
β-Arrestin 2 as a Prognostic Indicator and Immunomodulatory Factor in Multiple Myeloma
 ,
,  ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Animal and Clinical Study Approval
2.2. Clinical Data Collection and Analysis
2.3. Gene Expression Profile and Clinical Data
2.4. Reagents
2.5. Immunofluorescent Staining of Bone Marrow ARRB2 and Quantification
2.6. ARRB2 Knockout Mice
2.7. Isolation, Activation, and In Vitro Culture of Murine T Cells and MDSC Cells
2.8. Flow Cytometry Analysis
2.9. Immunoblotting
2.10. siRNA Knockdown
2.11. Statistical Analysis
3. Results
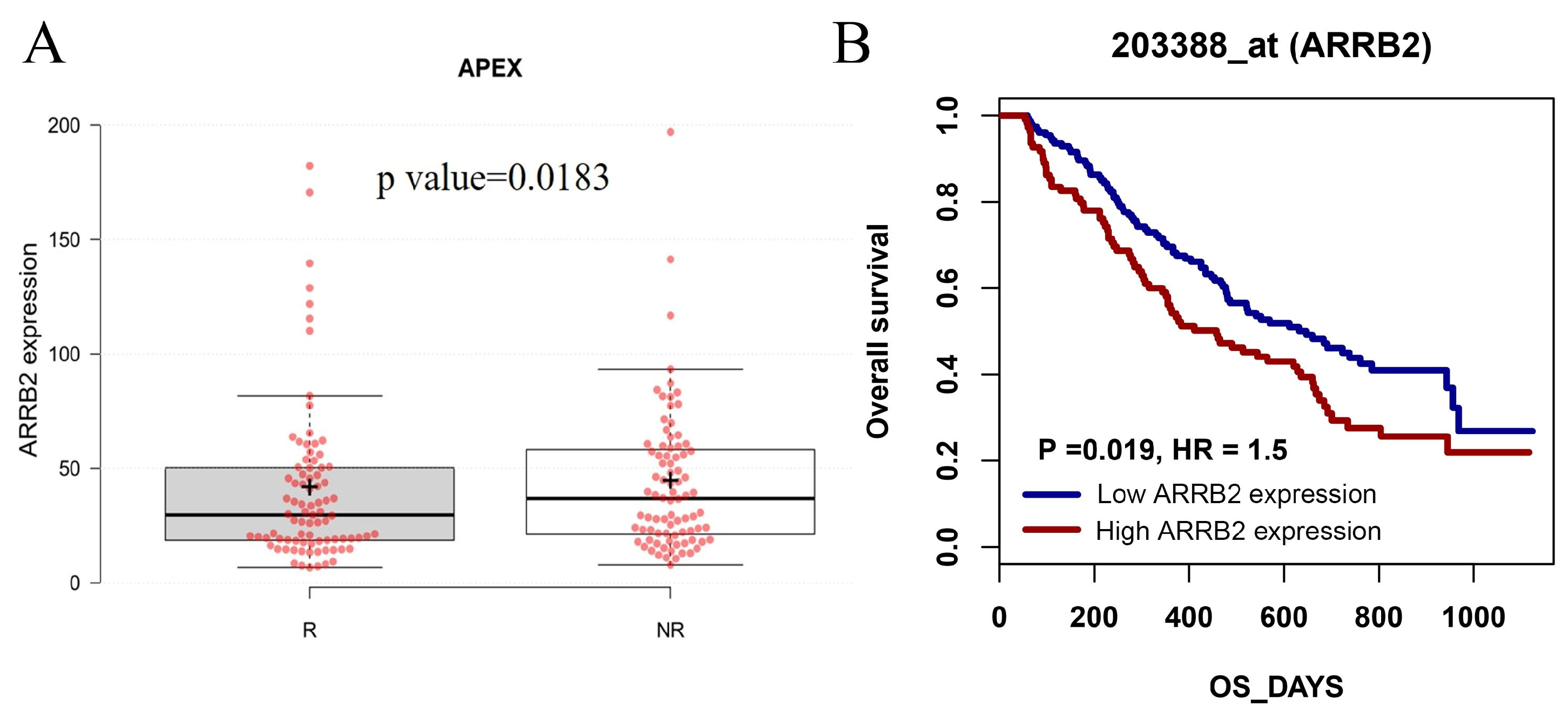
3.1. ARRB2 Expression Is Upregulated in Patients with MM, and ARRB2 Overexpression Is Associated with Poor Survival: An In-Silico Analysis
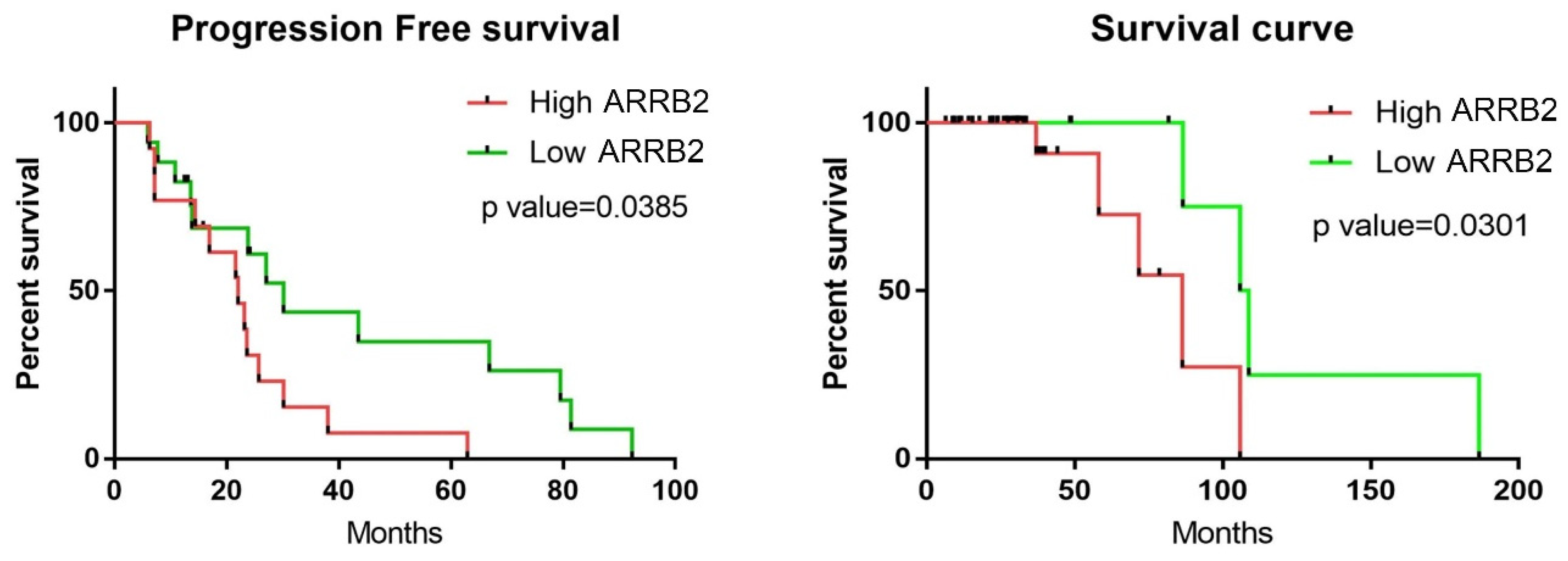
3.2. Higher ARRB2 Expression Correlates with Disease Progression and Poor Survival in Newly Diagnosed Myeloma (NDMM) Patients
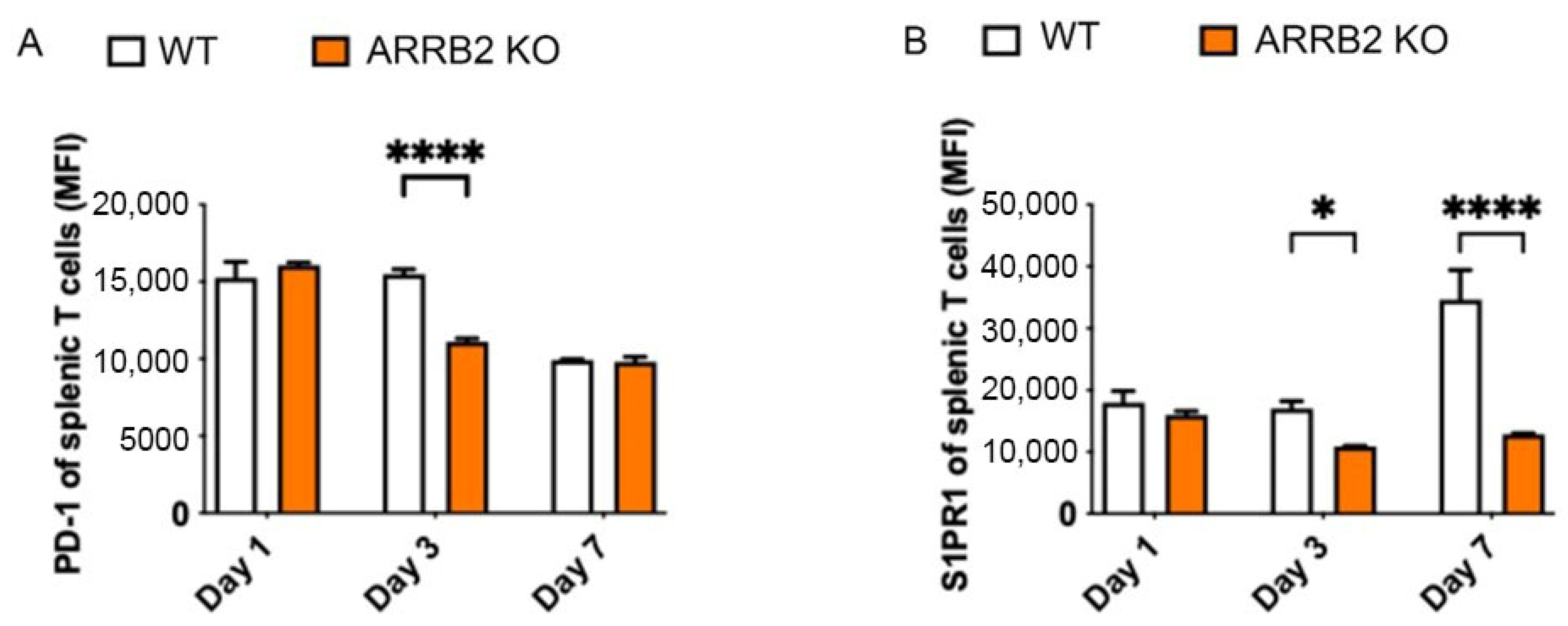
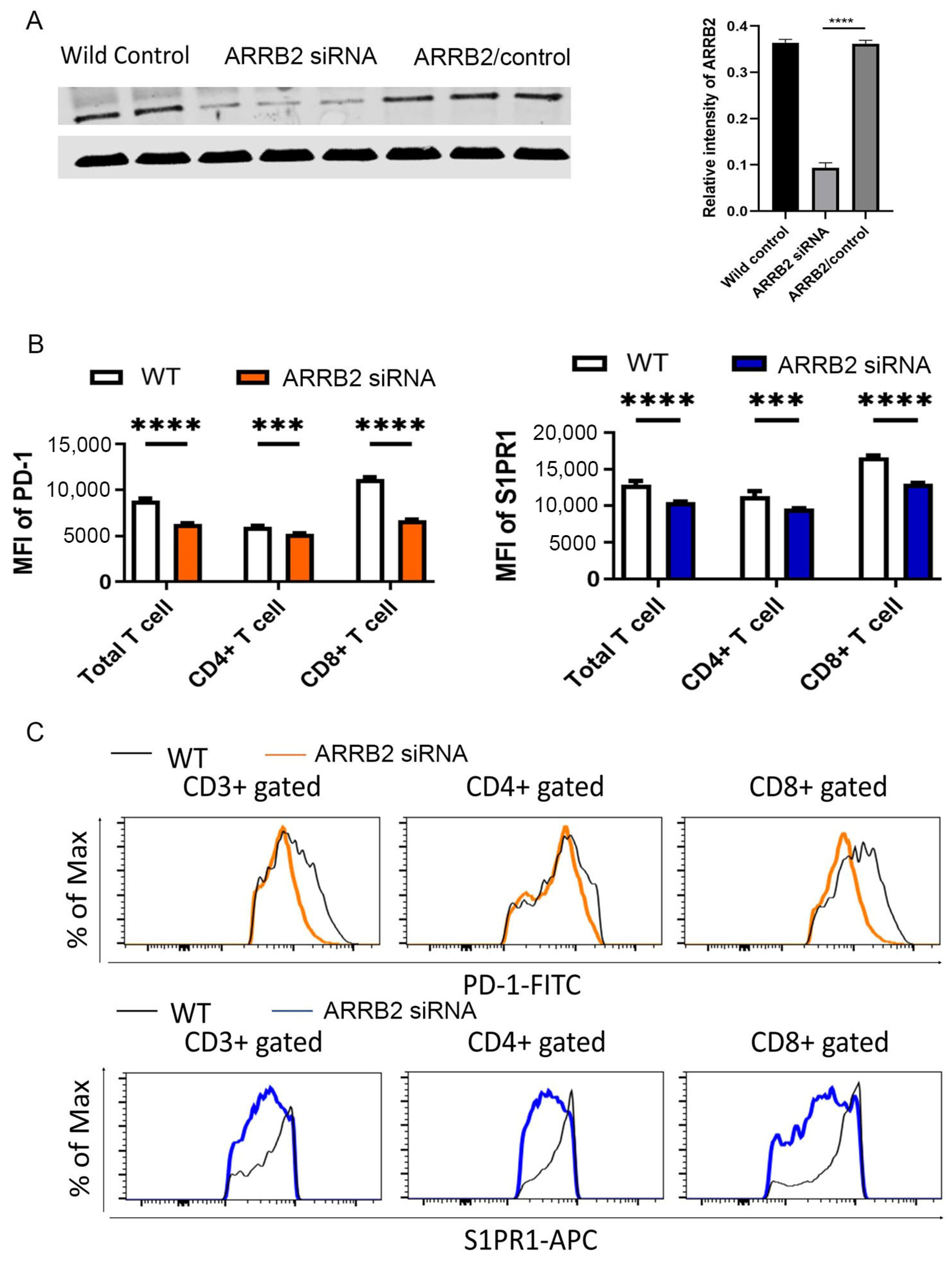
3.3. ARRB2 Is Involved in the Regulation of PD-1 Expression in T Cells
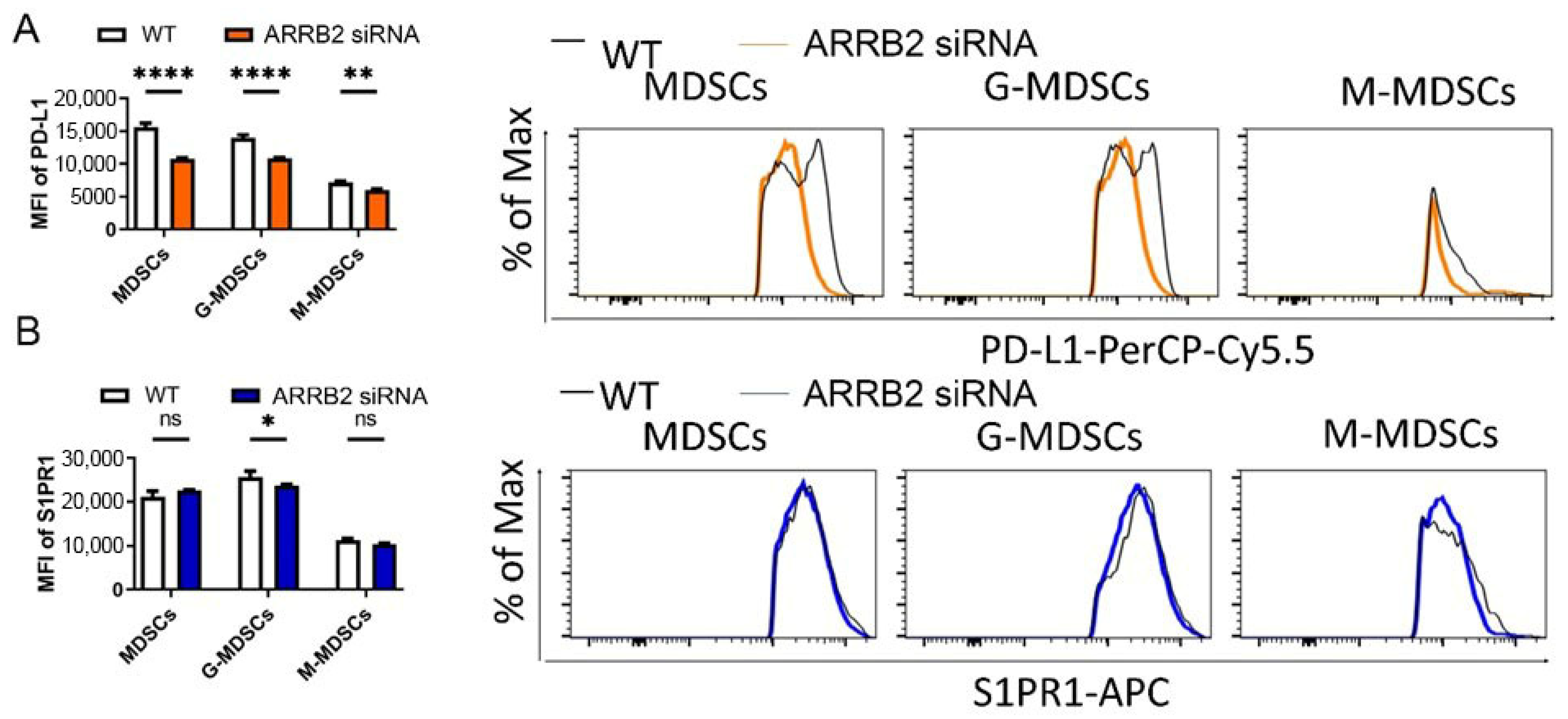
3.4. ARRB2 Regulates PD-L1 Expression in Myeloid-Derived Suppressor Cells (MDSCs)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MM | Multiple myeloma. |
| GPCRs | G protein-coupled receptors. |
| ARRB2 | β-arrestin 2. |
| PD-1 | programmed cell death protein-1. |
| PD-L1 | programmed death-ligand 1. |
| MDSC | Myeloid-derived suppressor cells. |
| BMBX | Bone marrow biopsy. |
| NDMM | Newly diagnosed multiple myeloma. |
References
- Cytryn, R.; Bickell, N.; Yagnik, R.; Jagannath, S.; Lin, J.J. What Affects Treatment Underuse in Multiple Myeloma in the United States: A Qualitative Study. Cancers 2023, 15, 2369. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.; Kang, Y. Chimeric antigen receptor (CAR) T-cell therapy for multiple myeloma. Pharmacol. Ther. 2022, 232, 108007. [Google Scholar] [PubMed]
- Chifotides, H.T.; Masarova, L.; Verstovsek, S. SOHO State of the Art Updates and Next Questions: Novel Therapeutic Strategies in Development for Myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2023, 23, 219–231. [Google Scholar]
- Feinberg, D.; Paul, B.; Kang, Y. The promise of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Cell Immunol. 2019, 345, 103964. [Google Scholar] [PubMed]
- Habrowska-Górczyńska, D.E.; Kozieł, M.J.; Kowalska, K.; Piastowska-Ciesielska, A.W. FOXO3a and Its Regulators in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 12530. [Google Scholar] [CrossRef]
- Botticelli, E.; Salazar Intriago, M.S.; Piovesana, R.; Tata, A.M. Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology. Life 2022, 12, 211. [Google Scholar] [CrossRef]
- SD, A.; Pasumarthi, D.; Pasha, A.; Doneti, R.; Botlagunta, M.; Pawar, S.C. Identification of Differentially Expressed Genes in Cervical Cancer Patients by Comparative Transcriptome Analysis. Biomed. Res. Int. 2021, 2021, 8810074. [Google Scholar]
- Liu, X.; Zhao, X.; Zeng, X.; Bossers, K.; Swaab, D.F.; Zhao, J.; Pei, G. beta-arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res. 2013, 23, 351–365. [Google Scholar]
- Lee, M.-H.; Appleton, K.M.; Strungs, E.G.; Kwon, J.Y.; Morinelli, T.A.; Peterson, Y.K.; Laporte, S.A.; Luttrell, L.M. The conformational signature of beta-arrestin2 predicts its trafficking and signalling functions. Nature 2016, 531, 665–668. [Google Scholar]
- Melkes, B.; Markova, V.; Hejnova, L.; Novotny, J. beta-Arrestin 2 and ERK1/2 Are Important Mediators Engaged in Close Cooperation between TRPV1 and micro-Opioid Receptors in the Plasma Membrane. Int. J. Mol. Sci. 2020, 21, 4626. [Google Scholar]
- Fletcher-Jones, A.; Hildick, K.L.; Evans, A.J.; Nakamura, Y.; Henley, J.M.; Wilkinson, K.A. Protein Interactors and Trafficking Pathways That Regulate the Cannabinoid Type 1 Receptor (CB1R). Front. Mol. Neurosci. 2020, 13, 108. [Google Scholar]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through beta-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Viswanathan, G.; Sellig, M.; Jassal, C.; Choi, I.; Garikipati, A.; Xiong, X.; Nazo, N.; Rajagopal, S. beta-Arrestin-Mediated Angiotensin II Type 1 Receptor Activation Promotes Pulmonary Vascular Remodeling in Pulmonary Hypertension. JACC Basic Transl. Sci. 2021, 6, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, B.; Partenheimer, A.; Jeschke, U.; von Schönfeldt, V.; Mayr, D.; Mahner, S.; Burges, A.; Simoni, M.; Melli, B.; Benevelli, R.; et al. beta-arrestin 2 Is a Prognostic Factor for Survival of Ovarian Cancer Patients Upregulating Cell Proliferation. Front. Endocrinol. 2020, 11, 554733. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Song, S.P. beta-Arrestin 1/2 Aggravates Podocyte Apoptosis of Diabetic Nephropathy via Wnt/beta-Catenin Pathway. Med. Sci. Monit. 2018, 24, 1724–1732. [Google Scholar] [CrossRef]
- Yin, D.; Yang, X.; Li, H.; Fan, H.; Zhang, X.; Feng, Y.; Stuart, C.; Hu, D.; Caudle, Y.; Xie, N.; et al. beta-Arrestin 2 Promotes Hepatocyte Apoptosis by Inhibiting Akt Protein. J. Biol. Chem. 2016, 291, 605–612. [Google Scholar] [CrossRef]
- Gentilini, A.; Caligiuri, A.; Raggi, C.; Rombouts, K.; Pinzani, M.; Lori, G.; Correnti, M.; Invernizzi, P.; Rovida, E.; Navari, N.; et al. CXCR7 contributes to the aggressive phenotype of cholangiocarcinoma cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2246–2256. [Google Scholar] [CrossRef]
- Zhou, J.; Zhu, Z.; Bai, C.; Sun, H.; Wang, X. Proteomic profiling of lymphocytes in autoimmunity, inflammation and cancer. J. Transl. Med. 2014, 12, 6. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Nishida, M.; Kim, K.-M.; Kurose, H. beta-arrestin2 in infiltrated macrophages inhibits excessive inflammation after myocardial infarction. PLoS ONE 2013, 8, e68351. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Li, L.; Qian, G.; Wang, Y.; Chen, Z.; Liu, J.; Fang, C.; Huang, F.; Guo, D.; et al. beta-arrestin 2 as an activator of cGAS-STING signaling and target of viral immune evasion. Nat. Commun. 2020, 11, 6000. [Google Scholar] [CrossRef]
- Rosanò, L.; Cianfrocca, R.; Masi, S.; Spinella, F.; Di Castro, V.; Biroccio, A.; Salvati, E.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Beta-arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2806–2811. [Google Scholar] [CrossRef] [PubMed]
- Masannat, J.; Purayil, H.T.; Zhang, Y.; Russin, M.; Mahmud, I.; Kim, W.; Liao, D.; Daaka, Y. βArrestin2 Mediates Renal Cell Carcinoma Tumor Growth. Sci. Rep. 2018, 8, 4879. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-Y.; Hu, S.-S.; Wu, J.-J.; Huang, Q.; Ma, Y.; Wang, Q.-T.; Chen, J.-Y.; Wei, W. Down-regulation of β-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 2016, 6, 35609. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Ji, Q.; Li, Q. The role and mechanism of beta-arrestins in cancer invasion and metastasis (Review). Int. J. Mol. Med. 2018, 41, 631–639. [Google Scholar]
- Oakley, R.H.; Revollo, J.; Cidlowski, J.A. Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 17591–17596. [Google Scholar]
- Shintani, Y.; Hayata-Takano, A.; Moriguchi, K.; Nakazawa, T.; Ago, Y.; Kasai, A.; Seiriki, K.; Shintani, N.; Hashimoto, H. beta-Arrestin1 and 2 differentially regulate PACAP-induced PAC1 receptor signaling and trafficking. PLoS ONE 2018, 13, e0196946. [Google Scholar] [CrossRef]
- Rein, L.A.; Wisler, J.W.; Kim, J.; Theriot, B.; Huang, L.; Price, T.; Yang, H.; Chen, M.; Chen, W.; Sipkins, D.; et al. beta-Arrestin2 mediates progression of murine primary myelofibrosis. JCI Insight 2017, 2, e98094. [Google Scholar] [CrossRef]
- Paul, B.; Kang, S.; Zheng, Z.; Kang, Y. The challenges of checkpoint inhibition in the treatment of multiple myeloma. Cell Immunol. 2018, 334, 87–98. [Google Scholar] [CrossRef]
- Liu, D.; Wen, C.; Chen, L.; Ye, M.; Liu, H.; Sun, X.; Liang, L.; Zhang, J.; Chang, S.; Liu, J. The emerging roles of PD-L1 subcellular localization in tumor immune evasion. Biochem. Pharmacol. 2024, 220, 115984. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Verzoni, E.; Grassi, P.; Ratta, R. Chapter 38—Emerging Immunotherapies for Renal Cell Carcinoma. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 541–549. [Google Scholar]
- Bae, J.; Nguyen, B.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Munshi, N.C.; Anderson, K.C. Function and expression of checkpoint inhibitors and immune agonists on immune cells in monoclonal gammopathy of undetermined significance (MGUS), smoldering multiple myeloma (SMM) and MM and tumor-specific T lymphocytes. J. Clin. Oncol. 2017, 35, 11577. [Google Scholar]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [PubMed]
- Menon, S.; Shin, S.; Dy, G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar]
- Mateos, M.-V.; Blacklock, H.; Schjesvold, F.; Rocafiguera, A.O.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Sherbenou, D.W.; Avivi, I.; et al. A phase 3 randomized study of pembrolizumab (Pembro) plus pomalidomide (Pom) and dexamethasone (Dex) for relapsed/refractory multiple myeloma (RRMM): KEYNOTE-183. J. Clin. Oncol. 2018, 36, 8021. [Google Scholar]
- Kyle, R.A.; Rajkumar, S.V. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009, 23, 3–9. [Google Scholar]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar]
- Greipp, P.R.; Miguel, J.S.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [PubMed]
- Munshi, N.C.; Anderson, K.C.; Bergsagel, P.L.; Shaughnessy, J.; Palumbo, A.; Durie, B.; Fonseca, R.; Stewart, A.K.; Harousseau, J.-L.; Dimopoulos, M.; et al. Consensus recommendations for risk stratification in multiple myeloma: Report of the International Myeloma Workshop Consensus Panel 2. Blood 2011, 117, 4696–4700. [Google Scholar] [PubMed]
- Bond, R.A.; Garcia-Rojas, E.Y.L.; Hegde, A.; Walker, J.K.L. Therapeutic Potential of Targeting ss-Arrestin. Front. Pharmacol. 2019, 10, 124. [Google Scholar]
- Kohout, T.A.; Lin, F.T.; Perry, S.J.; Conner, D.A.; Lefkowitz, R.J. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. USA 2001, 98, 1601–1606. [Google Scholar]
- Mohammadpour, H.; MacDonald, C.R.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A. β2-adrenergic receptor signaling regulates metabolic pathways critical to myeloid-derived suppressor cell function within the TME. Cell Rep. 2021, 37, 109883. [Google Scholar]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar]
- Wu, L.; Jiang, C.; Zhu, Z.; Sun, Y.; Zhang, T. Prognostic role of PD-L1 expression in patients with salivary gland carcinoma: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0272080. [Google Scholar]
- Ota, T.; Fukui, T.; Nakahara, Y.; Takeda, T.; Uchino, J.; Mouri, T.; Kudo, K.; Nakajima, S.; Suzumura, T.; Fukuoka, M. Serum immune modulators during the first cycle of anti-PD-1 antibody therapy in non-small cell lung cancer: Perforin as a biomarker. Thorac. Cancer 2020, 11, 3223–3233. [Google Scholar]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar]
- Singh, S.; Barik, D.; Arukha, A.P.; Prasad, S.; Mohapatra, I.; Singh, A.; Singh, G. Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment. Biomedicines 2023, 11, 2621. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, H.; Jihu, R.; Zhou, J.; Zeng, R.; Yan, H. Novel Characterization of Myeloid-Derived Suppressor Cells in Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 698532. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liang, H.L.; Yu, X.; Liu, Z.; Cao, X.; Rao, E.; Huang, X.; Wang, L.; Li, L.; Bugno, J.; et al. Radiotherapy and immunotherapy converge on elimination of tumor-promoting erythroid progenitor cells through adaptive immunity. Sci. Transl. Med. 2021, 13, eabb0130. [Google Scholar] [CrossRef] [PubMed]
- Wess, J.; Oteng, A.B.; Rivera-Gonzalez, O.; Gurevich, E.V.; Gurevich, V.V. beta-Arrestins: Structure, Function, Physiology, and Pharmacological Perspectives. Pharmacol. Rev. 2023, 75, 854–884. [Google Scholar]
- Bai, Z.; Shi, E.; Wang, Q.; Dong, Z.; Xu, P. A potential panel of two-long non-coding RNA signature to predict recurrence of patients with laryngeal cancer. Oncotarget 2017, 8, 69641–69650. [Google Scholar]
- Santhanam, B.; Sluter, M.; Babu, M.M. Exploring GPCR signaling pathway networks as cancer therapeutic targets. Cell Genom. 2024, 4, 100560. [Google Scholar] [CrossRef]
- Wang, X.; Tan, X.; Zhang, J.; Wu, J.; Shi, H. The emerging roles of MAPK-AMPK in ferroptosis regulatory network. Cell Commun. Signal. 2023, 21, 200. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Zeller, M.; Cottin, Y.; Vergely, C. Antitumoral Activity of Molecular Hydrogen and Proton in the Treatment of Glioblastoma: An Atypical Pharmacology? Brain. Sci. 2023, 13, 1168. [Google Scholar] [CrossRef]
- Oprita, A.; Staicu, G.A.; Baloi, C.; Dricu, A.; Artene, S.A. The Effect of Beta-Arrestin2 Overexpression Regarding Viability and Temozolomide Treatment in High-Grade Glioma Cells. Curr. Health Sci. J. 2022, 48, 407–412. [Google Scholar]
- Wojciech, L.; Tan, K.S.W.; Gascoigne, N.R.J. Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells. Int. J. Mol. Sci. 2020, 21, 7740. [Google Scholar] [CrossRef]
- Kee, T.R.; Khan, S.A.; Neidhart, M.B.; Masters, B.M.; Zhao, V.K.; Kim, Y.K.; Percy, K.C.M.; Woo, J.-A.A. The multifaceted functions of β-arrestins and their therapeutic potential in neurodegenerative diseases. Exp. Mol. Med. 2024, 56, 129–141. [Google Scholar] [PubMed]
- Ahn, K.H.; Mahmoud, M.M.; Shim, J.-Y.; Kendall, D.A. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J. Biol. Chem. 2013, 288, 9790–9800. [Google Scholar] [PubMed]
- Haider, R.S.; Matthees, E.S.F.; Drube, J.; Reichel, M.; Zabel, U.; Inoue, A.; Chevigné, A.; Krasel, C.; Deupi, X.; Hoffmann, C. β-arrestin1 and 2 exhibit distinct phosphorylation-dependent conformations when coupling to the same GPCR in living cells. Nat. Commun. 2022, 13, 5638. [Google Scholar] [PubMed]
- Wess, J. The Two β-Arrestins Regulate Distinct Metabolic Processes: Studies with Novel Mutant Mouse Models. Int. J. Mol. Sci. 2022, 23, 495. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar]
- Richardson, P.G.; Jacobus, S.J.; Weller, E.A.; Hassoun, H.; Lonial, S.; Raje, N.S.; Medvedova, E.; McCarthy, P.L.; Libby, E.N.; Voorhees, P.M.; et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N. Engl. J. Med. 2022, 387, 132–147. [Google Scholar]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar]
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar]
- Lefkowitz, R.J.; Rajagopal, K.; Whalen, E.J. New Roles for β-Arrestins in Cell Signaling: Not Just for Seven-Transmembrane Receptors. Mol. Cell 2006, 24, 643–652. [Google Scholar]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [PubMed]
- Szenasi, T.; Turu, G.; Hunyady, L. Interactions between beta-arrestin proteins and the cytoskeletal system, and their relevance to neurodegenerative disorders. Front. Endocrinol. 2023, 14, 957981. [Google Scholar]
- Yoon, S.; Rossi, J.J. Aptamers: Uptake mechanisms and intracellular applications. Adv. Drug Deliv. Rev. 2018, 134, 22–35. [Google Scholar] [PubMed]
- Tan, Y.; Li, Y.; Tang, F. Nucleic Acid Aptamer: A Novel Potential Diagnostic and Therapeutic Tool for Leukemia. Onco Targets Ther. 2019, 12, 10597–10613. [Google Scholar]
- Abiraj, K.; Ursillo, S.; Tamma, M.L.; Rylova, S.N.; Waser, B.; Constable, E.C.; Fani, M.; Nicolas, G.P.; Reubi, J.C.; Maecke, H.R. The tetraamine chelator outperforms HYNIC in a new technetium-99m-labelled somatostatin receptor 2 antagonist. EJNMMI Res. 2018, 8, 75. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| High ARRB2 (%) (n = 25) | Low ARRB2 (%) (n = 22) | p-Value | |
|---|---|---|---|
| Mean Age | 59.8 | 55.9 | 0.25 |
| Gender | |||
| Male | 11 (44) | 11 (50) | 0.77 |
| Female | 14 (56) | 11 (50) | |
| Race | |||
| Caucasian | 15 (60) | 14 (63.6) | >0.99 |
| African American | 10 (40) | 8 (36.4) | |
| MM Subtype | |||
| IgA-K | 4 (16) | 3 (13.6) | 0.81 |
| IgA-L | 3 (12) | 5 (22.7) | |
| IgG-K | 11 (44) | 9 (40.9) | |
| IgG-L | 2 (8) | 4 (18.2) | |
| Lambda LC | 3 (12) | 1 (4.5) | |
| Kappa LC | 1 (4) | 0 | |
| Unknown/Biclonal | 1 (4) | 0 | |
| Cytogenetics | |||
| Standard risk | 19 (76) | 15 (68.2) | 0.89 |
| High risk | 3 (12) | 1 (4.5) | |
| intermediate | 2 (8) | 3 (13.6) | |
| unknown | 1 (4) | 3 (13.6) | |
| ISS stage | |||
| 1 | 4 (16) | 6 (27.3) | 0.78 |
| 2 | 5 (20) | 3 (13.6) | |
| 3 | 8 (32) | 7 (31.8) | |
| Unknown | 8 (32) | 6 (27.3) | |
| Response | 0.0293 | ||
| CR | 4 (16) | 6 (27.3) | |
| VGPR | 6 (24) | 4 (18.2) | |
| PR | 4 (16) | 8 (36.4) | |
| SD | 0 | 2 (9.1) | |
| PD | 6 (24) | 1 (4.5) | |
| Unknown | 5 (20) | 1 (4.5) | |
| IMiD | |||
| Lenalidomide | 19 (76) | 21 (95) | 0.64 |
| Pomalidomide | 2 (8) | 6 (27) | |
| Thalidomide | 2 (8) | 3 (13) | |
| Unknown | 5 (20) | 0 | |
| PI | |||
| Bortezomid | 20 (80) | 19 (100) | 0.72 |
| Carfilzomib | 4 (16) | 4 (21) | |
| Ixazomib | 1 (4) | 0 | |
| Unknown | 5 (20) | 0 | |
| HSCT | |||
| Autologous | 8 (32) | 15 (68) | 0.042 |
| Tandem-Auto | 1 (4) | 0 (0) | |
| None | 12 (48) | 6 (27) | |
| Unknown | 4 (16) | 1 (4) | |
| Overall response rate | 56 | 81.8 | 0.0293 |
| Median survival (months) | 86.4 | 107.3 | 0.0301 |
| Progression-free survival (months) | 22 | 30.13 | 0.0385 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathews, P.; Wang, X.; Wu, J.; Jabbar, S.; Burcher, K.; Rein, L.; Kang, Y. β-Arrestin 2 as a Prognostic Indicator and Immunomodulatory Factor in Multiple Myeloma. Cells 2025, 14, 496. https://doi.org/10.3390/cells14070496
Mathews P, Wang X, Wu J, Jabbar S, Burcher K, Rein L, Kang Y. β-Arrestin 2 as a Prognostic Indicator and Immunomodulatory Factor in Multiple Myeloma. Cells. 2025; 14(7):496. https://doi.org/10.3390/cells14070496
Chicago/Turabian StyleMathews, Parker, Xiaobei Wang, Jian Wu, Shaima Jabbar, Kimberly Burcher, Lindsay Rein, and Yubin Kang. 2025. "β-Arrestin 2 as a Prognostic Indicator and Immunomodulatory Factor in Multiple Myeloma" Cells 14, no. 7: 496. https://doi.org/10.3390/cells14070496
APA StyleMathews, P., Wang, X., Wu, J., Jabbar, S., Burcher, K., Rein, L., & Kang, Y. (2025). β-Arrestin 2 as a Prognostic Indicator and Immunomodulatory Factor in Multiple Myeloma. Cells, 14(7), 496. https://doi.org/10.3390/cells14070496