
Control of Replication Stress Response by Cytosolic Fe-S Cluster Assembly (CIA) Machinery


Abstract

1. Introduction
2. The Cellular Response to Replication Stress
2.1. DNA Replication and DNA Polymerases
2.2. Replication Stress and the Replication Checkpoint
2.3. The Control of dNTPs by the Replication Checkpoint
2.4. Fork Remodeling After Replication Stress
3. The Iron–Sulfur Machinery
3.1. Structure and Function of Fe-S Clusters
3.2. Biogenesis of Mitochondrial, Cytosolic and Nuclear Fe-S Cluster Proteins
3.3. The CIA Targeting Complex: Structure and Activity
3.3.1. Cia1
3.3.2. Cia2
3.3.3. Met18
3.4. Assembly of CIA Targeting Complex and Target Maturation
4. Role of CIA Machinery in Response to Replication Stress
4.1. CIA and the Control of DNA Polymerases
4.2. CIA and the Restart of Replication Forks
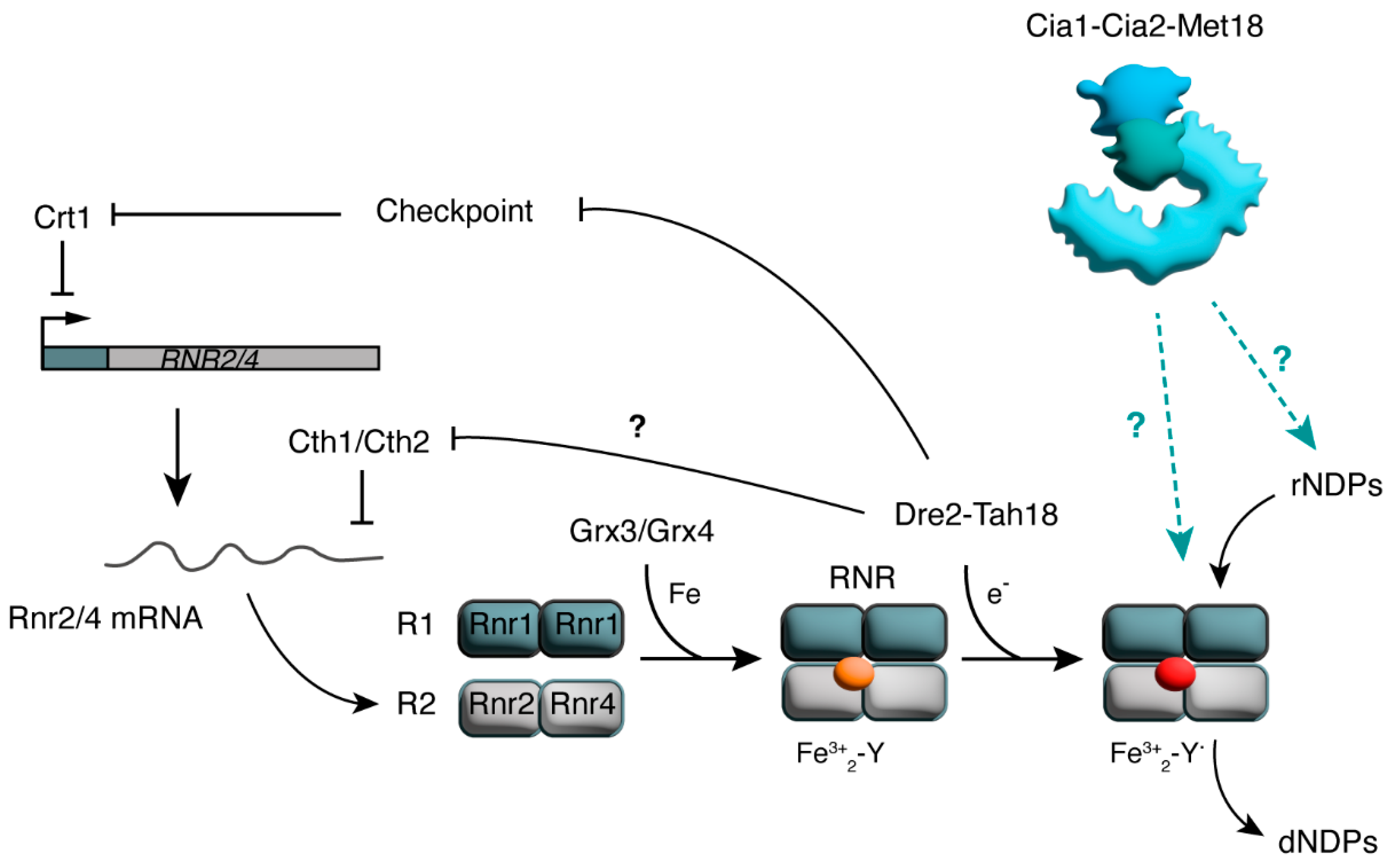
4.3. CIA and the Control of dNTPs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Saxena, S.; Zou, L. Hallmarks of DNA replication stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Branzei, D. S-phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell. Mol. Life Sci. 2017, 74, 2361–2380. [Google Scholar] [CrossRef] [PubMed]
- Pasero, P.; Vindigni, A. Nucleases acting at stalled forks: How to reboot the replication program with a few shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1528–1539. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Duong, V.N.; Babayeva, N.D.; Zhang, Y.; Pavlov, Y.I.; Anderson, K.S.; Tahirov, T.H. Activity and fidelity of human DNA polymerase α depend on primer structure. J. Biol. Chem. 2018, 293, 6824–6843. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Gambus, A.; Van Deursen, F.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef]
- Kang, Y.H.; Farina, A.; Bermudez, V.P.; Tappin, I.; Du, F.; Galal, W.C.; Hurwitz, J. Interaction between human Ctf4 and the Cdc45/Mcm2-7/GINS (CMG) replicative helicase. Proc. Natl. Acad. Sci. USA 2013, 110, 19760–19765. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.C.; Zhou, J.C.; Perera, R.L.; van Deursen, F.; Evrin, C.; Ivanova, M.E.; Kilkenny, M.L.; Renault, L.; Kjaer, S.; Matak-Vinković, D.; et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 2014, 510, 293–297. [Google Scholar] [CrossRef]
- Villa, F.; Simon, A.C.; Bazan, M.A.O.; Kilkenny, M.L.; Wirthensohn, D.; Wightman, M.; Matak-Vinković, D.; Pellegrini, L.; Labib, K. Ctf4 is a hub in the eukaryotic replisome that links multiple CIP-box proteins to the CMG helicase. Mol. Cell 2016, 63, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Kokoska, R.J.; Stefanovic, L.; DeMai, J.; Petes, T.D. Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase δ or by decreases in the cellular levels of DNA polymerase δ. Mol. Cell. Biol. 2000, 20, 362–373. [Google Scholar] [CrossRef]
- Netz, D.J.; Stith, C.M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Araki, H.; Clark, A.B.; Hamatake, R.K.; Sugino, A. A third essential DNA polymerase in S. cerevisiae. Cell 1990, 62, 1143–1151. [Google Scholar] [CrossRef]
- Burgers, P.M. Eukaryotic DNA polymerases in DNA replication and DNA repair. Chromosoma 1998, 107, 218–227. [Google Scholar] [CrossRef]
- Zhou, J.C.; Janska, A.; Goswami, P.; Renault, L.; Abid Ali, F.; Kotecha, A.; Diffley, J.F.X.; Costa, A. CMG–Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome. Proc. Natl. Acad. Sci. USA 2017, 114, 4141–4146. [Google Scholar] [CrossRef]
- Hogg, M.; Johansson, E. DNA polymerase ε. Eukaryot. Replisome Guide Protein Struct. Funct. 2012, 62, 237–257. [Google Scholar]
- Klinge, S.; Hirst, J.; Maman, J.D.; Krude, T.; Pellegrini, L. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat. Struct. Mol. Biol. 2007, 14, 875–877. [Google Scholar] [CrossRef]
- Weiner, B.E.; Huang, H.; Dattilo, B.M.; Nilges, M.J.; Fanning, E.; Chazin, W.J. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. J. Biol. Chem. 2007, 282, 33444–33451. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Carvalho, C.M.; Hastings, P.J.; Lupski, J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012, 22, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Segurado, M.; Tercero, J.A. The S-phase checkpoint: Targeting the replication fork. Biol. Cell 2009, 101, 617–627. [Google Scholar] [CrossRef]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef]
- Pizzul, P.; Casari, E.; Gnugnoli, M.; Rinaldi, C.; Corallo, F.; Longhese, M.P. The DNA damage checkpoint: A tale from budding yeast. Front. Genet. 2022, 13, 995163. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Pardo, B.; Crabbé, L.; Pasero, P. Signaling pathways of replication stress in yeast. FEMS Yeast Res. 2017, 17, fow101. [Google Scholar] [CrossRef]
- Bando, M.; Katou, Y.; Komata, M.; Tanaka, H.; Itoh, T.; Sutani, T.; Shirahige, K. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 2009, 284, 34355–34365. [Google Scholar] [CrossRef]
- Ciardo, D.; Goldar, A.; Marheineke, K. On the interplay of the DNA replication program and the intra-S phase checkpoint pathway. Genes 2019, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Sanvisens, N.; de Llanos, R.; Puig, S. Function and regulation of yeast ribonucleotide reductase: Cell cycle, genotoxic stress, and iron bioavailability. Biomed. J. 2013, 36, 51–58. [Google Scholar]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Yagüe-Capilla, M.; Rudd, S.G. Understanding the interplay between dNTP metabolism and genome stability in cancer. Dis. Models Mech. 2024, 17, dmm050775. [Google Scholar] [CrossRef] [PubMed]
- Elledge, S.J.; Davis, R.W. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 1990, 4, 740–751. [Google Scholar] [CrossRef]
- Domkin, V.; Thelander, L.; Chabes, A. Yeast DNA damage-inducible Rnr3 has a very low catalytic activity strongly stimulated after the formation of a cross-talking Rnr1/Rnr3 complex. J. Biol. Chem. 2002, 277, 18574–18578. [Google Scholar] [CrossRef]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martínez-Pastor, M.T. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Psakhye, I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016, 40, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Atkinson, J.; McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009, 37, 3475–3492. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Janska, A.; Early, A.; Diffley, J.F. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef]
- Petronek, M.S.; Allen, B.G. Maintenance of genome integrity by the late-acting cytoplasmic iron-sulfur assembly (CIA) complex. Front. Genet. 2023, 14, 1152398. [Google Scholar] [CrossRef]
- Lill, R.; Mühlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Dutkiewicz, R.; Elsässer, H.P.; Hausmann, A.; Netz, D.J.; Pierik, A.J.; Stehling, O.; Urzica, E.; Mühlenhoff, U. Mechanisms of iron–sulfur protein maturation in mitochondria, cytosol, and nucleus of eukaryotes. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Rydz, L.; Wróbel, M.; Jurkowska, H. Sulfur administration in Fe–S cluster homeostasis. Antioxidants 2021, 10, 1738. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Hou, W.; Wang, Z.Q.; Xu, X. Biogenesis of iron–sulfur clusters and their role in DNA metabolism. Front. Cell Dev. Biol. 2021, 9, 735678. [Google Scholar] [CrossRef]
- White, M.F.; Dillingham, M.S. Iron–sulfur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012, 22, 94–100. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Iron–sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1493–1512. [Google Scholar] [CrossRef]
- Valasatava, Y.; Rosato, A.; Banci, L.; Andreini, C. MetalPredator: A web server to predict iron-sulfur cluster binding proteomes. Bioinformatics 2016, 32, 2850–2852. [Google Scholar] [CrossRef]
- Wehrspan, Z.J.; McDonnell, R.T.; Elcock, A.H. Identification of iron-sulfur (Fe-S) cluster and zinc (Zn) binding sites within proteomes predicted by DeepMind’s AlphaFold2 program dramatically expands the metalloproteome. J. Mol. Biol. 2022, 434, 167377. [Google Scholar] [CrossRef]
- Vallières, C.; Benoit, O.; Guittet, O.; Huang, M.E.; Lepoivre, M.; Golinelli-Cohen, M.P.; Vernis, L. Iron-sulfur protein odyssey: Exploring their cluster functional versatility and challenging identification. Metallomics 2024, 16, mfae025. [Google Scholar] [CrossRef]
- Netz, D.J.; Mascarenhas, J.; Stehling, O.; Pierik, A.J.; Lill, R. Maturation of cytosolic and nuclear iron–sulfur proteins. Trends Cell Biol. 2014, 24, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Castro, C.; Saini, A.; Outten, F.W. Fe-S cluster assembly pathways in bacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Mettert, E.L.; Kiley, P.J. Fe–S proteins that regulate gene expression. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1284–1293. [Google Scholar] [CrossRef]
- Wu, Y.; Brosh, R.M., Jr. DNA helicase and helicase–nuclease enzymes with a conserved iron–sulfur cluster. Nucleic Acids Res. 2012, 40, 4247–4260. [Google Scholar] [CrossRef]
- Adrover, M.; Howes, B.D.; Iannuzzi, C.; Smulevich, G.; Pastore, A. Anatomy of an iron-sulfur cluster scaffold protein: Understanding the determinants of [2Fe–2S] cluster stability on IscU. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Uzarska, M.A.; Grochowina, I.; Soldek, J.; Jelen, M.; Schilke, B.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. During FeS cluster biogenesis, ferredoxin and frataxin use overlapping binding sites on yeast cysteine desulfurase Nfs1. J. Biol. Chem. 2022, 298, 101570. [Google Scholar] [CrossRef]
- Webert, H.; Freibert, S.A.; Gallo, A.; Heidenreich, T.; Linne, U.; Amlacher, S.; Hurt, E.; Mühlenhoff, U.; Banci, L.; Lill, R. Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat. Commun. 2014, 5, 5013. [Google Scholar] [CrossRef]
- Galeano, B.K.; Ranatunga, W.; Gakh, O.; Smith, D.Y., IV; Thompson, J.R.; Isaya, G. Zinc and the iron donor frataxin regulate oligomerization of the scaffold protein to form new Fe–S cluster assembly centers. Metallomics 2017, 9, 773–801. [Google Scholar] [CrossRef]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron–sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef]
- Voisine, C.; Cheng, Y.C.; Ohlson, M.; Schilke, B.; Hoff, K.; Beinert, H.; Marszalek, J.; Craig, E.A. Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2001, 98, 1483–1488. [Google Scholar] [CrossRef]
- Melber, A.; Na, U.; Vashisht, A.; Weiler, B.D.; Lill, R.; Wohlschlegel, J.A.; Winge, D.R. Role of Nfu1 and Bol3 in iron-sulfur cluster transfer to mitochondrial clients. Elife 2016, 5, e15991. [Google Scholar] [CrossRef]
- Li, P.; Hendricks, A.L.; Wang, Y.; Villones, R.L.E.; Lindkvist-Petersson, K.; Meloni, G.; Cowan, J.A.; Wang, K.; Gourdon, P. Structures of Atm1 provide insight into [2Fe-2S] cluster export from mitochondria. Nat. Commun. 2022, 13, 4339. [Google Scholar] [CrossRef] [PubMed]
- Netz, D.J.; Pierik, A.J.; Stümpfig, M.; Bill, E.; Sharma, A.K.; Pallesen, L.J.; Walden, W.E.; Lill, R. A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. J. Biol. Chem. 2012, 287, 12365–12378. [Google Scholar] [CrossRef] [PubMed]
- Netz, D.J.; Stümpfig, M.; Doré, C.; Mühlenhoff, U.; Pierik, A.J.; Lill, R. Tah18 transfers electrons to Dre2 in cytosolic iron-sulfur protein biogenesis. Nat. Chem. Biol. 2010, 6, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Lill, R. The role of mitochondria in cellular iron–sulfur protein biogenesis: Mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar]
- Mühlenhoff, U.; Braymer, J.J.; Christ, S.; Rietzschel, N.; Uzarska, M.A.; Weiler, B.D.; Lill, R. Glutaredoxins and iron-sulfur protein biogenesis at the interface of redox biology and iron metabolism. Biol. Chem. 2020, 401, 1407–1428. [Google Scholar] [CrossRef]
- Lill, R. From the discovery to molecular understanding of cellular iron-sulfur protein biogenesis. Biol. Chem. 2020, 401, 855–876. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Vo, A.T.; Fleischman, N.M.; Marquez, M.D.; Camire, E.J.; Esonwune, S.U.; Grossman, J.D.; Gay, K.A.; Cosman, J.A.; Perlstein, D.L. Defining the domains of Cia2 required for its essential function in vivo and in vitro. Metallomics 2017, 9, 1645–1654. [Google Scholar] [CrossRef]
- Srinivasan, V.; Netz, D.J.; Webert, H.; Mascarenhas, J.; Pierik, A.J.; Michel, H.; Lill, R. Structure of the yeast WD40 domain protein Cia1, a component acting late in iron-sulfur protein biogenesis. Structure 2007, 15, 1246–1257. [Google Scholar] [CrossRef]
- Vo, A.; Fleischman, N.M.; Froehlich, M.J.; Lee, C.Y.; Cosman, J.A.; Glynn, C.A.; Hassan, Z.O.; Perlstein, D.L. Identifying the protein interactions of the cytosolic iron–sulfur cluster targeting complex essential for its assembly and recognition of apo-targets. Biochemistry 2018, 57, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Wang, J.; Tommerup, N.; Vissing, H.; Roberts, T.; Shi, Y. Ciao 1 is a novel WD40 protein that interacts with the tumor suppressor protein WT1. J. Biol. Chem. 1998, 273, 10880–10887. [Google Scholar] [CrossRef]
- Balk, J.; Aguilar Netz, D.J.; Tepper, K.; Pierik, A.J.; Lill, R. The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol. Cell. Biol. 2005, 25, 10833–10841. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Orbach, R.; Zaharieva, I.T.; Töpf, A.; Donkervoort, S.; Munot, P.; Mueller, J.; Willis, T.; Verma, S.; Peric, S.; et al. CIAO1 loss of function causes a neuromuscular disorder with compromise of nucleocytoplasmic Fe-S enzymes. J. Clin. Investig. 2024, 134, e179559. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Stehling, O.; Stümpfig, M.; Rösser, R.; Spantgar, F.; Blinn, C.M.; Mühlenhoff, U.; Pierik, A.J.; Lill, R. Requirements for the biogenesis of [2Fe-2S] proteins in the human and yeast cytosol. Proc. Natl. Acad. Sci. USA 2024, 121, e2400740121. [Google Scholar] [CrossRef]
- Stehling, O.; Mascarenhas, J.; Vashisht, A.A.; Sheftel, A.D.; Niggemeyer, B.; Rösser, R.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 2013, 18, 187–198. [Google Scholar] [CrossRef]
- Maione, V.; Cantini, F.; Severi, M.; Banci, L. Investigating the role of the human CIA2A-CIAO1 complex in the maturation of aconitase. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1980–1987. [Google Scholar] [CrossRef]
- Ito, S.; Tan, L.J.; Andoh, D.; Narita, T.; Seki, M.; Hirano, Y.; Narita, K.; Kuraoka, I.; Hiraoka, Y.; Tanaka, K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol. Cell 2010, 39, 632–640. [Google Scholar] [CrossRef]
- Zhang, X.H.; Chen, X.J.; Yan, Y.B. Fam96b recruits brain-type creatine kinase to fuel mitotic spindle formation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2023, 1870, 119410. [Google Scholar] [CrossRef]
- Lev, I.; Volpe, M.; Goor, L.; Levinton, N.; Emuna, L.; Ben-Aroya, S. Reverse PCA, a systematic approach for identifying genes important for the physical interaction between protein pairs. PLoS Genet. 2013, 9, e1003838. [Google Scholar] [CrossRef]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; León Ortiz, A.M.; Borel, V.; Flynn, H.; Skehel, J.M.; Boulton, S.J. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 2012, 337, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Kassube, S.A.; Thomä, N.H. Structural insights into Fe–S protein biogenesis by the CIA targeting complex. Nat. Struct. Mol. Biol. 2020, 27, 735–742. [Google Scholar] [CrossRef]
- Vasquez, S.; Marquez, M.D.; Brignole, E.J.; Vo, A.; Kong, S.; Park, C.; Perlstein, D.L.; Drennan, C.L. Structural and biochemical investigations of a HEAT-repeat protein involved in the cytosolic iron-sulfur cluster assembly pathway. Commun. Biol. 2023, 6, 1276. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Takeda, Y.; Iwai, K.; Tanaka, K. IOP1 protein is an external component of the human cytosolic iron-sulfur cluster assembly (CIA) machinery and functions in the MMS19 protein-dependent CIA pathway. J. Biol. Chem. 2013, 288, 16680–16689. [Google Scholar] [CrossRef]
- Prakash, L.; Prakash, S. Three additional genes involved in pyrimidine dimer removal in Saccharomyces cerevisiae: RAD7, RAD14, and MMS19. Mol. Gen. Genet. (MGG) 1979, 176, 351–359. [Google Scholar] [CrossRef]
- Chang, M.; Bellaoui, M.; Boone, C.; Brown, G.W. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc. Natl. Acad. Sci. USA 2002, 99, 16934–16939. [Google Scholar] [CrossRef]
- Kou, H.; Zhou, Y.; Gorospe, R.C.; Wang, Z. Mms19 protein functions in nucleotide excision repair by sustaining an adequate cellular concentration of the TFIIH component Rad3. Proc. Natl. Acad. Sci. USA 2008, 105, 15714–15719. [Google Scholar] [CrossRef]
- Stehling, O.; Vashisht, A.A.; Mascarenhas, J.; Jonsson, Z.O.; Sharma, T.; Netz, D.J.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 2012, 337, 195–199. [Google Scholar] [CrossRef]
- Redwood, A.B.; Zhang, X.; Seth, S.B.; Ge, Z.; Bindeman, W.E.; Zhou, X.; Sinha, V.C.; Heffernan, T.P.; Piwnica-Worms, H. The cytosolic iron–sulfur cluster assembly (CIA) pathway is required for replication stress tolerance of cancer cells to Chk1 and ATR inhibitors. NPJ Breast Cancer 2021, 7, 152. [Google Scholar] [CrossRef]
- Corcoles-Saez, I.; Dong, K.; Johnson, A.L.; Waskiewicz, E.; Costanzo, M.; Boone, C.; Cha, R.S. Essential function of Mec1, the budding yeast ATM/ATR checkpoint-response kinase, in protein homeostasis. Dev. Cell 2018, 46, 495–503. [Google Scholar] [CrossRef]
- de Oliveira, F.M.B.; Kim, D.; Cussiol, J.R.; Das, J.; Jeong, M.C.; Doerfler, L.; Schmidt, K.H.; Yu, H.; Smolka, M.B. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol. Cell 2015, 57, 1124–1132. [Google Scholar] [CrossRef]
- Lanz, M.C.; Oberly, S.; Sanford, E.J.; Sharma, S.; Chabes, A.; Smolka, M.B. Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev. 2018, 32, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Forey, R.; Poveda, A.; Sharma, S.; Barthe, A.; Padioleau, I.; Renard, C.; Lambert, R.; Skrzypczak, M.; Ginalski, K.; Lengronne, A.; et al. Mec1 is activated at the onset of normal S phase by low-dNTP pools impeding DNA replication. Mol. Cell 2020, 78, 396–410. [Google Scholar] [CrossRef]
- Giot, L.; Chanet, R.; Simon, M.; Facca, C.; Faye, G. Involvement of the yeast DNA polymerase δ in DNA repair in vivo. Genetics 1997, 146, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Siebler, H.M.; Pavlov, Y.I.; Tahirov, T.H. Iron–sulfur clusters in DNA polymerases and primases of eukaryotes. Methods Enzymol. 2018, 599, 1–20. [Google Scholar]
- Jain, R.; Rice, W.J.; Malik, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Cryo-EM structure and dynamics of eukaryotic DNA polymerase δ holoenzyme. Nat. Struct. Mol. Biol. 2019, 26, 955–962. [Google Scholar] [CrossRef]
- Jain, R.; Vanamee, E.S.; Dzikovski, B.G.; Buku, A.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. An iron–sulfur cluster in the polymerase domain of yeast DNA polymerase ε. J. Mol. Biol. 2014, 426, 301–308. [Google Scholar] [CrossRef]
- Ter Beek, J.; Parkash, V.; Bylund, G.O.; Osterman, P.; Sauer-Eriksson, A.E.; Johansson, E. Structural evidence for an essential Fe–S cluster in the catalytic core domain of DNA polymerase ϵ. Nucleic Acids Res. 2019, 47, 5712–5722. [Google Scholar] [CrossRef]
- Lisova, A.E.; Baranovskiy, A.G.; Morstadt, L.M.; Babayeva, N.D.; Stepchenkova, E.I.; Tahirov, T.H. The iron-sulfur cluster is essential for DNA binding by human DNA polymerase ε. Sci. Rep. 2022, 12, 17436. [Google Scholar] [CrossRef]
- Barton, J.K.; Silva, R.M.; O’Brien, E. Redox chemistry in the genome: Emergence of the [4Fe4S] cofactor in repair and replication. Annu. Rev. Biochem. 2019, 88, 163–190. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Holt, M.E.; Thompson, M.K.; Salay, L.E.; Ehlinger, A.C.; Chazin, W.J.; Barton, J.K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355, eaag1789. [Google Scholar] [CrossRef]
- Bartels, P.L.; Stodola, J.L.; Burgers, P.M.; Barton, J.K. A redox role for the [4Fe4S] cluster of yeast DNA polymerase δ. J. Am. Chem. Soc. 2017, 139, 18339–18348. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.N.; Ter Beek, J.; Ekanger, L.A.; Johansson, E.; Barton, J.K. The [4Fe4S] cluster of yeast DNA polymerase ε is redox active and can undergo DNA-mediated signaling. J. Am. Chem. Soc. 2021, 143, 16147–16153. [Google Scholar] [CrossRef]
- Huang, M.E.; Facca, C.; Fatmi, Z.; Baïlle, D.; Bénakli, S.; Vernis, L. DNA replication inhibitor hydroxyurea alters Fe-S centers by producing reactive oxygen species in vivo. Sci. Rep. 2016, 6, 29361. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.E.; Whitted, J.E.; Mihelich, M.N.; Reitman, H.J.; Timmerman, A.J.; Schauer, G.D. Revised Mechanism of Hydroxyurea Induced Cell Cycle Arrest and an Improved Alternative. Proc. Natl. Acad. Sci. USA 2024, 121, e2404470121. [Google Scholar] [CrossRef]
- Jozwiakowski, S.K.; Kummer, S.; Gari, K. Human DNA polymerase delta requires an iron–sulfur cluster for high-fidelity DNA synthesis. Life Sci. Alliance 2019, 2, e201900321. [Google Scholar] [CrossRef]
- Chanet, R.; Baïlle, D.; Golinelli-Cohen, M.P.; Riquier, S.; Guittet, O.; Lepoivre, M.; Huang, M.E.; Vernis, L. Fe-S coordination defects in the replicative DNA polymerase delta cause deleterious DNA replication in vivo and subsequent DNA damage in the yeast Saccharomyces cerevisiae. G3 2021, 11, jkab124. [Google Scholar] [CrossRef]
- Kiktev, D.A.; Dominska, M.; Zhang, T.; Dahl, J.; Stepchenkova, E.I.; Mieczkowski, P.; Burgers, P.M.; Lujan, S.; Burkholder, A.; Kunkel, T.A.; et al. The fidelity of DNA replication, particularly on GC-rich templates, is reduced by defects of the Fe–S cluster in DNA polymerase δ. Nucleic Acids Res. 2021, 49, 5623–5636. [Google Scholar] [CrossRef]
- Fuss, J.O.; Tsai, C.L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim. Biophys. Acta 2015, 1853, 1253–1271. [Google Scholar] [CrossRef]
- Rossi, S.E.; Foiani, M.; Giannattasio, M. Dna2 processes behind the fork long ssDNA flaps generated by Pif1 and replication-dependent strand displacement. Nat. Commun. 2018, 9, 4830. [Google Scholar] [CrossRef]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Cammack, R.; Dillingham, M.S. An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. J. Biol. Chem. 2009, 284, 7746–7755. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.; Campbell, J.L. Cross talk between the nuclease and helicase activities of Dna2: Role of an essential iron–sulfur cluster domain. Nucleic Acids Res. 2012, 40, 7821–7830. [Google Scholar] [CrossRef]
- Zhou, C.; Pourmal, S.; Pavletich, N.P. Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife 2015, 4, e09832. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Wild, S.; Brunoldi, G.; Piceni, A.; Ceppi, I.; Kummer, S.; Lutz, R.E.; Cejka, P.; Gari, K. The iron–sulfur cluster in human DNA2 is required for all biochemical activities of DNA2. Commun. Biol. 2020, 3, 322. [Google Scholar] [CrossRef]
- van der Lelij, P.; Chrzanowska, K.H.; Godthelp, B.C.; Rooimans, M.A.; Oostra, A.B.; Stumm, M.; Zdzienicka, M.Z.; Joenje, H.; de Winter, J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010, 86, 262–266. [Google Scholar] [CrossRef]
- Jegadesan, N.K.; Branzei, D. DDX11 loss causes replication stress and pharmacologically exploitable DNA repair defects. Proc. Natl. Acad. Sci. USA 2021, 118, e2024258118. [Google Scholar] [CrossRef]
- Calì, F.; Bharti, S.K.; Perna, R.D.; Brosh, R.M., Jr.; Pisani, F.M. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res. 2016, 44, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Cortone, G.; Zheng, G.; Pensieri, P.; Chiappetta, V.; Tatè, R.; Malacaria, E.; Pichierri, P.; Yu, H.; Pisani, F.M. Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork-protection factor Timeless promotes sister chromatid cohesion. PLoS Genet. 2018, 14, e1007622. [Google Scholar] [CrossRef]
- Simon, A.K.; Kummer, S.; Wild, S.; Lezaja, A.; Teloni, F.; Jozwiakowski, S.K.; Altmeyer, M.; Gari, K. The iron–sulfur helicase DDX11 promotes the generation of single-stranded DNA for CHK1 activation. Life Sci. Alliance 2020, 3, e201900547. [Google Scholar] [CrossRef] [PubMed]
- Pellicanò, G.; Al Mamun, M.; Jurado-Santiago, D.; Villa-Hernández, S.; Yin, X.; Giannattasio, M.; Lanz, M.C.; Smolka, M.B.; Yeeles, J.; Shirahige, K.; et al. Checkpoint-mediated DNA polymerase ε exonuclease activity curbing counteracts resection-driven fork collapse. Mol. Cell 2021, 81, 2778–2792. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Wu, X.; An, X.; Stubbe, J.; Huang, M. Investigation of in vivo diferric tyrosyl radical formation in Saccharomyces cerevisiae Rnr2 protein: Requirement of Rnr4 and contribution of Grx3/4 and Dre2 proteins. J. Biol. Chem. 2011, 286, 41499–41509. [Google Scholar] [CrossRef]
- Mühlenhoff, U.; Molik, S.; Godoy, J.R.; Uzarska, M.A.; Richter, N.; Seubert, A.; Zhang, Y.; Stubbe, J.; Pierrel, F.; Herrero, E.; et al. Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron-sulfur cluster. Cell Metab. 2010, 12, 373–385. [Google Scholar] [CrossRef]
- Netz, D.J.; Genau, H.M.; Weiler, B.D.; Bill, E.; Pierik, A.J.; Lill, R. The conserved protein Dre2 uses essential [2Fe–2S] and [4Fe–4S] clusters for its function in cytosolic iron–sulfur protein assembly. Biochem. J. 2016, 473, 2073–2085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.; Zhang, C.; An, X.; Liu, L.; Stubbe, J.; Huang, M. Conserved electron donor complex Dre2–Tah18 is required for ribonucleotide reductase metallocofactor assembly and DNA synthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E1695–E1704. [Google Scholar] [CrossRef]
- Li, H.; Stümpfig, M.; Zhang, C.; An, X.; Stubbe, J.; Lill, R.; Huang, M. The diferric-tyrosyl radical cluster of ribonucleotide reductase and cytosolic iron-sulfur clusters have distinct and similar biogenesis requirements. J. Biol. Chem. 2017, 292, 11445–11451. [Google Scholar] [CrossRef]
- Furukawa, T.; Naitoh, Y.; Kohno, H.; Tokunaga, R.; Taketani, S. Iron deprivation decreases ribonucleotide reductase activity and DNA synthesis. Life Sci. 1992, 50, 2059–2065. [Google Scholar] [CrossRef]
- Sanvisens, N.; Bañó, M.C.; Huang, M.; Puig, S. Regulation of ribonucleotide reductase in response to iron deficiency. Mol. Cell 2011, 44, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Mishra, P.; Williams, L.G.; Moskalenko, R.; Sharma, S.; Nilsson, A.K.; Watt, D.L.; Andersson, P.; Bergh, A.; Pursell, Z.F.; et al. Altered dNTP pools accelerate tumour formation in mice. Nucleic Acids Res. 2024, 52, 12475–12486. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.; Méndez, J. DNA replication stress: From molecular mechanisms to human disease. Chromosoma 2017, 126, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Spitz, D.R.; Allen, B.G. Iron–sulfur cluster biogenesis as a critical target in cancer. Antioxidants 2021, 10, 1458. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Targeting iron-sulfur clusters in cancer: Opportunities and challenges for ferroptosis-based therapy. Cancers 2023, 15, 2694. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yeast | Human | Function | |
|---|---|---|---|
| ISC system components | Isu | ISCU | Major scaffold protein for Fe-S cluster assembly |
| Nfs1 | NFS1 | Cysteine desulfurase that provides sulfur to Isu/ISCU protein | |
| Yah1 | FDX2 | Ferredoxin involved in electron transport chain from NADH to sulfur | |
| Arh1 | FDXR | Ferredoxin reductase that participates in electron transport chain from NADH to sulfur | |
| Yfh1 | Frataxin | Promotes the delivery of iron to Isu/ISCU protein | |
| Ssq1 | HSPA9 | Chaperone that stabilizes scaffold protein and induces cluster release | |
| Jac1 | HSC20 | Chaperone that stabilizes scaffold protein and induces cluster release | |
| Isa1 | ISCA1 | Synthesizes [4Fe-4S]+ cluster from [2Fe-2S]2+ cluster | |
| Isa2 | ISCA2 | Synthesizes [4Fe-4S]+ cluster from [2Fe-2S]2+ cluster | |
| Iba57 | IBA57 | Synthesizes [4Fe-4S]+ cluster from [2Fe-2S]2+ cluster | |
| Nfu1 | NFU1 | Ensures transfer of Fe-S cluster to mitochondrial targets | |
| Atm1 | ABCB7 | ABC transporter that exports X-S from mitochondria | |
| CIA system components | Cfd1 | CFD1/NUBP2 | Forms complex with Nb35/NBP35 that coordinates [4Fe-4S]2+ clusters |
| Nbp35 | NBP35/NUBP1 | In complex with Cfd1/CFD1, coordinates [4Fe-4S]2+ clusters | |
| Tah18 | NDOR1 | Component of cytosolic electron transport chain | |
| Dre2 | CIAPIN | Component of cytosolic electron transport chain | |
| Grx3-Grx4 | PICOT | Promotes Fe-S cluster assembly on Dre2 | |
| Nar1 | IOP1 | Adapter between early- and late-acting CIA | |
| Cia1 | CIA1 | Inserts Fe-S cluster into target proteins | |
| Cia2 | CIA2B/FAM96B/MIP18 | Inserts Fe-S cluster into target proteins | |
| Met18 | MMS19 | Inserts Fe-S cluster into target proteins |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frigerio, C.; Galli, M.; Castelli, S.; Da Prada, A.; Clerici, M. Control of Replication Stress Response by Cytosolic Fe-S Cluster Assembly (CIA) Machinery. Cells 2025, 14, 442. https://doi.org/10.3390/cells14060442
Frigerio C, Galli M, Castelli S, Da Prada A, Clerici M. Control of Replication Stress Response by Cytosolic Fe-S Cluster Assembly (CIA) Machinery. Cells. 2025; 14(6):442. https://doi.org/10.3390/cells14060442
Chicago/Turabian StyleFrigerio, Chiara, Michela Galli, Sara Castelli, Aurora Da Prada, and Michela Clerici. 2025. "Control of Replication Stress Response by Cytosolic Fe-S Cluster Assembly (CIA) Machinery" Cells 14, no. 6: 442. https://doi.org/10.3390/cells14060442
APA StyleFrigerio, C., Galli, M., Castelli, S., Da Prada, A., & Clerici, M. (2025). Control of Replication Stress Response by Cytosolic Fe-S Cluster Assembly (CIA) Machinery. Cells, 14(6), 442. https://doi.org/10.3390/cells14060442