qiao, J.; Shen, Y.; Davis, A.K.; Dunne, E.; Berndt, M.C.; Gardiner, E.E.; Andrews, R.K. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J. Thromb. Haemost. 2012, 10, 1133–1141. [Google Scholar] [CrossRef]
- Arthur, J.F.; Shen, Y.; Gardiner, E.E.; Coleman, L.; Murphy, D.; Kenny, D.; Andrews, R.K.; Berndt, M.C. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J. Thromb. Haemost. 2011, 9, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Carrim, N.; Arthur, J.F.; Hamilton, J.R.; Gardiner, E.E.; Andrews, R.K.; Moran, N.; Berndt, M.C.; Metharom, P. Thrombin-induced reactive oxygen species generation in platelets: A novel role for protease-activated receptor 4 and GPIbα. Redox Biol. 2015, 6, 640–647. [Google Scholar] [CrossRef]
- Wang, L.; Soe, N.N.; Sowden, M.; Xu, Y.; Modjeski, K.; Baskaran, P.; Kim, Y.; Smolock, E.M.; Morrell, C.N.; Berk, B.C. Cyclophilin A is an important mediator of platelet function by regulating integrin αIIbβ3 bidirectional signalling. Thromb. Haemost. 2014, 111, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Smolock, E.M.; Kim, Y.; Nigro, P.; Berk, B.C. Cyclophilin A is required for angiotensin II-induced p47phox translocation to caveolae in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2147–2153. [Google Scholar] [CrossRef]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal. 2005, 7, 560–577. [Google Scholar] [CrossRef]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Watson, S.P.; Auger, J.M.; McCarty, O.J.; Pearce, A.C. GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost. 2005, 3, 1752–1762. [Google Scholar] [CrossRef]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Wang, S.B.; Park, S.J.; Oh, G.T.; Lee, S.H.; Ho, Y.S.; Chang, T.S. Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Revisiting the reactions of superoxide with glutathione and other thiols. Arch. Biochem. Biophys. 2016, 595, 68–71. [Google Scholar] [CrossRef] [PubMed]
- van Gorp, R.M.; van Dam-Mieras, M.C.; Hornstra, G.; Heemskerk, J.W. Effect of membrane-permeable sulfhydryl reagents and depletion of glutathione on calcium mobilisation in human platelets. Biochem. Pharmacol. 1997, 53, 1533–1542. [Google Scholar] [CrossRef]
- Essex, D.W.; Li, M. Redox control of platelet aggregation. Biochemistry 2003, 42, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ball, C.; Vijayan, K.V.; Nguyen, T.; Anthony, K.; Bray, P.F.; Essex, D.W.; Dong, J.F. Glutathione regulates integrin alpha(IIb)beta(3)-mediated cell adhesion under flow conditions. Thromb. Haemost. 2008, 100, 857–863. [Google Scholar] [CrossRef]
- Yan, B.; Smith, J.W. Mechanism of integrin activation by disulfide bond reduction. Biochemistry 2001, 40, 8861–8867. [Google Scholar] [CrossRef] [PubMed]
- Véricel, E.; Januel, C.; Carreras, M.; Moulin, P.; Lagarde, M. Diabetic patients without vascular complications display enhanced basal platelet activation and decreased antioxidant status. Diabetes 2004, 53, 1046–1051. [Google Scholar] [CrossRef]
- Guidi, G.; Schiavon, R.; Sheiban, I.; Perona, G. Platelet glutathione peroxidase activity is impaired in patients with coronary heart disease. Scand. J. Clin. Lab. Investig. 1986, 46, 549–551. [Google Scholar] [CrossRef]
- Kamhieh-Milz, J.; Bal, G.; Sterzer, V.; Kamhieh-Milz, S.; Arbach, O.; Salama, A. Reduced antioxidant capacities in platelets from patients with autoimmune thrombocytopenia purpura (ITP). Platelets 2012, 23, 184–194. [Google Scholar] [CrossRef]
- Wang, B.; Yee Aw, T.; Stokes, K.Y. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018, 14, 218–228. [Google Scholar] [CrossRef]
- De Bona, K.S.; Bellé, L.P.; Sari, M.H.; Thomé, G.; Schetinger, M.R.; Morsch, V.M.; Boligon, A.; Athayde, M.L.; Pigatto, A.S.; Moretto, M.B. Syzygium cumini extract decrease adenosine deaminase, 5′nucleotidase activities and oxidative damage in platelets of diabetic patients. Cell. Physiol. Biochem. 2010, 26, 729–738. [Google Scholar] [CrossRef]
- Jain, K.; Tyagi, T.; Patell, K.; Xie, Y.; Kadado, A.J.; Lee, S.H.; Yarovinsky, T.; Du, J.; Hwang, J.; Martin, K.A.; et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: Implications for CVD risk patients. EBioMedicine 2019, 44, 28–40. [Google Scholar] [CrossRef]
- Barrachina, M.N.; Hermida-Nogueira, L.; Moran, L.A.; Casas, V.; Hicks, S.M.; Sueiro, A.M.; Di, Y.; Andrews, R.K.; Watson, S.P.; Gardiner, E.E.; et al. Phosphoproteomic Analysis of Platelets in Severe Obesity Uncovers Platelet Reactivity and Signaling Pathways Alterations. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 478–490. [Google Scholar] [CrossRef]
- Barrachina, M.N.; Sueiro, A.M.; Izquierdo, I.; Hermida-Nogueira, L.; Guitián, E.; Casanueva, F.F.; Farndale, R.W.; Moroi, M.; Jung, S.M.; Pardo, M.; et al. GPVI surface expression and signalling pathway activation are increased in platelets from obese patients: Elucidating potential anti-atherothrombotic targets in obesity. Atherosclerosis 2019, 281, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Bernardo, E.; Sabaté, M.; Jimenez-Quevedo, P.; Costa, M.A.; Palazuelos, J.; Hernández-Antolin, R.; Moreno, R.; Escaned, J.; Alfonso, F.; et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J. Am. Coll. Cardiol. 2007, 50, 1541–1547. [Google Scholar] [CrossRef]
- Naseem, K.M.; Goodall, A.H.; Bruckdorfer, K.R. Differential effects of native and oxidatively modified low-density lipoproteins on platelet function. Platelets 1997, 8, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.C.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef]
- Chan, H.C.; Ke, L.Y.; Chu, C.S.; Lee, A.S.; Shen, M.Y.; Cruz, M.A.; Hsu, J.F.; Cheng, K.H.; Chan, H.C.B.; Lu, J.; et al. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef]
- Wang, X.; Fu, Y.F.; Liu, X.; Feng, G.; Xiong, D.; Mu, G.F.; Chen, F.P. ROS Promote Ox-LDL-Induced Platelet Activation by Up-Regulating Autophagy Through the Inhibition of the PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2018, 50, 1779–1793. [Google Scholar] [CrossRef]
- Berger, M.; Wraith, K.; Woodward, C.; Aburima, A.; Raslan, Z.; Hindle, M.S.; Moellmann, J.; Febbraio, M.; Naseem, K.M. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase C gamma 2-dependent reactive oxygen species generation. Platelets 2019, 30, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.L.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Assinger, A.; Koller, F.; Schmid, W.; Zellner, M.; Koller, E.; Volf, I. Hypochlorite-oxidized LDL induces intraplatelet ROS formation and surface exposure of CD40L-A prominent role of CD36. Atherosclerosis 2010, 213, 129–134. [Google Scholar] [CrossRef]
- Krause, M.; Dent, E.W.; Bear, J.E.; Loureiro, J.J.; Gertler, F.B. Ena/VASP proteins: Regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 2003, 19, 541–564. [Google Scholar] [CrossRef] [PubMed]
- Urban, D.; Pöss, J.; Böhm, M.; Laufs, U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 2013, 62, 1401–1408. [Google Scholar] [CrossRef]
- Leander, K.; Mälarstig, A.; Van’t Hooft, F.M.; Hyde, C.; Hellénius, M.L.; Troutt, J.S.; Konrad, R.J.; Öhrvik, J.; Hamsten, A.; de Faire, U. Circulating Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Predicts Future Risk of Cardiovascular Events Independently of Established Risk Factors. Circulation 2016, 133, 1230–1239. [Google Scholar] [CrossRef]
- Shi, P.; Zhang, L.; Zhang, M.; Yang, W.; Wang, K.; Zhang, J.; Otsu, K.; Huang, G.; Fan, X.; Liu, J. Platelet-Specific p38α Deficiency Improved Cardiac Function After Myocardial Infarction in Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e185–e196. [Google Scholar] [CrossRef]
- Cammisotto, V.; Pastori, D.; Nocella, C.; Bartimoccia, S.; Castellani, V.; Marchese, C.; Scavalli, A.S.; Ettorre, E.; Viceconte, N.; Violi, F.; et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants 2020, 9, 296. [Google Scholar] [CrossRef]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.K.R.; Sangle, G.V.; Xie, X.P.; Stelmack, G.L.; Halayko, A.J.; Shen, G.X. Effects of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Yang, M.; Huang, W.X.; Chen, W.J.; Zhao, Y.Q.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Ding, Z.F.; Liu, S.J.; Wang, X.W.; Dai, Y.; Khaidakov, M.; Deng, X.Y.; Fan, Y.B.; Xiang, D.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628. [Google Scholar] [CrossRef]
- Yesilbursa, D.; Serdar, A.; Senturk, T.; Serdar, Z.; Sağ, S.; Cordan, J. Effect of N-acetylcysteine on oxidative stress and ventricular function in patients with myocardial infarction. Heart Vessel. 2006, 21, 33–37. [Google Scholar] [CrossRef]
- Pasupathy, S.; Tavella, R.; Grover, S.; Raman, B.; Procter, N.E.K.; Du, Y.T.; Mahadavan, G.; Stafford, I.; Heresztyn, T.; Holmes, A.; et al. Early Use of N-acetylcysteine With Nitrate Therapy in Patients Undergoing Primary Percutaneous Coronary Intervention for ST-Segment-Elevation Myocardial Infarction Reduces Myocardial Infarct Size (the NACIAM Trial [N-acetylcysteine in Acute Myocardial Infarction]). Circulation 2017, 136, 894–903. [Google Scholar] [CrossRef]
- Arstall, M.A.; Yang, J.; Stafford, I.; Betts, W.H.; Horowitz, J.D. N-acetylcysteine in combination with nitroglycerin and streptokinase for the treatment of evolving acute myocardial infarction. Safety and biochemical effects. Circulation 1995, 92, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Porro, B.; Aldini, G.; Colli, S.; Banfi, C. N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin. Antioxidants 2022, 11, 445. [Google Scholar] [CrossRef]
- Borgström, L.; Kågedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J. Neurochem. 2004, 89, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Munno, M.; Atlas, D.; Banfi, C. N-acetylcysteine Amide AD4/NACA and Thioredoxin Mimetic Peptides Inhibit Platelet Aggregation and Protect against Oxidative Stress. Antioxidants 2023, 12, 1395. [Google Scholar] [CrossRef] [PubMed]
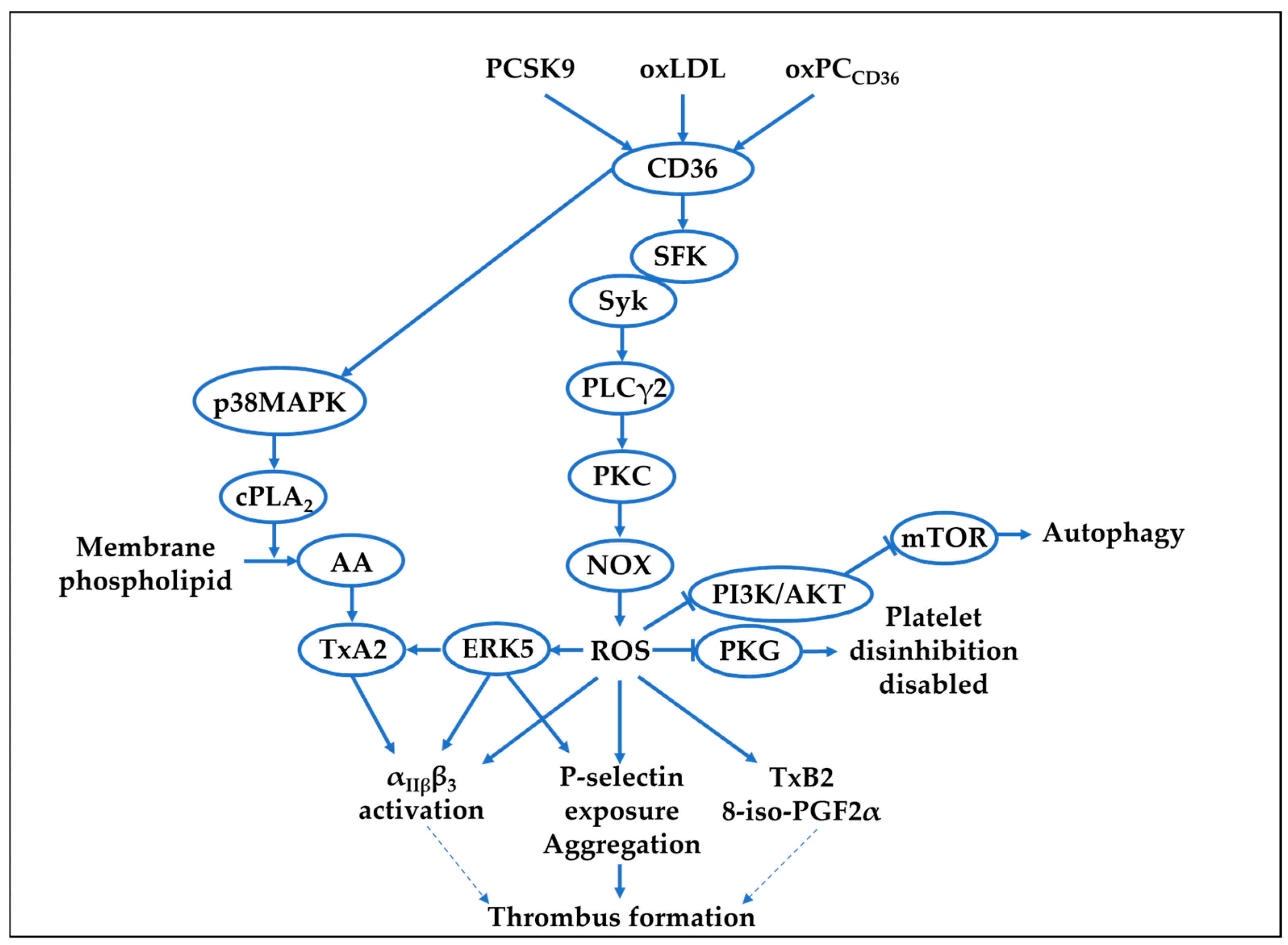

{kind=link}
| Agonists | Mice/ Inhibitor | Observations | Conclusions | Ref. | |
|---|---|---|---|---|---|
| CRP (0.5 µg/mL) Thrombin (0.018 or 0.025 U/mL) | NOX1−/− | ROS Aggregation ATP secretion Ca2+ mobilisation | Thrombin ↓ CRP ↔ | NOX1 is involved in GPCR-induced platelet activation | [21] |
| NOX2−/− | ROS Aggregation ATP secretion Ca2+ mobilisation | Thrombin partial CRP ↓ | NOX2 is involved in GPCR- and GPVI-induced platelet activation | ||
| NOX1−/− and NOX2−/− | Carotid artery occlusion Tail bleeding | NOX1−/− ↔ NOX2−/− ↓ NOX1−/− ↔ NOX2−/− ↔ | NOX2 is involved in thrombosis NOX1 and NOX2 are dispensable for haemostasis | ||
| Collagen (3 µg/mL) Thrombin (0.1 U/mL) | NOX1−/− | Aggregation | Thrombin ↔ Collagen ↓ | NOX1 is involved in GPVI-induced platelet activation | [23] |
| NOX2−/− | Aggregation | Thrombin ↓ Collagen ↔ | NOX2 is involved in GPCR-induced platelet activation | ||
| NOX1−/− and NOX2−/− | Carotid artery occlusion Tail bleeding | NOX1−/− ↓ NOX2−/− ↔ NOX1−/− ↔ NOX2−/− ↔ | NOX1 is involved in thrombosis NOX1 and NOX2 are dispensable for haemostasis | ||
| 2-APT (NOX1 inhibitor) | Superoxide anion Aggregation Static adhesion over collagen Thrombus formation under flow Carotid artery occlusion Tail bleeding | Thrombin ↓ Collagen ↓ Thrombin marginally↓ Collagen ↓ ↓ ↓ ↓ ↔ | NOX1 inhibition impairs GPVI-induced platelet activation without affecting GPCR responses | ||
| Collagen (10 µg/mL) CRP (5 µg/mL) Thrombin (0.25U or 1U/mL) | NOX1/2/4−/− | Superoxide anion Aggregation αIIbβ3 P-selectin PS exposure Carotid artery occlusion Tail bleeding | Thrombin ↓ Collagen /CRP ↓ Thrombin ↔ CRP ↓ ↓ ↔ | NOXs are critical for GPVI-induced platelet activation. GPCR-associated integrin activation and platelet aggregation are NOX-dependent, whilst P-selectin and PS exposure are NOXs independent | [17] |
| NOX1−/− | Aggregation Thrombus formation over collagen | Thrombin ↔ Collagen ↓ ↔ | NOX1 is involved in GPVI- and GPCR-induced platelet activation | ||
| NOX2−/− | Aggregation Thrombus formation over collagen | Thrombin ↓ Collagen ↔ ↓ | NOX2 is involved in GPCR-induced platelet activation | ||
| NOX4−/− | Aggregation Thrombus formation over collagen | Thrombin ↔ Collagen ↔ ↔ | NOX4 has negligible role in platelet regulation | ||
| Hyperlipidaemic Insult | Platelet Function Downstream of ROS Production | ROS Related Inhibitors Used | Pathway Involved | Ref. |
|---|---|---|---|---|
| oxLDL | αIIbβ3 activation, TxB2 production, thrombosis under shear stress | -Gp91ds-tat | CD36/LOX1-p38MAPK/PKC-NOX2 | [90] |
| oxLDL | aggregation, P-selectin, adhesion under shear stress | NAC | PI3K-AKT-mTOR | [91] |
| oxPCCD36 | P-selectin | NAC Gp91ds-tat ML171 | CD36-Src-PLCγ2-NOX2 | [92] |
| oxLDL | aggregation | VAS2870 DPTA-NONOate PEG-catalase | CD36-Src-NOX-ERK5 | [93] |
| oxLDL oxPCCD36 | diminished sensitivity to inhibitory NO-cGMP signalling | TEMPOL MnTMPyP Gp91ds-tat | CD36-Src-Syk-PLCγ2-PKC-NOX2 | [18] |
| PCSK9 | thrombin-induced platelet aggregation | VAS2870 | CD36-Src-ERK5-JNK-ROS-p38MAPK/cPLA2/COX-1/TxA2 | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheah, L.T.; Hindle, M.S.; Khalil, J.S.; Duval, C.; Unsworth, A.J.; Naseem, K.M. Platelet Reactive Oxygen Species, Oxidised Lipid Stress, Current Perspectives, and an Update on Future Directions. Cells 2025, 14, 500. https://doi.org/10.3390/cells14070500
Cheah LT, Hindle MS, Khalil JS, Duval C, Unsworth AJ, Naseem KM. Platelet Reactive Oxygen Species, Oxidised Lipid Stress, Current Perspectives, and an Update on Future Directions. Cells. 2025; 14(7):500. https://doi.org/10.3390/cells14070500
Chicago/Turabian StyleCheah, Lih T., Matthew S. Hindle, Jawad S. Khalil, Cedric Duval, Amanda J. Unsworth, and Khalid M. Naseem. 2025. "Platelet Reactive Oxygen Species, Oxidised Lipid Stress, Current Perspectives, and an Update on Future Directions" Cells 14, no. 7: 500. https://doi.org/10.3390/cells14070500
APA StyleCheah, L. T., Hindle, M. S., Khalil, J. S., Duval, C., Unsworth, A. J., & Naseem, K. M. (2025). Platelet Reactive Oxygen Species, Oxidised Lipid Stress, Current Perspectives, and an Update on Future Directions. Cells, 14(7), 500. https://doi.org/10.3390/cells14070500