
Harnessing the Power of Metabolomics for Precision Oncology: Current Advances and Future Directions

Abstract
1. Introduction
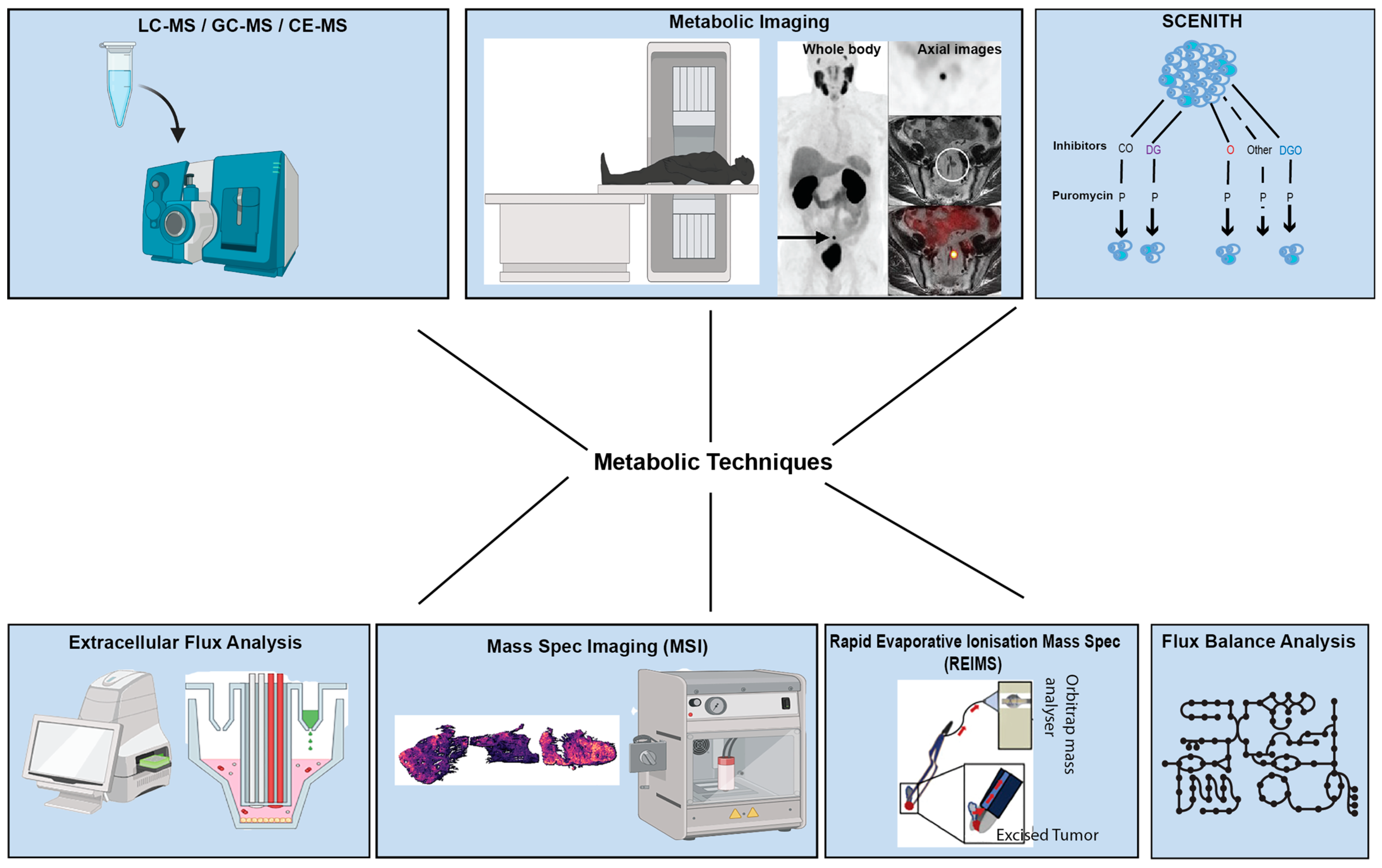
2. An Overview of Technologies and Approaches in Studying Tumour Metabolism
2.1. Nuclear Magnetic Resonance (NMR) Spectroscopy and Mass Spectrometric Methods Remain the Cornerstone of Metabolomic Profiling in Cancer
2.2. Metabolic Imaging Enables the Examination of Metabolism In Vivo
2.3. Matrix-Assisted Laser Desorption/Ionisation (MALDI) and DESI Preserve Spatial Data During Metabolomic Profiling
2.4. Extracellular Flux Analysis (EFA) Facilitates Investigation into Metabolic Phenotypes
2.5. SCENITH Can Be Used for Immuno-Metabolic Profiling Applications
2.6. Rapid Evaporative Ionisation Mass Spectrometry (REIMS) Permits Real-Time Analysis of Surgical Samples
2.7. Flux Balance Analysis (FBA) Allows a near Genome-Wide View of Cancer Metabolism
2.8. Comparison of Different Metabolomics Techniques
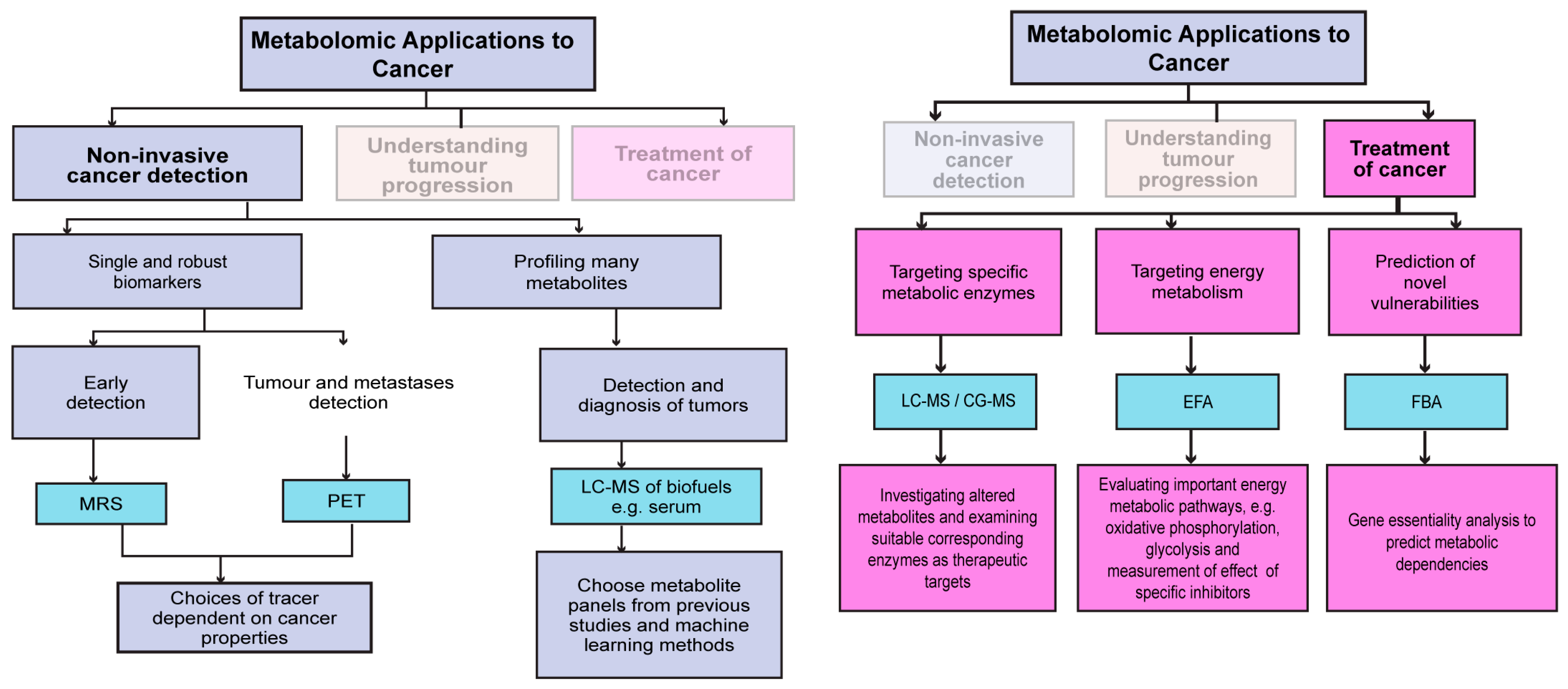
3. The Application of Metabolomics in the Diagnosis and Treatment of Cancer
3.1. Non-Invasive Metabolic Tracking for Tumour Detection and Response to Therapy
3.2. Metabolomic Profiling of Clinical Specimens to Understand Tumour Progression
3.3. Metabolomics Applied to Therapeutic Settings
4. Metabolomics in Multimodal Profiling of Clinical Cohorts
4.1. Fudan Cohort
4.2. Glioblastomas
5. Future Directions
5.1. Using Multi-Omics Tools and Machine Learning to Examine Tumour Metabolism and Predict Therapy Response
5.2. Analytic Techniques Can Aid in Discovering Metabolic Biomarkers or Vulnerabilities
5.3. Single-Cell and Spatial Metabolomics Technologies
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, R.; Maeda, K.; Aki, S.; Osawa, T. Metabolic adaptations of cancer in extreme tumor microenvironments. Cancer Sci. 2023, 114, 1200–1207. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Demicco, M.; Liu, X.Z.; Leithner, K.; Fendt, S.M. Metabolic heterogeneity in cancer. Nat. Metab. 2024, 6, 18–38. [Google Scholar] [CrossRef]
- Danzi, F.; Pacchiana, R.; Mafficini, A.; Scupoli, M.T.; Scarpa, A.; Donadelli, M.; Fiore, A. To metabolomics and beyond: A technological portfolio to investigate cancer metabolism. Signal Transduct. Target. Ther. 2023, 8, 1–22. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef]
- Benedetti, E.; Liu, E.M.; Tang, C.; Kuo, F.; Buyukozkan, M.; Park, T.; Park, J.; Correa, F.; Hakimi, A.A.; Intlekofer, A.M.; et al. A multimodal atlas of tumour metabolism reveals the architecture of gene–metabolite covariation. Nat. Metab. 2023, 5, 1029–1044. [Google Scholar] [CrossRef]
- Trifonova, O.P.; Maslov, D.L.; Balashova, E.E.; Lokhov, P.G. Current State and Future Perspectives on Personalized Metabolomics. Metabolites 2023, 13, 67. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass Spectrometry-Based Metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef]
- Bhinderwala, F.; Wase, N.; DiRusso, C.; Powers, R. Combining Mass Spectrometry and NMR Improves Metabolite Detection and Annotation. J. Proteome Res. 2018, 17, 4017–4022. [Google Scholar] [CrossRef] [PubMed]
- Mayberry, C.L.; Wilson, J.J.; Sison, B.; Chang, C.H. Protocol to assess bioenergetics and mitochondrial fuel usage in murine autoreactive immunocytes using the Seahorse Extracellular Flux Analyzer. STAR Protoc. 2024, 5, 102971. [Google Scholar] [CrossRef] [PubMed]
- Gatidis, S.; Hepp, T.; Früh, M.; La Fougère, C.; Nikolaou, K.; Pfannenberg, C.; Schölkopf, B.; Küstner, T.; Cyran, C.; Rubin, D. A whole-body FDG-PET/CT Dataset with manually annotated Tumor Lesions. Sci. Data 2022, 9, 601. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Inglese, P.; Ghanate, A.; Gjelaj, E.; Wu, V.; Panina, Y.; Mendez-Lucas, A.; MacLachlan, C.; Patani, N.; Hubert, C.B.; et al. Vitamin B5 supports MYC oncogenic metabolism and tumor progression in breast cancer. Nat. Metab. 2023, 5, 1870–1886. [Google Scholar] [CrossRef]
- Liesenfeld, D.B.; Habermann, N.; Owen, R.W.; Scalbert, A.; Ulrich, C.M. Review of mass spectrometry-based metabolomics in cancer research. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2182–2201. [Google Scholar] [CrossRef] [PubMed]
- Moco, S. Studying Metabolism by NMR-Based Metabolomics. Front. Mol. Biosci. 2022, 9, 882487. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Nagana Gowda, G.A.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Raja, G.; Jung, Y.; Jung, S.H.; Kim, T.J. 1H-NMR-based metabolomics for cancer targeting and metabolic engineering—A review. Process Biochem. 2020, 99, 112–122. [Google Scholar] [CrossRef]
- Cortés, I.; Cuadrado, C.; Hernández Daranas, A.; Sarotti, A.M. Machine learning in computational NMR-aided structural elucidation. Front. Nat. Prod. 2023, 2, 1122426. [Google Scholar] [CrossRef]
- Emwas, A.H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar] [CrossRef]
- Bouziani, A.; Yahya, M.; Bouziani, A.; Yahya, M. Mass Spectrometry Coupled with chromatography toward separation and identification of organic mixtures. In Biodegradation Technology of Organic and Inorganic Pollutants; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Bajo-Fernández, M.; Souza-Silva, É.A.; Barbas, C.; Rey-Stolle, M.F.; García, A. GC-MS-based metabolomics of volatile organic compounds in exhaled breath: Applications in health and disease. A review. Front. Mol. Biosci. 2023, 10, 1295955. [Google Scholar] [CrossRef] [PubMed]
- Krone, N.; Hughes, B.A.; Lavery, G.G.; Stewart, P.M.; Arlt, W.; Shackleton, C.H.L. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J. Steroid Biochem. Mol. Biol. 2010, 121, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Komal Pramod, S.; Amol Navnath, K.; Mahesh Pramod, S.; Shri Babanrao, H.; Komal Pramod Hon Shri Babanrao Pachpute Vichardhara, S. A Review On Gas Chromatography-Mass Spectrometry (GC-MS). World J. Pharm. Res. Factor 2021, 10, 741–763. [Google Scholar]
- Pitt, J.J. Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry. Clin. Biochem. Rev. 2009, 30, 19–34. [Google Scholar]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef]
- Ramautar, R. Capillary Electrophoresis-Mass Spectrometry for Clinical Metabolomics. Adv. Clin. Chem. 2016, 74, 1–34. [Google Scholar] [CrossRef]
- Ramautar, R.; Somsen, G.W.; de Jong, G.J. CE-MS for metabolomics: Developments and applications in the period 2016–2018. Electrophoresis 2019, 40, 165–179. [Google Scholar] [CrossRef]
- Emwas, A.H.; Szczepski, K.; Al-Younis, I.; Lachowicz, J.I.; Jaremko, M. Fluxomics—New Metabolomics Approaches to Monitor Metabolic Pathways. Front. Pharmacol. 2022, 13, 805782. [Google Scholar] [CrossRef]
- Pantel, A.R.; Ackerman, D.; Lee, S.C.; Mankoff, D.A.; Gade, T.P. Imaging Cancer Metabolism: Underlying Biology and Emerging Strategies. J. Nucl. Med. 2018, 59, 1340–1349. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Goyal, A. Fludeoxyglucose (18F). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Kawada, K.; Iwamoto, M.; Sakai, Y. Mechanisms underlying 18F-fluorodeoxyglucose accumulation in colorectal cancer. World J. Radiol. 2016, 8, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.Y.; Soloviev, D.; Brindle, K.M. Imaging Tumor Metabolism Using Positron Emission Tomography. Cancer J. 2015, 21, 129–136. [Google Scholar] [CrossRef]
- Kasem, J.; Wazir, U.; Mokbel, K. Sensitivity, Specificity and the Diagnostic Accuracy of PET/CT for Axillary Staging in Patients With Stage I-III Cancer: A Systematic Review of The Literature. In Vivo 2021, 35, 23–30. [Google Scholar] [CrossRef]
- Moses, W.W. Fundamental Limits of Spatial Resolution in PET. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2011, 648 (Suppl. S1), S236–S240. [Google Scholar] [CrossRef] [PubMed]
- Keshari, K.R.; Wilson, D.M. Chemistry and biochemistry of 13C hyperpolarized magnetic resonance using dynamic nuclear polarization. Chem. Soc. Rev. 2014, 43, 1627–1659. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef]
- Brindle, K.M. Imaging cancer metabolism using magnetic resonance. npj Imaging 2024, 2, 1. [Google Scholar] [CrossRef]
- Buchberger, A.R.; DeLaney, K.; Johnson, J.; Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem. 2018, 90, 240–265. [Google Scholar] [CrossRef]
- Aichler, M.; Walch, A. MALDI Imaging mass spectrometry: Current frontiers and perspectives in pathology research and practice. Lab. Investig. 2015, 95, 422–431. [Google Scholar] [CrossRef]
- Núñez-Delgado, A.; Zhang, Z.; Bontempi, E.; Coccia, M.; Race, M.; Zhou, Y.; Wu, J.; Rong, Z.; Xiao, P.; Li, Y. Imaging Method by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS) for Tissue or Tumor: A Mini Review. Processes 2022, 10, 388. [Google Scholar] [CrossRef]
- Takáts, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef]
- Calligaris, D.; Caragacianu, D.; Liu, X.; Norton, I.; Thompson, C.J.; Richardson, A.L.; Golshan, M.; Easterling, M.L.; Santagata, S.; Dillon, D.A.; et al. Application of desorption electrospray ionization mass spectrometry imaging in breast cancer margin analysis. Proc. Natl. Acad. Sci. USA 2014, 111, 15184–15189. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, K.; Caprioli, R.M. Molecular imaging by mass spectrometry—Looking beyond classical histology. Nat. Rev. Cancer 2010, 10, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, L.S.; Ferreira, C.R.; Dill, A.L.; Ifa, D.R.; Cooks, R.G. Desorption electrospray ionization mass spectrometry for lipid characterization and biological tissue imaging. Biochim. Biophys. Acta 2011, 1811, 946–960. [Google Scholar] [CrossRef] [PubMed]
- He, M.J.; Pu, W.; Wang, X.; Zhang, W.; Tang, D.; Dai, Y. Comparing DESI-MSI and MALDI-MSI Mediated Spatial Metabolomics and Their Applications in Cancer Studies. Front. Oncol. 2022, 12, 891018. [Google Scholar] [CrossRef]
- Schwaiger-Haber, M.; Stancliffe, E.; Anbukumar, D.S.; Sells, B.; Yi, J.; Cho, K.; Adkins-Travis, K.; Chheda, M.G.; Shriver, L.P.; Patti, G.J. Using mass spectrometry imaging to map fluxes quantitatively in the tumor ecosystem. Nat. Commun. 2023, 14, 2876. [Google Scholar] [CrossRef]
- Dueñas, M.E.; Larson, E.A.; Lee, Y.J. Toward mass spectrometry imaging in the metabolomics scale: Increasing metabolic coverage through multiple on-tissue chemical modifications. Front. Plant Sci. 2019, 10, 860. [Google Scholar]
- Ren, J.L.; Zhang, A.H.; Kong, L.; Wang, X.J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv. 2018, 8, 22335–22350. [Google Scholar] [CrossRef]
- Caines, J.K.; Barnes, D.A.; Berry, M.D. The Use of Seahorse XF Assays to Interrogate Real-Time Energy Metabolism in Cancer Cell Lines. Methods Mol. Biol. 2022, 2508, 225–234. [Google Scholar] [CrossRef]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef]
- Mercier-Letondal, P.; Marton, C.; Godet, Y.; Galaine, J. Validation of a method evaluating T cell metabolic potential in compliance with ICH Q2 (R1). J. Transl. Med. 2021, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.W.; Throckmorton, H.; Burroughs, S.E. Targeting Energy Metabolism for Cancer Therapeutic Discovery Using Agilent Seahorse XF Technology—Drug Discovery World (DDW); Agilent: Santa Clara, CA, USA, 2019. [Google Scholar]
- Purohit, V.; Wagner, A.; Yosef, N.; Kuchroo, V.K. Systems-based approaches to study immunometabolism. Cell. Mol. Immunol. 2022, 19, 409–420. [Google Scholar] [CrossRef]
- Argüello, R.J.; Combes, A.J.; Char, R.; Gigan, J.P.; Baaziz, A.I.; Bousiquot, E.; Camosseto, V.; Samad, B.; Tsui, J.; Yan, P.; et al. SCENITH: A flow cytometry based method to functionally profile energy metabolism with single cell resolution. Cell Metab. 2020, 32, 1063.e7–1075.e7. [Google Scholar] [CrossRef] [PubMed]
- Vaysse, P.M.; Demers, I.; van den Hout, M.F.C.M.; van de Worp, W.; Anthony, I.G.M.; Baijens, L.W.J.; Tan, B.I.; Lacko, M.; Vaassen, L.A.A.; van Mierlo, A.; et al. Evaluation of the Sensitivity of Metabolic Profiling by Rapid Evaporative Ionization Mass Spectrometry: Toward More Radical Oral Cavity Cancer Resections. Anal. Chem. 2022, 94, 6939–6947. [Google Scholar] [CrossRef] [PubMed]
- Barlow, R.S.; Fitzgerald, A.G.; Hughes, J.M.; McMillan, K.E.; Moore, S.C.; Sikes, A.L.; Tobin, A.B.; Watkins, P.J. Rapid Evaporative Ionization Mass Spectrometry: A Review on Its Application to the Red Meat Industry with an Australian Context. Metabolites 2021, 11, 171. [Google Scholar] [CrossRef]
- Koundouros, N.; Karali, E.; Tripp, A.; Valle, A.; Inglese, P.; Perry, N.J.S.; Magee, D.J.; Anjomani Virmouni, S.; Elder, G.A.; Tyson, A.L.; et al. Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids. Cell 2020, 181, 1596–1611.e1527. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.-E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef]
- Colarusso, A.V.; Goodchild-Michelman, I.; Rayle, M.; Zomorrodi, A.R. Computational modeling of metabolism in microbial communities on a genome-scale. Curr. Opin. Syst. Biol. 2021, 26, 46–57. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Heiden, M.G.V.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef] [PubMed]
- Armitage, E.G.; Barbas, C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef]
- Kdadra, M.; Höckner, S.; Leung, H.; Kremer, W.; Schiffer, E. Metabolomics Biomarkers of Prostate Cancer: A Systematic Review. Diagnostics 2019, 9, 21. [Google Scholar] [CrossRef]
- Long, N.P.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Lim, D.K.; Hong, Y.J.; Hong, S.S.; Kwon, S.W. A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics 2018, 14, 1–26. [Google Scholar] [CrossRef]
- Poli, D.; Carbognani, P.; Corradi, M.; Goldoni, M.; Acampa, O.; Balbi, B.; Bianchi, L.; Rusca, M.; Mutti, A. Exhaled volatile organic compounds in patients with non-small cell lung cancer: Cross sectional and nested short-term follow-up study. Respir. Res. 2005, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, B.M.; Stirdivant, S.M.; Mitchell, M.W.; Wulff, J.E.; McDunn, J.E.; Li, Z.; Dennis-Barrie, A.; Neri, B.P.; Milburn, M.V.; Lotan, Y.; et al. Bladder Cancer Biomarker Discovery Using Global Metabolomic Profiling of Urine. PLoS ONE 2014, 9, e115870. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.; Zhao, W.; Deng, K.; Wang, Z.; Yang, C.; Ma, L.; Openkova, M.S.; Hou, Y.; Li, K.; et al. Metabolomics for biomarker discovery in the diagnosis, prognosis, survival and recurrence of colorectal cancer: A systematic review. Oncotarget 2017, 8, 35460–35472. [Google Scholar] [CrossRef]
- Weber, W.A. Use of PET for monitoring cancer therapy and for predicting outcome. J. Nucl. Med. 2005, 46, 983–995. [Google Scholar]
- Almuhaideb, A.; Papathanasiou, N.; Bomanji, J. 18F-FDG PET/CT Imaging In Oncology. Ann. Saudi Med. 2011, 31, 3–13. [Google Scholar] [CrossRef]
- Pu, Y.; Wang, C.; Zhao, S.; Xie, R.; Zhao, L.; Li, K.; Yang, C.; Zhang, R.; Tian, Y.; Tan, L.; et al. The clinical application of 18F-FDG PET/CT in pancreatic cancer: A narrative review. Transl. Cancer Res. 2021, 10, 3560–3575. [Google Scholar] [CrossRef]
- Griffeth, L.K. Use of PET/CT scanning in cancer patients: Technical and practical considerations. Bayl. Univ. Med. Cent. Proc. 2005, 18, 321–330. [Google Scholar] [CrossRef]
- Verger, A.; Kas, A.; Darcourt, J.; Guedj, E. PET Imaging in Neuro-Oncology: An Update and Overview of a Rapidly Growing Area. Cancers 2022, 14, 1103. [Google Scholar] [CrossRef]
- Cohen, A.S.; Grudzinski, J.; Smith, G.T.; Peterson, T.E.; Whisenant, J.G.; Hickman, T.L.; Ciombor, K.K.; Cardin, D.; Eng, C.; Goff, L.W.; et al. First-in-Human PET Imaging and Estimated Radiation Dosimetry of l-[5-11C]-Glutamine in Patients with Metastatic Colorectal Cancer. J. Nucl. Med. 2022, 63, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Q.; Zhu, Q.; Zhan, Y.; Li, Y.; Huang, X. Role and therapeutic targeting of glutamine metabolism in non-small cell lung cancer (Review). Oncol. Lett. 2023, 25, 159. [Google Scholar] [CrossRef] [PubMed]
- Miner, M.W.; Liljenbäck, H.; Virta, J.; Merisaari, J.; Oikonen, V.; Westermarck, J.; Li, X.G.; Roivainen, A. (2S, 4R)-4-[18F]Fluoroglutamine for In vivo PET Imaging of Glioma Xenografts in Mice: An Evaluation of Multiple Pharmacokinetic Models. Mol. Imaging Biol. 2020, 22, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Umbehr, M.H.; Müntener, M.; Hany, T.; Sulser, T.; Bachmann, L.M. The Role of 11C-Choline and 18F-Fluorocholine Positron Emission Tomography (PET) and PET/CT in Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2013, 64, 106–117. [Google Scholar] [CrossRef]
- Wu, Z.; Zha, Z.; Li, G.; Lieberman, B.P.; Choi, S.R.; Ploessl, K.; Kung, H.F. [18F](2S,4S)-4-(3-Fluoropropyl)glutamine as a tumor imaging agent. Mol. Pharm. 2014, 11, 3852–3866. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Najac, C.; Ronen, S.M. Studies of Metabolism Using (13)C MRS of Hyperpolarized Probes. Methods Enzymol. 2015, 561, 1–71. [Google Scholar] [CrossRef]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; Van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef]
- Albers, M.J.; Bok, R.; Chen, A.P.; Cunningham, C.H.; Zierhut, M.L.; Zhang, V.Y.; Kohler, S.J.; Tropp, J.; Hurd, R.E.; Yen, Y.F.; et al. Hyperpolarized 13C Lactate, Pyruvate, and Alanine: Noninvasive Biomarkers for Prostate Cancer Detection and Grading. Cancer Res. 2008, 68, 8607–8615. [Google Scholar] [CrossRef] [PubMed]
- Day, S.E.; Kettunen, M.I.; Gallagher, F.A.; Hu, D.E.; Lerche, M.; Wolber, J.; Golman, K.; Ardenkjaer-Larsen, J.H.; Brindle, K.M. Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med. 2007, 13, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- de Kouchkovsky, I.; Chen, H.Y.; Ohliger, M.A.; Wang, Z.J.; Bok, R.A.; Gordon, J.W.; Larson, P.E.Z.; Frost, M.; Okamoto, K.; Cooperberg, M.R.; et al. Hyperpolarized 1-[13C]-Pyruvate Magnetic Resonance Imaging Detects an Early Metabolic Response to Immune Checkpoint Inhibitor Therapy in Prostate Cancer. Eur. Urol. 2022, 81, 219–221. [Google Scholar] [CrossRef]
- Mair, R.; Wright, A.J.; Ros, S.; Hu, D.e.; Booth, T.; Kreis, F.; Rao, J.; Watts, C.; Brindle, K.M. Metabolic imaging detects low levels of glycolytic activity that vary with levels of C-MyC expression in patient-derived xenograft models of glioblastoma. Cancer Res. 2018, 78, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Fala, M.; Ros, S.; Sawle, A.; Rao, J.U.; Tsyben, A.; Tronci, L.; Frezza, C.; Mair, R.; Brindle, K.M. The role of branched-chain aminotransferase 1 in driving glioblastoma cell proliferation and invasion varies with tumor subtype. Neuro-Oncol. Adv. 2023, 5, vdad120. [Google Scholar] [CrossRef]
- Gallagher, F.A.; Kettunen, M.I.; Hu, D.E.; Jensen, P.R.; Zandt, R.I.t.; Karlsson, M.; Gisselsson, A.; Nelson, S.K.; Witney, T.H.; Bohndiek, S.E.; et al. Production of hyperpolarized [1,4-13C2]malate from [1,4-13C2]fumarate is a marker of cell necrosis and treatment response in tumors. Proc. Natl. Acad. Sci. USA 2009, 106, 19801–19806. [Google Scholar] [CrossRef]
- Barrio, J.R.; Huang, S.C.; Satyamurthy, N.; Scafoglio, C.S.; Yu, A.S.; Alavi, A.; Krohn, K.A. Does 2-FDG PET Accurately Reflect Quantitative In Vivo Glucose Utilization? J. Nucl. Med. 2020, 61, 931–937. [Google Scholar] [CrossRef]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572.e555. [Google Scholar] [CrossRef]
- Lin, A.; Andronesi, O.; Bogner, W.; Choi, I.Y.; Coello, E.; Cudalbu, C.; Juchem, C.; Kemp, G.J.; Kreis, R.; Krššák, M.; et al. Minimum Reporting Standards for in vivo Magnetic Resonance Spectroscopy (MRSinMRS): Experts’ consensus recommendations. NMR Biomed. 2021, 34, e4484. [Google Scholar] [CrossRef]
- Park, J.; Shin, Y.; Kim, T.H.; Kim, D.H.; Lee, A. Plasma metabolites as possible biomarkers for diagnosis of breast cancer. PLoS ONE 2019, 14, e0225129. [Google Scholar] [CrossRef]
- Miyagi, Y.; Higashiyama, M.; Gochi, A.; Akaike, M.; Ishikawa, T.; Miura, T.; Saruki, N.; Bando, E.; Kimura, H.; Imamura, F.; et al. Plasma Free Amino Acid Profiling of Five Types of Cancer Patients and Its Application for Early Detection. PLoS ONE 2011, 6, e24143. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, B.; Zhao, Y.; Shao, X.; Wang, M.; Ma, F.; Yang, L.; Nie, M.; Jin, P.; Yao, K.; et al. Metabolomic machine learning predictor for diagnosis and prognosis of gastric cancer. Nat. Commun. 2024, 15, 1657. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, A.; Zhou, Y.; Chen, P.; Wang, X.; Huang, J.; Gao, J.; Wang, X.; Shu, L.; Lu, J.; et al. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 2023, 14, 2692. [Google Scholar] [CrossRef]
- Halma, M.T.J.; Tuszynski, J.A.; Marik, P.E. Cancer Metabolism as a Therapeutic Target and Review of Interventions. Nutrients 2023, 15, 4245. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, T.J.; Xu, Y.; Ding, R.; Wang, Y.P.; Jiang, Y.Z.; Shao, Z.M. Emerging therapies in cancer metabolism. Cell Metab. 2023, 35, 1283–1303. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef]
- Rohde, J.M.; Karavadhi, S.; Pragani, R.; Liu, L.; Fang, Y.; Zhang, W.; McIver, A.; Zheng, H.; Liu, Q.; Davis, M.I.; et al. Discovery and optimization of 2H-1λ2-pyridin-2-one inhibitors of mutant isocitrate dehydrogenase 1 for the treatment of cancer. J. Med. Chem. 2021, 64, 4913–4946. [Google Scholar] [CrossRef]
- Gill, K.S.; Fernandes, P.; O’Donovan, T.R.; McKenna, S.L.; Doddakula, K.K.; Power, D.G.; Soden, D.M.; Forde, P.F. Glycolysis inhibition as a cancer treatment and its role in an anti-tumour immune response. Biochim. Biophys. Acta (BBA) 2016, 1866, 87–105. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef]
- McGrail, K.; Granado-Martínez, P.; Esteve-Puig, R.; García-Ortega, S.; Ding, Y.; Sánchez-Redondo, S.; Ferrer, B.; Hernandez-Losa, J.; Canals, F.; Manzano, A.; et al. BRAF activation by metabolic stress promotes glycolysis sensitizing NRASQ61-mutated melanomas to targeted therapy. Nat. Commun. 2022, 13, 7113. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Herrera Juarez, M.; Tolosa Ortega, P.; Sanchez De Torre, A.; Ciruelos Gil, E. Biology of the Triple-Negative Breast Cancer: Immunohistochemical, RNA, and DNA Features. Breast Care 2020, 15, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Ellis, P.; Tutt, A. Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr. Opin. Oncol. 2011, 23, 566–577. [Google Scholar] [CrossRef]
- Sharmni Vishnu, K.; Win, T.T.; Aye, S.N.; Basavaraj, A.K. Combined atezolizumab and nab-paclitaxel in the treatment of triple negative breast cancer: A meta-analysis on their efficacy and safety. BMC Cancer 2022, 22, 1139. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e425. [Google Scholar] [CrossRef]
- Yang, F.; Xiao, Y.; Ding, J.H.; Jin, X.; Ma, D.; Li, D.Q.; Shi, J.X.; Huang, W.; Wang, Y.P.; Jiang, Y.Z.; et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023, 35, 84–100.e108. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Yang, Y.S.; Yang, F.; Ding, J.H.; Gong, Y.; Jiang, L.; Ge, L.P.; Wu, S.Y.; Yu, Q.; et al. Comprehensive metabolomics expands precision medicine for triple-negative breast cancer. Cell Res. 2022, 32, 477–490. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Lombardi, M.Y.; Assem, M. Glioblastoma Genomics: A Very Complicated Story. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 3–25. [Google Scholar] [CrossRef]
- Sidaway, P. Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- García-Montaño, L.A.; Licón-Muñoz, Y.; Martinez, F.J.; Keddari, Y.R.; Ziemke, M.K.; Chohan, M.O.; Piccirillo, S.G.M. Dissecting Intra-tumor Heterogeneity in the Glioblastoma Microenvironment Using Fluorescence-Guided Multiple Sampling. Mol. Cancer Res. 2023, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Holland, E.; DeAngelis, L.M. Glioblastoma: Molecular analysis and clinical implications. Annu. Rev. Med. 2013, 64, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.K.; Morrow, D.; Bayley, N.A.; Fernandez, E.G.; Salinas, J.J.; Tse, C.; Zhu, H.; Su, B.; Plawat, R.; Jones, A.; et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell 2023, 41, 1048–1060.e1049. [Google Scholar] [CrossRef]
- Hu, T.; Allam, M.; Cai, S.; Henderson, W.; Yueh, B.; Garipcan, A.; Ievlev, A.V.; Afkarian, M.; Beyaz, S.; Coskun, A.F. Single-cell spatial metabolomics with cell-type specific protein profiling for tissue systems biology. Nat. Commun. 2023, 14, 8260. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Technique | Advantage | Disadvantage |
|---|---|---|
| GC-MS |
|
|
| LC-MS |
|
|
| Metabolic imaging—PET |
|
|
| Metabolic Imaging—MRS |
|
|
| Mass Spectrometry Imaging |
|
|
| Extracellular Flux Analysis |
|
|
| Category | Fudan Cohort | Glioblastomas |
|---|---|---|
| Type of cancer |
|
|
| Metabolomics performed |
|
|
| Metabolomic subtypings |
|
|
| Metabolite-transcriptomic associations |
|
|
| Therapeutic insights gained |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohli, M.; Poulogiannis, G. Harnessing the Power of Metabolomics for Precision Oncology: Current Advances and Future Directions. Cells 2025, 14, 402. https://doi.org/10.3390/cells14060402
Kohli M, Poulogiannis G. Harnessing the Power of Metabolomics for Precision Oncology: Current Advances and Future Directions. Cells. 2025; 14(6):402. https://doi.org/10.3390/cells14060402
Chicago/Turabian StyleKohli, Manas, and George Poulogiannis. 2025. "Harnessing the Power of Metabolomics for Precision Oncology: Current Advances and Future Directions" Cells 14, no. 6: 402. https://doi.org/10.3390/cells14060402
APA StyleKohli, M., & Poulogiannis, G. (2025). Harnessing the Power of Metabolomics for Precision Oncology: Current Advances and Future Directions. Cells, 14(6), 402. https://doi.org/10.3390/cells14060402