1. Introduction
The replication of the mammalian mitochondrial genome (mtDNA) depends on the heterotrimeric enzyme complex DNA polymerase γ (Pol-γ). Loss or reduced functionality of Pol-γ directly affects mtDNA synthesis and impairs mtDNA replication, which disrupts the expression of mtDNA-encoded proteins and protein subunits and leads to a constricted function of the respiratory chain and reduced provision of ATP [
1,
2]. The catalytic subunit of Pol-γ is encoded by the nuclear gene
POLG. Mutations in
POLG result in POLG-spectrum disorders, mitochondriopathies characterized by progressive mtDNA depletion, mitochondrial dysfunction and early onset neurodegeneration [
3,
4,
5]. Clinically, the detrimental neurogenetic diseases lead to severe epilepsy, multi-organ failure and premature death (age range from 30 days to 64 years, median age 3.5 years) [
5]. The two most common pathogenic POLG mutations result in the amino acid substitutions p.A467T and p.W748S. Patients with these mutations show significant residual (~5%) Pol-γ activity [
5,
6,
7]. There is neither a cure nor an effective treatment available for POLG-SDs. Disease mechanisms are understudied, and therapeutic strategies are lacking.
The human neuroblastoma cell line SH-SY5Y is derived from the parental cell line SK-N-SH, which originates from a metastatic bone tumor biopsy [
8,
9]. SH-SY5Y cultures contain both epithelial-like (“S”-type) and neuroblast-like (“N”-type) cells [
9,
10]. “N”-type SH-SY5Y cells can be differentiated into more mature, neuron-like phenotype cells by modifying components of the cell culture medium [
11]. Neuron-like SH-SY5Y cells (SHC) differentiated by treatment with retinoic acid (RA) and brain-derived neurotrophic factor (BDNF) (“SH-SY5Y-derived neurons”) constitute an established in vitro model that is commonly used to study neurodegenerative disorders such as Alzheimer’s disease [
12,
13] or Parkinson’s disease [
14]. Besides its abilities relevant to neuronal differentiation, this cell line shows some notable advantages compared to human-induced pluripotent stem cells (hiPSCs), including ease of cultivation, rapid large-scale expansion and low costs in maintaining the culture [
15,
16].
The development of suitable human neuronal cell models is crucial for the in vitro mimicking of POLG-SDs in order to enable research in both the investigation of pathogenic mechanisms as well as the testing of potential treatment approaches. Still, there is a lack of cheap, easy-to-handle and established cell models for neuroscientific research of POLG-SDs, as hiPSC-based cell models are expensive, time-consuming to obtain and differentiate and often lack isogenic controls [
17]. As SHC have already been used for experiments on mitochondrial biogenesis and function [
18,
19,
20], we here test whether SHC are a suitable cell model for mitochondrial dysfunction as observed in POLG-SDs, and whether they can be a simple and easy-to-handle cell model which could be used for mimicking the phenotype in neuronal cell populations, which are the ones mostly affected. We report the generation of a stable
POLG knockdown SH-SY5Y cell line and characterize mitochondrially based phenotypes and neuronal differentiation impairments, implying that SHC are a versatile tool for studying POLG-SD in human neurons.
2. Materials and Methods
2.1. Molecular Biology
Small hairpin RNAs (shRNAs) were cloned into pLKO.1 puro vector (Addgene #8453). For this, the following shRNA oligonucleotides were ordered and annealed: POLG sh1+ (5′-CCGGGGTGCACAGACTTTATGTACTCGAGTACATAAAGTCTGTGCACCTTTTTG-3′), POLG sh1- (5′-AATTCAAAAAGGTGCACAGACTTTATGTACTCGAGTACATAAAGTCTGTGCACC-3′), POLG sh2+ (5′-CCGGGGATGGTAATAGCTGTAATCTCGAGATTACAGCTATTACCATCCTTTTTG-3′), POLG sh2- (5′-AATTCAAAAAGGATGGTAATAGCTGTAATCTCGAGATTACAGCTATTACCATCC-3′), POLG sh3+ (5′-CCGGGCTATTACCATCCTTGTGACTCGAGTCACAAGGATGGTAATAGCTTTTTG-3′), POLG sh3- (5′-AATTCAAAAAGCTATTACCATCCTTGTGACTCGAGTCACAAGGATGGTAATAGC-3′), POLG sh4+ (5′-CCGGTTAAACTGCATTAGTAAGCCTCGAGGCTTACTAATGCAGTTTAATTTTTG-3′), POLG sh4- (5′-AATTCAAAAATTAAACTGCATTAGTAAGCCTCGAGGCTTACTAATGCAGTTTAA-3′), POLG SCR+ (5′-CCGGTTGTCTTGCATTCGACTAACTCGAGTTAGTCGAATGCAAGACAATTTTTG-3′), POLG SCR- (5′-AATTCAAAAATTGTCTTGCATTCGACTAACTCGAGTTAGTCGAATGCAAGACAA-3′).
The pLKO.1 vector was digested with abcI and defII and ligated with annealed shRNA oligonucleotides using T4 ligase (New England Biolabs GmbH, Frankfurt-Höchst, Germany). For plasmid amplification, ligation mixture was transformed into chemocompetent bacteria (E. coli Stbl3TM, Thermo Fisher Scientific, Langenselbold, Germany (TFS)). Bacteria were cultivated on pre-warmed agar plates with 100 μg/mL ampicillin for 16–18 h at 37 °C; single colonies were then picked and cultivated in a liquid culture (LB-Medium with 100 μg/mL ampicillin). For plasmid isolation and purification from bacterial suspensions, the PureYieldTM MidiPrep Kit (ProMega, Walldorf, Germany) was used according to the manufacturer’s protocols. Plasmid concentration and purity were checked spectrophotometrically (NanoDropTM 100, TFS). Plasmids were stored at −20 °C.
2.2. Production of Viral Particles
On day zero, HEK293T cells were seeded on T75 plates such that confluency would reach 70–75% on the next day. On day one, the medium on the cells was removed, the cells were washed with 1X PBS and 5 mL of DMEM was added onto cells before transfection. Transfection solution was prepared using the following vectors: 10 μg of shPOLG-plko.1 vector, 9 μg of pCMV-ΔR8.2dvpr packaging vector and 1 μg of pCMV-VSVG envelope vector. Each transfection was conducted separately for each shRNA construct. A quantity of 20 μL of PEI was added into the vector mixture and the mixture was equalized to 400 μL total volume with Opti-MEM (Gibco, TFS). The transfection mix was incubated for 20 min and added onto the cells. At 16 h post-transfection, 3 mL of DMEM was added into the plate. On day three, the medium of the cells was changed. The lentiviral particles were ready to collect after 72 h of transfection; particles in the medium were collected on days four and five. The lentiviral particles were sterilized with a 0.45 μm filter and stored at −80 °C. All the work was performed following S2-safety regulations.
2.3. Transfection and Selection of Knockdown Cells
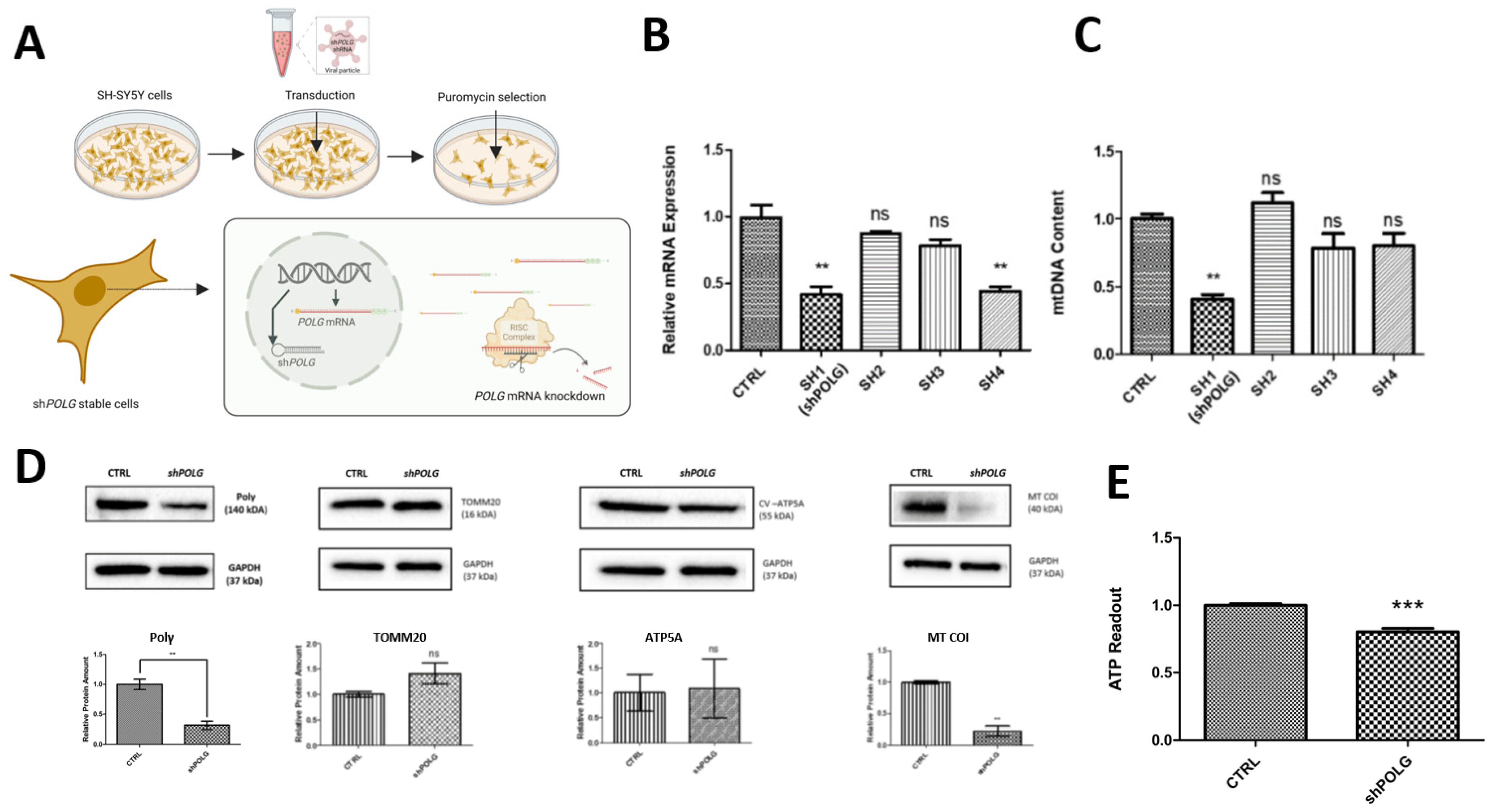
SH-SY5Y neuroblastoma cells were seeded on 6-well plates, with around 30 × 104 cells in each well, on day zero. The next day, the medium on the cells was removed and lentiviral particles (in DMEM) were given to the cells. On day 2, the lentiviral-particle-containing medium was removed and normal medium was given to the cells. On day 3, antibiotic selection was started and cells (including an untransfected control) were given 1 μg of puromycin in maintenance medium. Over the following days, the puromycin-containing medium was replenished every day. On day 8, when all the cells of the control group were dead, the cells successfully transduced with shRNA plasmids formed small cell colonies. Viable cells were distributed into 96-well plates in order to start a single-cell colony using the serial dilution method. These cells were observed daily, grown for about a month, and transferred into a larger dish as they grew. At the end of the single-cell colony expansion, cells were harvested and sequenced to confirm the presence of shRNA constructs.
2.4. SH-SY5Y Cultivation
A SH-SY5Y neuroblastoma cell line was kindly provided by Prof. Dr. Rudolf Wiesner (Institute of Veg. Physiology II, University Hospital Cologne, Cologne, Germany). SH-SY5Y cells were cultured in DMEM/F-12 GlutaMAX™ (#10565-018, TFS) supplemented with 10% fetal bovine serum (FBS, Biochrom AG, Berlin, Germany), Antibiotic–Antimycotic (1X Anti/Anti, #15240062, TFS) (referred to as ‘SHM-10’). For maintenance, SH-SY5Y cells were cultivated in uncoated T75 cell culture flasks (VWR) in a sterile incubator (Heraeus HeraCell 150, Kendro, Langenselbold, Germany) at a temperature of 37 °C, 95% air humidity and 5% CO2 concentration. Culture medium was changed once or twice per week, depending on cell density; the latter was monitored and images were taken with a brightfield light microscope (DM IL LED, Leica, Wetzlar, Germany), using objectives with 10× and 20× (air-based) magnification (Leica) and Las X imaging software (Leica). Cells were passaged for further cultivation or seeded for differentiation experiments at 70–80% confluency; for this, cells were washed with phosphate-buffered saline (PBS, TFS), trypsinized (0.05% Trypsin/EDTA, TFS) for 3–5 min, spun down at 1000× g for 3 min and resuspended in SHM-10. For passaging, the culture was diluted 1:10 and seeded again in an uncoated T75 flask. For long-term storage, trypsinized and spun-down cells were resuspended in FBS with 10% DMSO, cooled down at −1 °C/min in a cryo-container (Mr. FrostyTM, VWR, Darmstadt, Germany) and stored at −80 °C or in liquid nitrogen. For seeding, resuspended SH-SY5Y cells were counted with an automatic cell counter (TC20TM, Bio-Rad) and seeded at 3 × 104 cells/cm2 (experiments with undifferentiated SH-SY5Y cells) or 1 × 104 cells/cm2 (cells for differentiation with RA/BDNF) in uncoated 96-well plates, 24-well plates on glass coverslips (VWR) previously coated with 20 μg/mL Poly-D-lysine (PDL, AppliChem, Darmstadt), or uncoated 6-well plates.
2.5. SH-SY5Y Differentiation
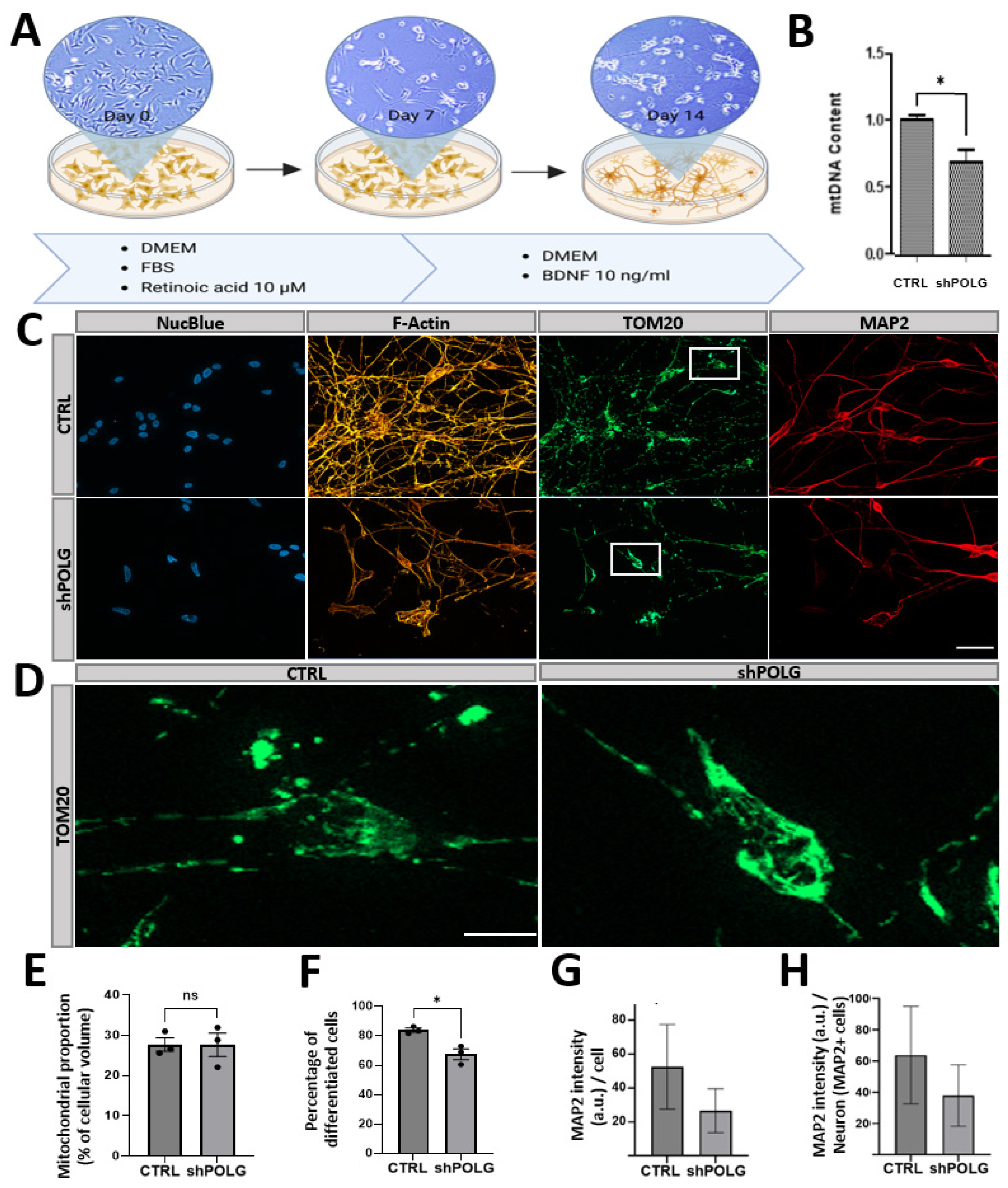
Differentiation of SH-SY5Y-derived neurons was conducted using a protocol applying retinoic acid (RA) and brain-derived neurotrophic factor (BDNF), as previously reported [
11]. In brief, differentiation was started 24 h after seeding at day 0 (d0). Fresh SHM-10 with 10 μM RA (Sigma-Aldrich, Taufkirchen, Germany) was added and replaced with fresh SHM-10 + 10 μM RA at d3 and d5. At d7, cells were washed once with PBS, and SHM-10 medium without FBS (referred to as ‘SHM-0’) with 10 ng/mL BDNF (Peprotech, TFS) was added and replaced with fresh SHM-0 + 10 ng/mL BDNF at d10. At d14, differentiation was completed, and cells were either fixed or harvested.
2.6. Measurement of Cell Viability and Drug Treatments
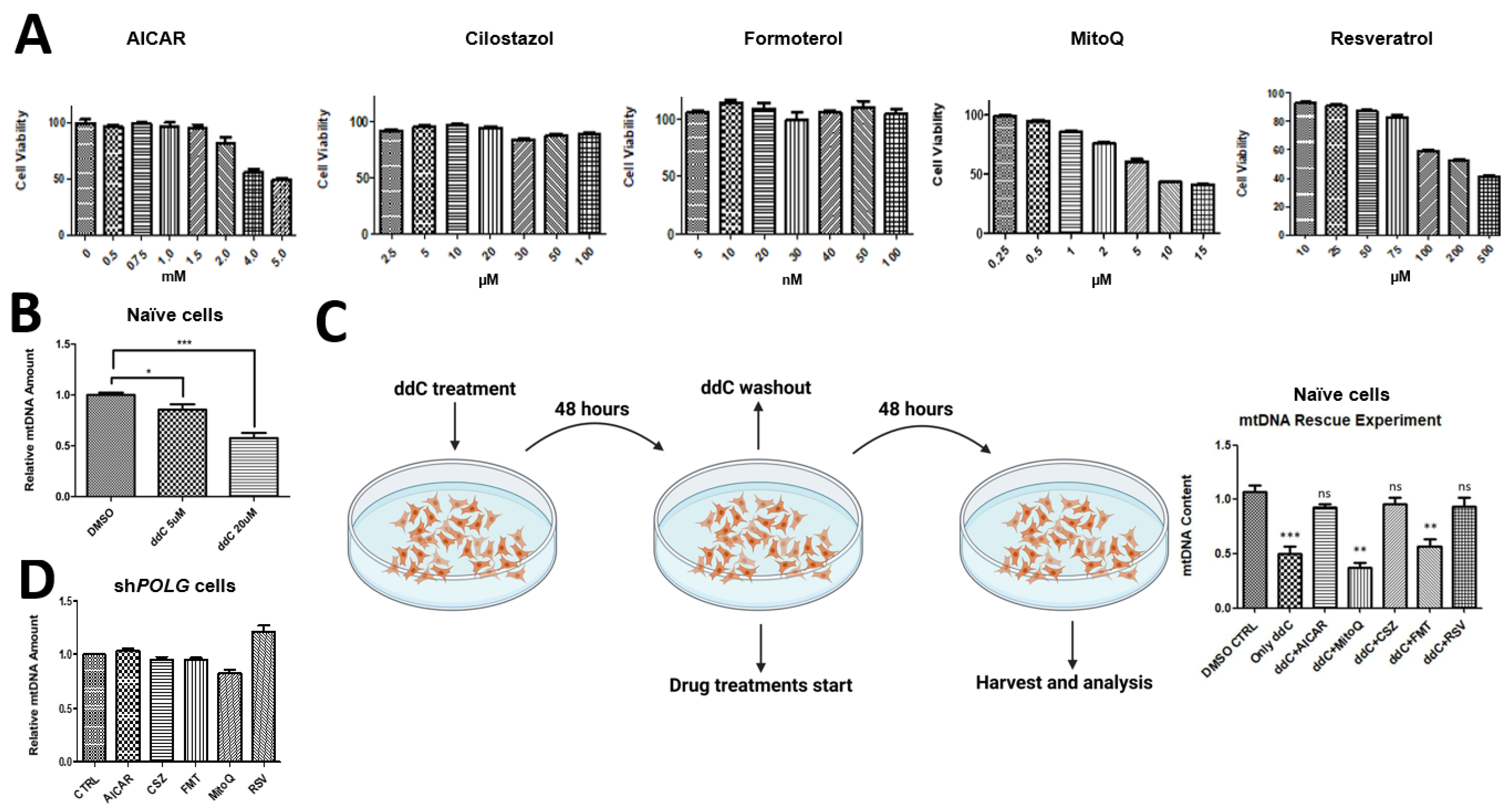
In order to select the correct drug concentration to administer on the SH-SY5Y cells, we conducted cell viability tests with the drugs we have tested, namely, AICAR (SelleckChem, München), cilostazol (Cayman, 1503, Hamburg, Germany), formoterol (Cayman, 15584, Hamburg, Germany), MitoQ (Cayman, 89950, Hamburg, Germany) and trans-resveratrol (Cayman, 70675, Hamburg, Germany), using a CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7570, Walldorf, Deutschland). SH-SY5Y cells were seeded on 24-well plates to test for each drug and corresponding vehicle control. The following concentrations were tested on SH-SY5Y cells: AICAR in H2O (0.5 mM, 0.75 mM, 1 mM, 1.5 mM, 2 mM, 4 mM and 5 mM) cilostazol in DMSO (2.5 μM, 5 μM, 10 μM, 20 μM, 30 μM, 50 μM and 100 μM), formoterol in DMSO (5 nM, 10 nM, 20 nM, 30 nM, 40 nM, 50 nM and 100 nM), MitoQ in DMSO (0.25 μM, 0.5 μM, 1 μM, 2 μM, 5 μM, 10 μM and 15 μM), trans-resveratrol in DMSO (10 μM, 25 μM, 50 μM, 75 μM, 100 μM, 200 μM and 500 μM), zalcitabine (ddC) in DMSO (5 μM, 20 μM). We determined, according to the manufacturer’s instructions, the highest nontoxic concentration for each drug to be as follows: AICAR 2 mM, cilostazol 20 μM, formoterol 30 nM, MitoQ 1 μM and trans-resveratrol 75 μM. For the rescue experiment, naïve SHC were treated with 20 μM ddC for 48 h and ddC-containing medium was replenished in the middle of the treatment, at around 24 h. After ddC treatment, ddC-containing medium was removed and cells were washed 3 times with PBS. After washing, drug treatments were administered with the following concentrations: AICAR 2 mM, cilostazol 20 μM, formoterol 30 nM, MitoQ 1 μM and trans-resveratrol 75 μM.
2.7. Quantification of Mitochondrial DNA
In order to calculate the mtDNA amount, we used the mtDNA/nDNA ratio by using two mitochondrial DNA-encoded (MT-CTYB and MT-3319R) and two nuclear DNA-encoded (APP, 18s rRNA) genes. The total DNA was isolated from harvested cells using a QIAamp DNA kit (Qiagen, 56304, Hilden, Germany). Quantitative Real-Time PCR (RT-qPCR) was performed with SYBR green master mix. For each reaction, 6.25 μL of SYBR green mix, 4.25 μL of ddH2O, 1 μL and 25 ng of total DNA and 1 μL of forward and reverse primer mix (10 μM) were mixed. All reactions had 3 technique replicates. The cycling protocol for the RT-qPCR reaction was as follows: denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s and elongation at 72 °C for 30 s, with 40 cycles. The relative mtDNA amount was calculated by measuring the mtDNA/nDNA ratio with the 2-ΔΔCT method.
2.8. Western Blot
Cells were seeded in 6-well plates, harvested with PBS and lysed with lysis buffer with 1× Phosphatase/Protease inhibitor. Protein concentrations were measured with a Bradford protein assay using Bradford Solution (AppliChem, Darmstadt, Germany) and a spectrometer (BioPhotometer RS 232 C, Eppendorf, Hamburg, Germany). For sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-Page), a gel casting kit (Mini-PROTEAN Tetra Cell, Bio-Rad, Dreieich, Germany) was used and 25 μg of samples (diluted in 5× Laemmli buffer) were loaded and run for 80 V for ~20 min; the current was then increased to 120 V and SDS-Page was run for approximately another 100 min until all samples had been run through the separating gel. For wet protein transfer to a polyvinylidene fluoride (PVDF) membrane (Merck, Darmstadt, Germany), the transfer was run at 30 V for ~16 h at 4 °C with constant stirring of buffer. After the transfer had finished, the transfer stack was disassembled, and the membrane was washed with Tris-buffered saline with Tween20 (TBS-T) for 1 min and then blocked for 1 h at room temperature in 5% milk in TBS-T. Membranes were incubated with the following primary antibodies for 24–48 h: anti-Pol-γ at 1:250 dilution in 5% milk in TBS-T (Santa Cruz, Heidelberg, Germany), anti-TOMM20 at 1:5000 dilution in 5% milk in TBS-T (Invitrogen, TFS), ATP5A (Mitococktail) at 1:5000 dilution in 5% milk in TBS-T (Abcam, VWR, Darmstadt, Germany) and anti-MT-COI at 1:1000 dilution in 5% milk in TBS-T (Invitrogen, TFS). After incubation with the primary antibody, the membrane was washed 3× with TBS-T (5 min each) and incubated with the corresponding secondary antibody (1:5000 dilution of anti-mouse antibody in 5% milk in TBS-T for anti-Pol-γ, ATP5A, anti-MT-COI antibodies; and 1:5000 dilution of anti-rabbit antibody in 5% milk in TBS-T for anti-TOMM20) for 1 h at room temperature. Before analysis, the membranes were washed 3× in TBS-T and incubated with ~150 μL 1:1 mixed solution of SuperSignal™ West Femto Kit (TFS). To take pictures, a Chemidoc MP Imaging System (Bio-Rad) was used, and for quantification, Image Lab 5.2.1 (Bio-Rad, Dreieich, Germany) was used.
2.9. ATP Measurement
ATP measurements were performed with a Perkin Elmer ATPlite Luminescence Assay System according to the manufacturer’s instructions. SH-SY5Y cells were seeded on black 96-well plates with 3 technical replicates and 40 × 103 cells in each well on day 0. The next day, ATP substrate solution was added to each well at a 1:1 ratio. After 5 min of darkness adaptation, luminescence was measured with Promega Luminex Luminometer.
2.10. Immunofluorescence and Microscopy
Cells were cultivated as stated above in 24-well plates on glass coverslips (VWR) previously coated with 20 μg/mL Poly-D-lysine (PDL, AppliChem, Darmstadt, Germany), fixed for 30 min at room temperature in 3.7% formaldehyde (in PBS) and permeabilized + blocked with 5% bovine serum albumin (BSA, Sigma-Aldrich, Darmstadt, Germany) and 0.2% Triton X-100 (AppliChem, Darmstadt, Germany) in PBS for 10 min. Incubation with the primary antibody (the following antibodies were used: rabbit anti-Tom20 (1:500 in PBS, sc-11415, SCBT), mouse anti-ds DNA (1:1000 in PBS, ab27156, Abcam, VWR, Darmstadt, Germany) and chicken anti-MAP2 (1:2000 in PBS, ab5392, Abcam)) was conducted for 16–18 h at 4 °C. The coverslips were washed thoroughly and incubated with the corresponding secondary antibody (coupled to AlexaFluorTM fluorophores, diluted in PBS) for 1–2 h at room temperature. Nuclei were stained with 1 drop/mL NucBlueTM (Hochest 33342, TFS) for 20–30 min and samples were mounted on objective slides (#145-0011, Bio-Rad, Dreieich, Germany) with aqueous PolyMountTM (#18606, Polysciences, Hirschberg an der Bergstraße, Germany), dried for 24 at room temperature in the dark and placed in long-term storage at 4 °C in the dark.
Immunostained cells were imaged with a widefield fluorescence microscope (Axioscope 5, Zeiss, Jena, Germany), using an LED excitation lamp (Colibri 7, Zeiss, Jena, Germany), a fluorescence camera device (Axiocam 705 mono, Zeiss, Jena, Germany), a 40× (oil-based) magnification objective (Zeiss, Jena, Germany), ApoTome.2 (Zeiss, Jena, Germany) and Zen imaging software (Zen 3.6 blue edition, Zeiss, Jena, Germany). For the analysis of protein expression levels, all images were taken with identical settings (laser intensity and exposure time) to ensure statistical comparability.
4. Discussion
The development of suitable neuronal cell models is crucial for investigating the pathogenic mechanisms of POLG spectrum disorders (POLG-SDs) and testing potential translatable treatments. This study presents a novel cell model for POLG-SDs using SH-SY5Y neuroblastoma cells (SHC) with stable POLG knockdown, which mimics key features of the disease in both naïve SHC and neuron-like states.
The shRNA-mediated knockdown of
POLG in SHC resulted in an approximately 50% reduction in
POLG mRNA and a 70% reduction in protein levels, which may fall short of the actual reductions in residual activity in POLG-SD when derived mutations are tested not in a cellular context, but approximates well the significant loss of enzyme activity suggested for all POLG-SDs [
6]. This level of knockdown led to several phenotypes characteristic of POLG-SDs, including reduced mtDNA content, decreased levels of mitochondrial-encoded proteins, lower ATP production and altered mitochondrial morphology and distribution. Considering that
POLG-KO cells are not viable, this knockdown approach is an excellent tool for the study of POLG-SD in human cells, without the need for cells with patient-derived mutations.
One striking finding of this study relates to the impaired neuronal differentiation capacity of
POLG-knockdown cells upon treatment with retinoic acid (RA) and brain-derived neurotrophic factor (BDNF), which in healthy cells allows SHC to differentiate in human neurons of several lineages [
21]. This observation has important implications for understanding the neurodevelopmental aspects of POLG-SDs. The reduced ability of POLG-knockdown cells to differentiate into neurons may reflect the neurodevelopmental defects observed in some POLG-SD patients with severe mutations [
22], but could also suggest impaired neurogenesis once sufficient mtDNA depletion has taken place, which would be consistent with a delayed age of onset of symptoms for loss-of-functions mutations of
POLG associated with less severe dysfunction or considerable residual activity.
Our data from differentiated
POLG-knockdown cells suggest that mitochondrial dysfunction can significantly impact neuronal differentiation capacity, with
POLG knockdown resulting in a significantly decreased number of neurons upon neuronal differentiation. We could not determine whether successfully differentiated cells showed significantly altered neuronal morphological features, but we did observe trends towards decreased MAP2 expression per cell and reduced complexity, which may, however, be due to reduced cell density. It is thus possible that successfully differentiated cells have found a mechanism to compensate for a reduced POLG function, which would be in line with the less severe effects on mtDNA levels observed in differentiated neurons. Noteworthy is the altered mitochondrial distribution observed in both undifferentiated and differentiated
POLG-knockdown cells. Control cells exhibited an interconnected network of tubular mitochondria with elongated and filamentous mitochondria evenly distributed throughout the cell. In contrast,
POLG-knockdown cells show perinuclear clustering of mitochondria which appear fragmented, smaller and possibly swollen. This perinuclear clustering, which has also been shown to play a role in other types of mitochondrial stress or dysfunction, such as oxidative stress and mitofusion impairment, may reflect disrupted mitochondrial transport along neurites—a process essential for maintaining proper neuronal function, or mitochondrial quality-control based prevention of mitochondrial trafficking into the periphery [
23,
24].
The study also demonstrated that POLG-knockdown cells are resistant to treatments that stimulate mitochondrial biosynthesis and protect against chemically induced mtDNA depletion. This resistance underscores the central role of POLG in mtDNA replication and maintenance, and suggests that therapeutic approaches for POLG-SDs may need to directly target the underlying genetic defect, rather than relying solely on general mitochondrial biogenesis stimulators. This may not hold true for POLG-SDs for which the underlying causes are mutations with considerable residual Pol-y function. There, slight stimulation may be sufficient to compensate for partial loss of enzyme function, but severe mutations will not be addressable with mitochondrial biosynthesis stimulators.
This SHC-based POLG-SD model offers several advantages for the study of these disorders. Compared to patient-derived induced pluripotent stem cells (iPSCs), SHC are easier to cultivate, expand, and maintain. The ability to differentiate SHC into neuron-like cells allows for the study of POLG-SD pathology in a more relevant cellular context, since POLG-SDs primarily affect neurons. Additionally, the use of scrambled shRNA controls provides a genetically matched comparison, reducing the variability due to genetic background differences.
This model provides valuable insights into POLG-SD pathology. As the ~50% reduction in POLG levels may not fully recapitulate the most severe cases of POLG SDs, in which there is only a little residual activity left, future studies could explore the effect of a more severe knockdown or pathogenic mutations that decrease POLG levels even more, as a complete knockout of
POLG has been difficult to achieve, despite efforts by several laboratories, and is likely not viable. Also, we note that RA, which was used here to differentiate SHC into neurons, could enhance mitochondrial function. RA is also known to alter cellular responses to oxidative stress and mitochondrial membrane potential (MMP) in SHC, potentially attenuating the severity of the phenotype, compared to models or differentiation protocols that are not RA-based [
19]. Therefore, these findings should be validated in other neuronal cell types to ensure generalizability. While the characterization we provide here is sufficient to attribute the observed effects to successful
POLG knockdown, future studies could provide for a more in-depth characterization (oxygen consumption, extracellular acidification, mitochondrial membrane potential, electron transport chain complex activity, mitochondrial metabolite analysis, mitochondrial calcium, reactive oxygen species detection, etc.). The comprehensive data we have collected strongly support the conclusion that the observed mitochondrial impairments are due to
POLG knockdown, and that Pol-γ dysfunction is the key factor driving the observed alterations in SH-SY5Y cells and the derived neurons. Nonetheless, future studies should also use different cell lines and investigate whether
POLG knockdown has similar effects in other cell lines or cell types.
In conclusion, the creation of a stable POLG knockdown SH-SY5Y cell line represents a significant step forward in modeling POLG-SDs. By mimicking the reduced Pol-γ activity seen in patients with common POLG mutations, this model effectively replicates key disease features, including impaired mtDNA content, resistance to mitochondrial biosynthesis stimulation, reduced ATP production, abnormal mitochondrial distribution, and hindered neuronal differentiation. The observed resistance to rescue by mitochondrial biosynthesis stimulators in POLG-knockdown cells underscores the critical role of functional Pol-γ in mtDNA maintenance. Furthermore, the model’s ability to recapitulate impaired neuronal differentiation and disrupted cytoskeletal organization in differentiated neurons highlights its relevance to the neurological deficits observed in POLG-SDs. This simple, cost-effective and human-derived platform overcomes limitations found in existing models and offers a valuable tool for the dissection of disease mechanisms and the screening of potential therapeutic interventions, thereby accelerating the search for effective treatments for these devastating neurogenetic disorders. The ease of use and scalability of this model will facilitate broader research efforts aimed at understanding—and combating—POLG-related mitochondrial dysfunction in human neurons.




{kind=link}
{kind=link}
{kind=link}
{kind=link}