Therapeutic Potential of Emricasan, a Pan-Caspase Inhibitor, in Reducing Cell Death and Extracellular Matrix Accumulation in Fuchs Endothelial Corneal Dystrophy

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. Cell Culture and Establishment of Cell Lines
2.4. Annexin V Assay
2.5. Caspase 3/7 Activity Assay
2.6. Immunoblotting
2.7. Immunofluorescence Staining
2.8. qPCR Analysis
2.9. siRNA
2.10. Preparation of Emricasan Eye Drops
2.11. High-Performance Liquid Chromatography
2.12. Contact Specular Microscopy Imaging in Col8a2Q455K/Q455K Mice
2.13. mRNA Library Preparation and Sequencing
2.14. RNA Sequencing Data Analysis
2.15. Multivariate Analysis
2.16. Pathway Analysis
2.17. Statistical Analysis
3. Results
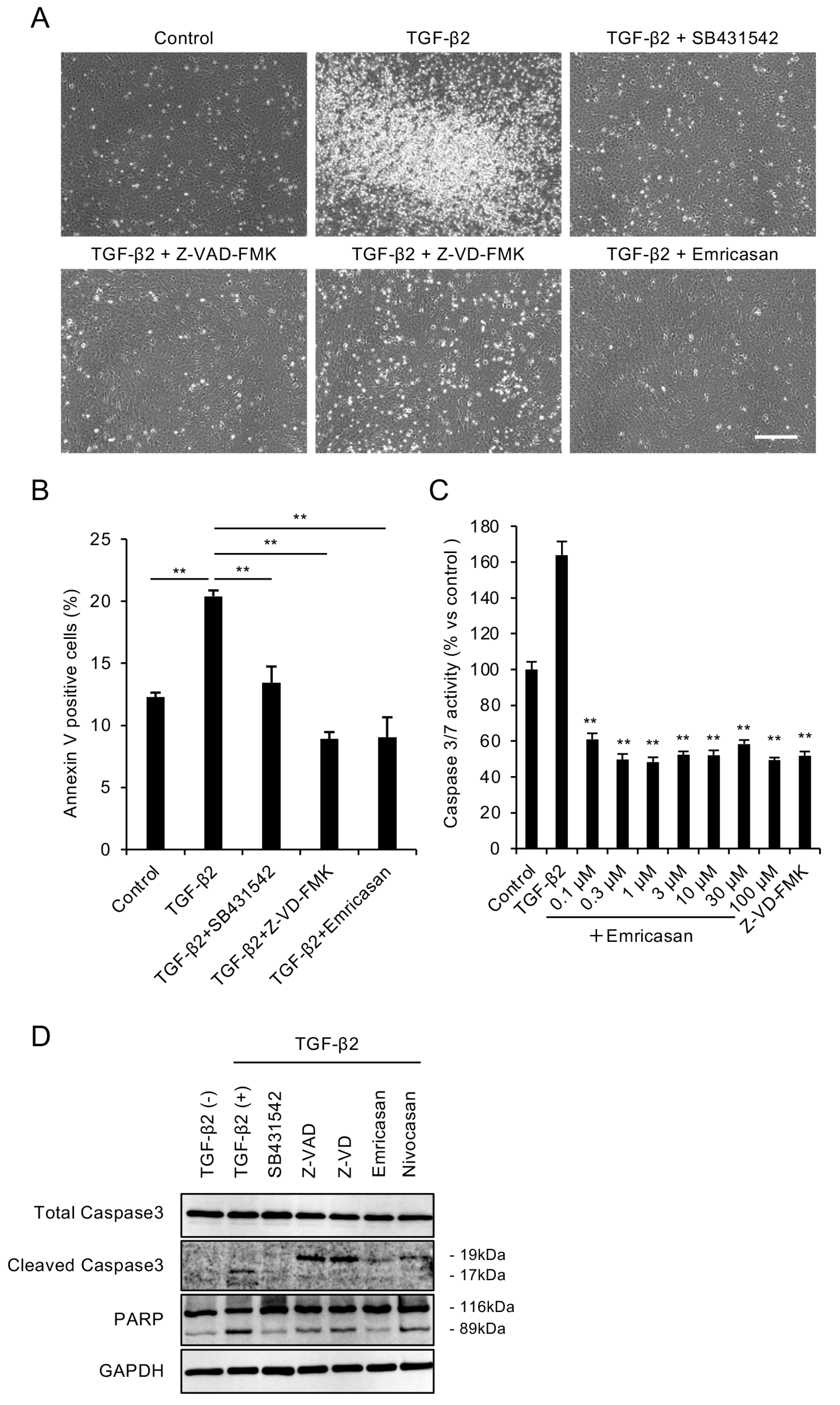
3.1. Caspase Inhibitors Attenuate TGF-β2-Induced Apoptosis in an FECD Cell Model
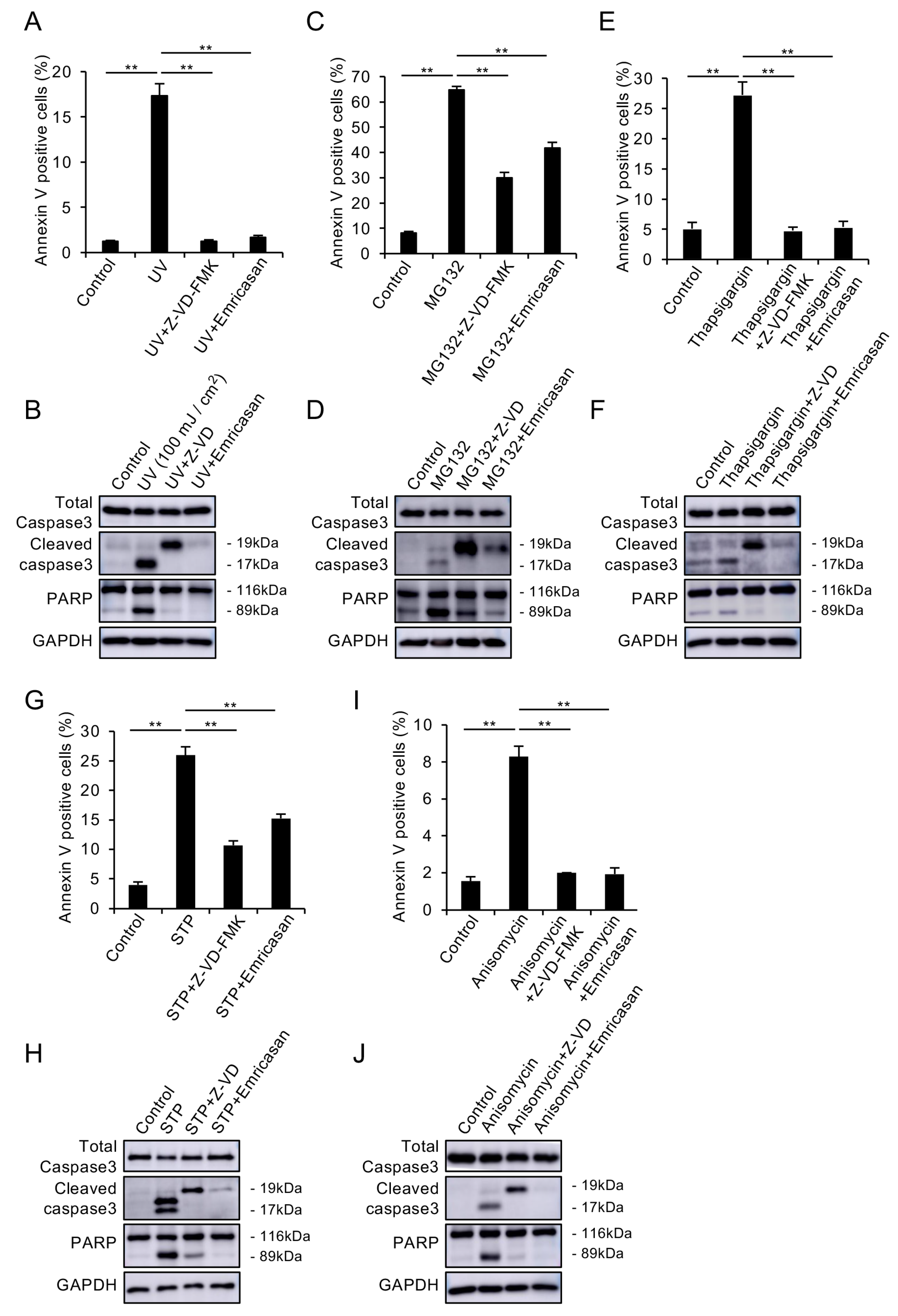
3.2. Caspase Inhibitors Suppress Various Stress-Induced Apoptosis in CECs
3.3. Emricasan Suppresses TGF-β2-Induced ECM Production and EMT
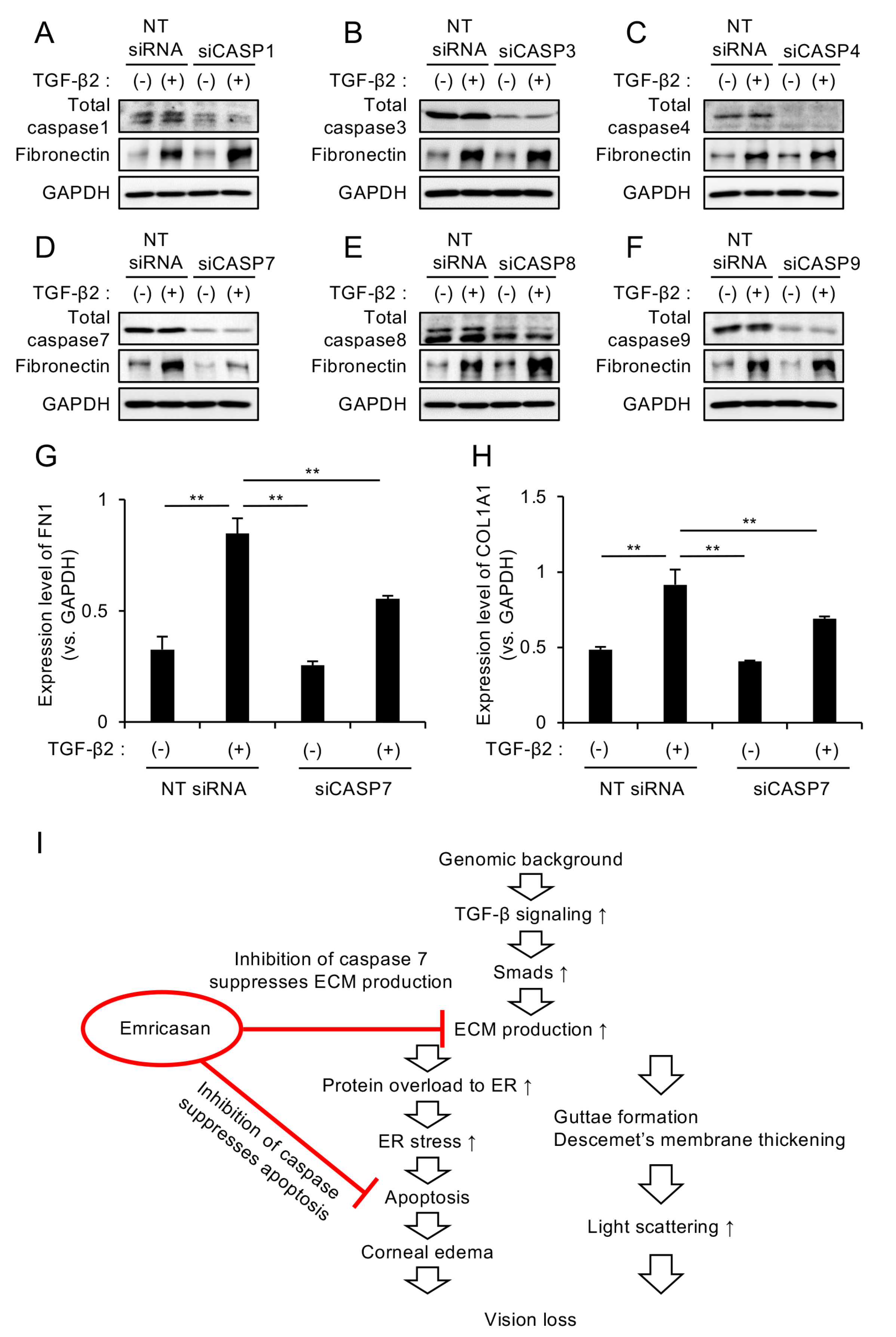
3.4. Caspase-7 Knockdown Specifically Suppresses TGF-β2-Induced ECM Expression
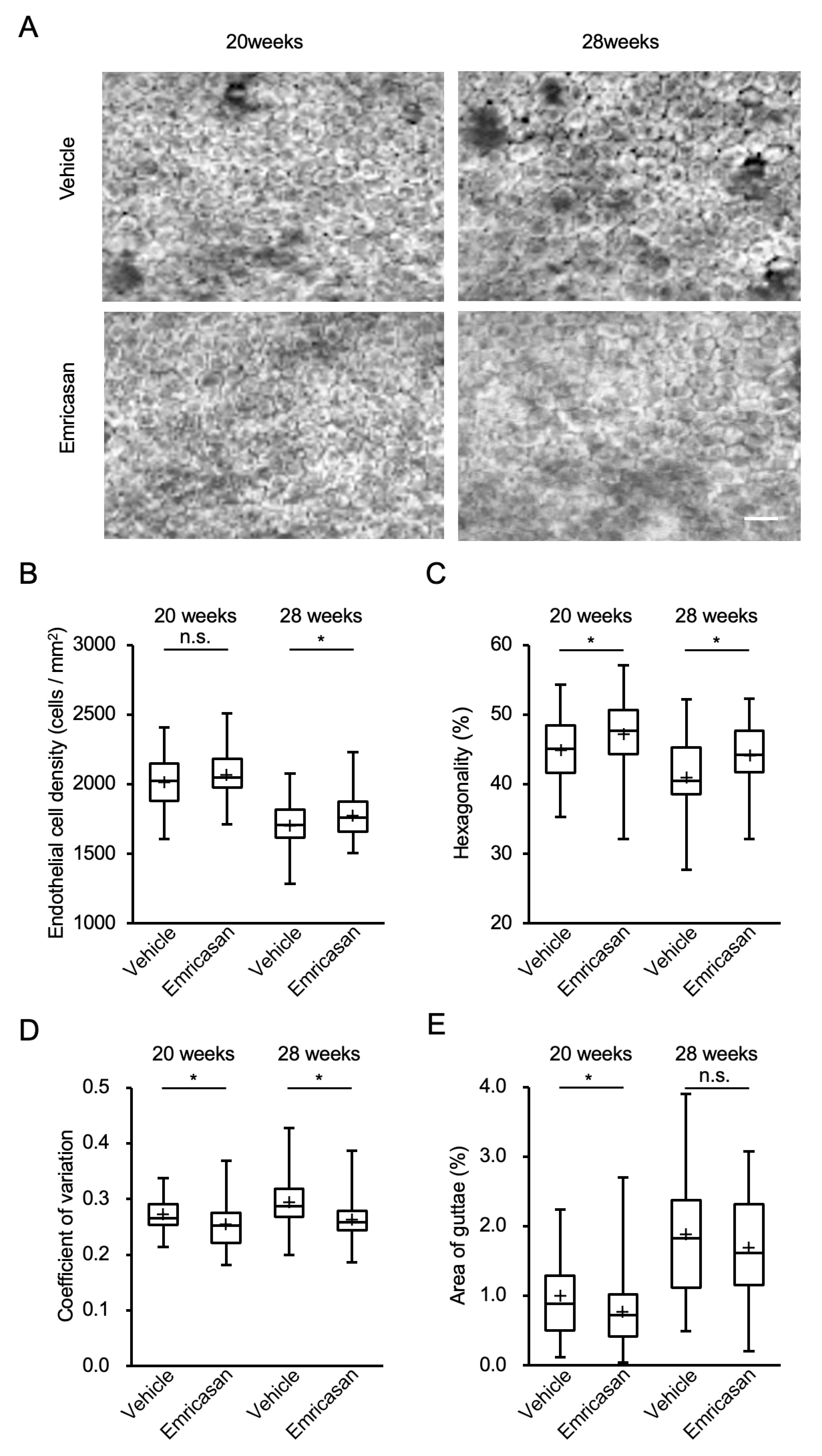
3.5. Effects of Topical Emricasan Administration on FECD Phenotype in Col8a2Q455K/Q455K Mice
3.6. Effects of Topical Emricasan Administration on Gene Expression in Corneal Endothelium of Col8a2Q455K/Q455K Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Fuchs Corneal Dystrophy. Prog. Mol. Biol. Transl. Sci. 2015, 134, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Fautsch, M.P.; Wieben, E.D.; Baratz, K.H.; Bhattacharyya, N.; Sadan, A.N.; Hafford-Tear, N.J.; Tuft, S.J.; Davidson, A.E. TCF4-mediated Fuchs endothelial corneal dystrophy: Insights into a common trinucleotide repeat-associated disease. Prog. Retin. Eye Res. 2021, 81, 100883. [Google Scholar] [CrossRef] [PubMed]
- Ong Tone, S.; Kocaba, V.; Bohm, M.; Wylegala, A.; White, T.L.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Prog. Retin. Eye Res. 2021, 80, 100863. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Oie, Y.; Fujimoto, H.; Soma, T.; Koh, S.; Tsujikawa, M.; Maeda, N.; Nishida, K. Relationship between Corneal Guttae and Quality of Vision in Patients with Mild Fuchs’ Endothelial Corneal Dystrophy. Ophthalmology 2015, 122, 2103–2109. [Google Scholar] [CrossRef]
- Wacker, K.; McLaren, J.W.; Amin, S.R.; Baratz, K.H.; Patel, S.V. Corneal High-Order Aberrations and Backscatter in Fuchs’ Endothelial Corneal Dystrophy. Ophthalmology 2015, 122, 1645–1652. [Google Scholar] [CrossRef]
- Patel, S.V.; Diehl, N.N.; Hodge, D.O.; Bourne, W.M. Donor risk factors for graft failure in a 20-year study of penetrating keratoplasty. Arch. Ophthalmol. 2010, 128, 418–425. [Google Scholar] [CrossRef]
- Patel, S.V. Graft survival and endothelial outcomes in the new era of endothelial keratoplasty. Exp. Eye Res. 2012, 95, 40–47. [Google Scholar] [CrossRef]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef]
- Hori, J.; Yamaguchi, T.; Keino, H.; Hamrah, P.; Maruyama, K. Immune privilege in corneal transplantation. Prog. Retin. Eye Res. 2019, 72, 100758. [Google Scholar] [CrossRef]
- Hos, D.; Matthaei, M.; Bock, F.; Maruyama, K.; Notara, M.; Clahsen, T.; Hou, Y.; Le, V.N.H.; Salabarria, A.C.; Horstmann, J.; et al. Immune reactions after modern lamellar (DALK, DSAEK, DMEK) versus conventional penetrating corneal transplantation. Prog. Retin. Eye Res. 2019, 73, 100768. [Google Scholar] [CrossRef]
- Jurkunas, U.V.; Bitar, M.S.; Funaki, T.; Azizi, B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am. J. Pathol. 2010, 177, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Azizi, B.; Ziaei, A.; Fuchsluger, T.; Schmedt, T.; Chen, Y.; Jurkunas, U.V. p53-regulated increase in oxidative-stress--induced apoptosis in Fuchs endothelial corneal dystrophy: A native tissue model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9291–9297. [Google Scholar] [CrossRef]
- Engler, C.; Kelliher, C.; Spitze, A.R.; Speck, C.L.; Eberhart, C.G.; Jun, A.S. Unfolded protein response in fuchs endothelial corneal dystrophy: A unifying pathogenic pathway? Am. J. Ophthalmol. 2010, 149, 194–202.e2. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Kitahara, M.; Okuda, H.; Hashimoto, K.; Ueda, E.; Nakahara, M.; Kinoshita, S.; Young, R.D.; Quantock, A.J.; Tourtas, T.; et al. Sustained Activation of the Unfolded Protein Response Induces Cell Death in Fuchs’ Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3697–3707. [Google Scholar] [CrossRef]
- Okumura, N.; Hashimoto, K.; Kitahara, M.; Okuda, H.; Ueda, E.; Watanabe, K.; Nakahara, M.; Sato, T.; Kinoshita, S.; Tourtas, T.; et al. Activation of TGF-β signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6801. [Google Scholar] [CrossRef]
- Kumar, V.; Jurkunas, U.V. Mitochondrial Dysfunction and Mitophagy in Fuchs Endothelial Corneal Dystrophy. Cells 2021, 10, 1888. [Google Scholar] [CrossRef]
- Kocaba, V.; Katikireddy, K.R.; Gipson, I.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Association of the Gutta-Induced Microenvironment With Corneal Endothelial Cell Behavior and Demise in Fuchs Endothelial Corneal Dystrophy. JAMA Ophthalmol. 2018, 136, 886–892. [Google Scholar] [CrossRef]
- Fadeel, B.; Orrenius, S.; Zhivotovsky, B. Apoptosis in human disease: A new skin for the old ceremony? Biochem. Biophys. Res. Commun. 1999, 266, 699–717. [Google Scholar]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Solary, E.; Dubrez, L.; Eymin, B. The role of apoptosis in the pathogenesis and treatment of diseases. Eur. Respir. J. 1996, 9, 1293–1305. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [PubMed]
- Zhang, W.; Wu, H.; Liao, Y.; Zhu, C.; Zou, Z. Caspase family in autoimmune diseases. Autoimmun. Rev. 2024, 24, 103714. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22. [Google Scholar]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar]
- Jan, R. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205. [Google Scholar]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar]
- Li, W.F.; Atalla, E.; Dong, J.; Konopleva, M. BCL2i-Based Therapies and Emerging Resistance in Chronic Lymphocytic Leukemia. Cells 2024, 13, 1922. [Google Scholar] [CrossRef]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar]
- Garcia-Tsao, G.; Fuchs, M.; Shiffman, M.; Borg, B.B.; Pyrsopoulos, N.; Shetty, K.; Gallegos-Orozco, J.F.; Reddy, K.R.; Feyssa, E.; Chan, J.L.; et al. Emricasan (IDN-6556) Lowers Portal Pressure in Patients With Compensated Cirrhosis and Severe Portal Hypertension. Hepatology 2019, 69, 717–728. [Google Scholar] [CrossRef]
- Frenette, C.T.; Morelli, G.; Shiffman, M.L.; Frederick, R.T.; Rubin, R.A.; Fallon, M.B.; Cheng, J.T.; Cave, M.; Khaderi, S.A.; Massoud, O. Emricasan improves liver function in patients with cirrhosis and high model for end-stage liver disease scores compared with placebo. Clin. Gastroenterol. Hepatol. 2019, 17, 774–783.e774. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.M.; Curry, M.P.; Frenette, C.T.; Regenstein, F.G.; Schiff, E.R.; Goodman, Z.D.; Robinson, J.M.; Chan, J.L.; Imperial, J.C.; Reddy, K.R. Multicenter, Double-Blind, Randomized Trial of Emricasan in Hepatitis C–Treated Liver Transplant Recipients With Residual Fibrosis or Cirrhosis. Liver Transplant. 2021, 27, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Jun, A.S.; Meng, H.; Ramanan, N.; Matthaei, M.; Chakravarti, S.; Bonshek, R.; Black, G.C.; Grebe, R.; Kimos, M. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum. Mol. Genet. 2012, 21, 384–393. [Google Scholar] [CrossRef]
- Meng, H.; Matthaei, M.; Ramanan, N.; Grebe, R.; Chakravarti, S.; Speck, C.L.; Kimos, M.; Vij, N.; Eberhart, C.G.; Jun, A.S. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1887–1897. [Google Scholar] [CrossRef]
- Dorado-Cortez, O.; Crouzet, E.; Trone, M.C.; Gain, P.; He, Z.; Vaitinadapoule, H.; Mentek, M.; Mascarelli, F.; Poinard, S.; Yasunaga, M.; et al. Change in Visual Acuity of Patients With Fuchs Endothelial Corneal Dystrophy Over 1 Year. Cornea 2024, 43, 1207–1215. [Google Scholar] [CrossRef]
- Okumura, N.; Kakutani, K.; Numata, R.; Nakahara, M.; Schlötzer-Schrehardt, U.; Kruse, F.; Kinoshita, S.; Koizumi, N. Laminin-511 and -521 enable efficient in vitro expansion of human corneal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2933–2942. [Google Scholar] [CrossRef]
- Nakahara, M.; Okumura, N.; Nakano, S.; Koizumi, N. Effect of a p38 Mitogen-Activated Protein Kinase Inhibitor on Corneal Endothelial Cell Proliferation. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4218–4227. [Google Scholar] [CrossRef]
- Okumura, N.; Minamiyama, R.; Ho, L.T.; Kay, E.P.; Kawasaki, S.; Tourtas, T.; Schlötzer-Schrehardt, U.; Kruse, F.E.; Young, R.D.; Quantock, A.J.; et al. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Investig. A J. Tech. Methods Pathol. 2015, 95, 1291–1304. [Google Scholar] [CrossRef]
- Okumura, N.; Yamada, S.; Nishikawa, T.; Narimoto, K.; Okamura, K.; Izumi, A.; Hiwa, S.; Hiroyasu, T.; Koizumi, N. U-Net Convolutional Neural Network for Segmenting the Corneal Endothelium in a Mouse Model of Fuchs Endothelial Corneal Dystrophy. Cornea 2022, 41, 901–907. [Google Scholar] [CrossRef]
- Brown, M.; Lowe, D.G. Automatic panoramic image stitching using invariant features. Int. J. Comput. Vis. 2007, 74, 59–73. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Weller, J.M.; Zenkel, M.; Schlotzer-Schrehardt, U.; Bachmann, B.O.; Tourtas, T.; Kruse, F.E. Extracellular matrix alterations in late-onset Fuchs’ corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3700–3708. [Google Scholar] [CrossRef]
- Tchatchouang, A.; Brunette, I.; Rochette, P.J.; Proulx, S. Expression and Impact of Fibronectin, Tenascin-C, Osteopontin, and Type XIV Collagen in Fuchs Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2024, 65, 38. [Google Scholar] [CrossRef]
- Katikireddy, K.R.; White, T.L.; Miyajima, T.; Vasanth, S.; Raoof, D.; Chen, Y.; Price, M.O.; Price, F.W.; Jurkunas, U.V. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free Radic. Biol. Med. 2018, 116, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [PubMed]
- Wilson, C.H.; Kumar, S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ. 2018, 25, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Manicardi, N.; Ortega-Ribera, M.; Maeso-Diaz, R.; Guixe-Muntet, S.; Fernandez-Iglesias, A.; Hide, D.; Garcia-Caldero, H.; Boyer-Diaz, Z.; Contreras, P.C.; et al. Emricasan Ameliorates Portal Hypertension and Liver Fibrosis in Cirrhotic Rats Through a Hepatocyte-Mediated Paracrine Mechanism. Hepatol. Commun. 2019, 3, 987–1000. [Google Scholar] [CrossRef]
- Chaintreuil, P.; Laplane, L.; Esnault, F.; Ghesquier, V.; Savy, C.; Furstoss, N.; Arcangeli, M.-L.; Cluzeau, T.; Robert, G.; Droin, N. Reprogramming monocyte-derived macrophages through caspase inhibition. Oncoimmunology 2022, 11, 2015859. [Google Scholar]
- Borderie, V.M.; Baudrimont, M.; Vallee, A.; Ereau, T.L.; Gray, F.; Laroche, L. Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2501–2505. [Google Scholar]
- Li, Q.J.; Ashraf, M.F.; Shen, D.; Green, W.R.; Stark, W.J.; Chan, C.-C.; O’Brien, T.P. The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Arch. Ophthalmol. 2001, 119, 1597–1604. [Google Scholar] [CrossRef]
- Halilovic, A.; Schmedt, T.; Benischke, A.S.; Hamill, C.; Chen, Y.; Santos, J.H.; Jurkunas, U.V. Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy. Antioxid. Redox Signal. 2016, 24, 1072–1083. [Google Scholar] [CrossRef]
- Bitar, M.S.; Liu, C.; Ziaei, A.; Chen, Y.; Schmedt, T.; Jurkunas, U.V. Decline in DJ-1 and decreased nuclear translocation of Nrf2 in Fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5806–5813. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Y.; Kochevar, I.E.; Jurkunas, U.V. Decreased DJ-1 leads to impaired Nrf2-regulated antioxidant defense and increased UV-A–induced apoptosis in corneal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5551–5560. [Google Scholar]
- Ke, Y.; Tang, H.; Ye, C.; Lei, C.-T.; Wang, Y.-M.; Su, H.; Zhang, C.; Jiang, H.-J. Role and association of inflammatory and apoptotic caspases in renal tubulointerstitial fibrosis. Kidney Blood Press. Res. 2016, 41, 643–653. [Google Scholar] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [PubMed]
- Kuranaga, E. Beyond apoptosis: Caspase regulatory mechanisms and functions in vivo. Genes Cells 2012, 17, 83–97. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimoto, S.; Endo, M.; Tonomura, S.; Tsuji, F.; Haraguchi, H.; Hasegawa, K.; Numao, T.; Izumi, A.; Tourtas, T.; Schlötzer-Schrehardt, U.; et al. Therapeutic Potential of Emricasan, a Pan-Caspase Inhibitor, in Reducing Cell Death and Extracellular Matrix Accumulation in Fuchs Endothelial Corneal Dystrophy. Cells 2025, 14, 498. https://doi.org/10.3390/cells14070498
Fujimoto S, Endo M, Tonomura S, Tsuji F, Haraguchi H, Hasegawa K, Numao T, Izumi A, Tourtas T, Schlötzer-Schrehardt U, et al. Therapeutic Potential of Emricasan, a Pan-Caspase Inhibitor, in Reducing Cell Death and Extracellular Matrix Accumulation in Fuchs Endothelial Corneal Dystrophy. Cells. 2025; 14(7):498. https://doi.org/10.3390/cells14070498
Chicago/Turabian StyleFujimoto, Sohya, Mako Endo, Shigehito Tonomura, Fuuga Tsuji, Hirotaka Haraguchi, Kanna Hasegawa, Taisuke Numao, Ayaka Izumi, Theofilos Tourtas, Ursula Schlötzer-Schrehardt, and et al. 2025. "Therapeutic Potential of Emricasan, a Pan-Caspase Inhibitor, in Reducing Cell Death and Extracellular Matrix Accumulation in Fuchs Endothelial Corneal Dystrophy" Cells 14, no. 7: 498. https://doi.org/10.3390/cells14070498
APA StyleFujimoto, S., Endo, M., Tonomura, S., Tsuji, F., Haraguchi, H., Hasegawa, K., Numao, T., Izumi, A., Tourtas, T., Schlötzer-Schrehardt, U., Kruse, F., Oyama, Y., Ikawa, M., Jun, A. S., Koizumi, N., & Okumura, N. (2025). Therapeutic Potential of Emricasan, a Pan-Caspase Inhibitor, in Reducing Cell Death and Extracellular Matrix Accumulation in Fuchs Endothelial Corneal Dystrophy. Cells, 14(7), 498. https://doi.org/10.3390/cells14070498