
Proinflammatory Cytokines in Chronic Respiratory Diseases and Their Management

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Classification of Cytokines
2.1. Interleukins
2.2. Tumor Necrosis Factor
2.3. Interferons
2.4. Colony-Stimulating Factor
2.5. Chemokines
2.6. Transforming Growth Factor
2.7. Other Cytokines
3. Pro-Inflammatory Cytokines in Respiratory Diseases
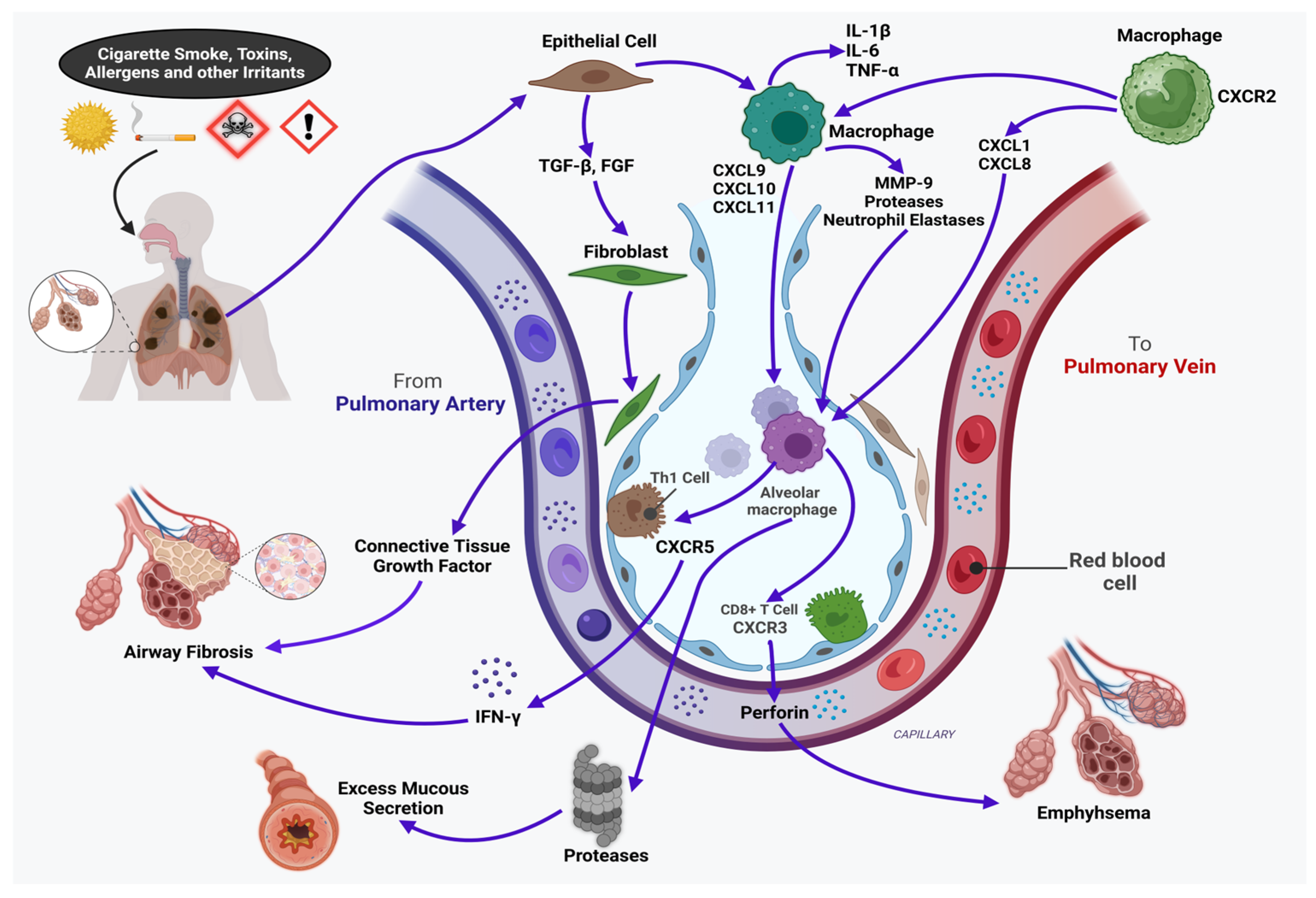
3.1. Chronic Obstructive Pulmonary Disease
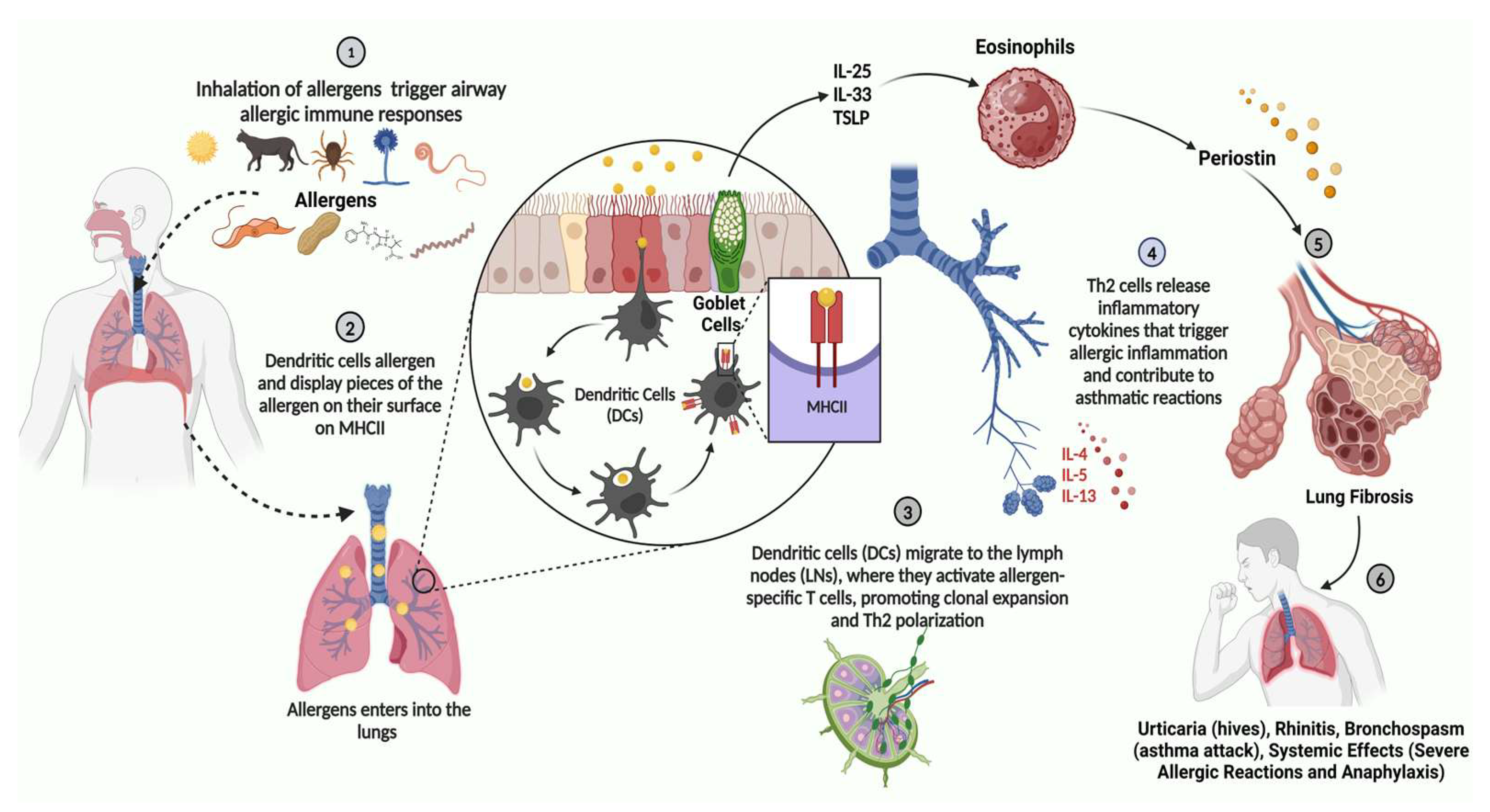
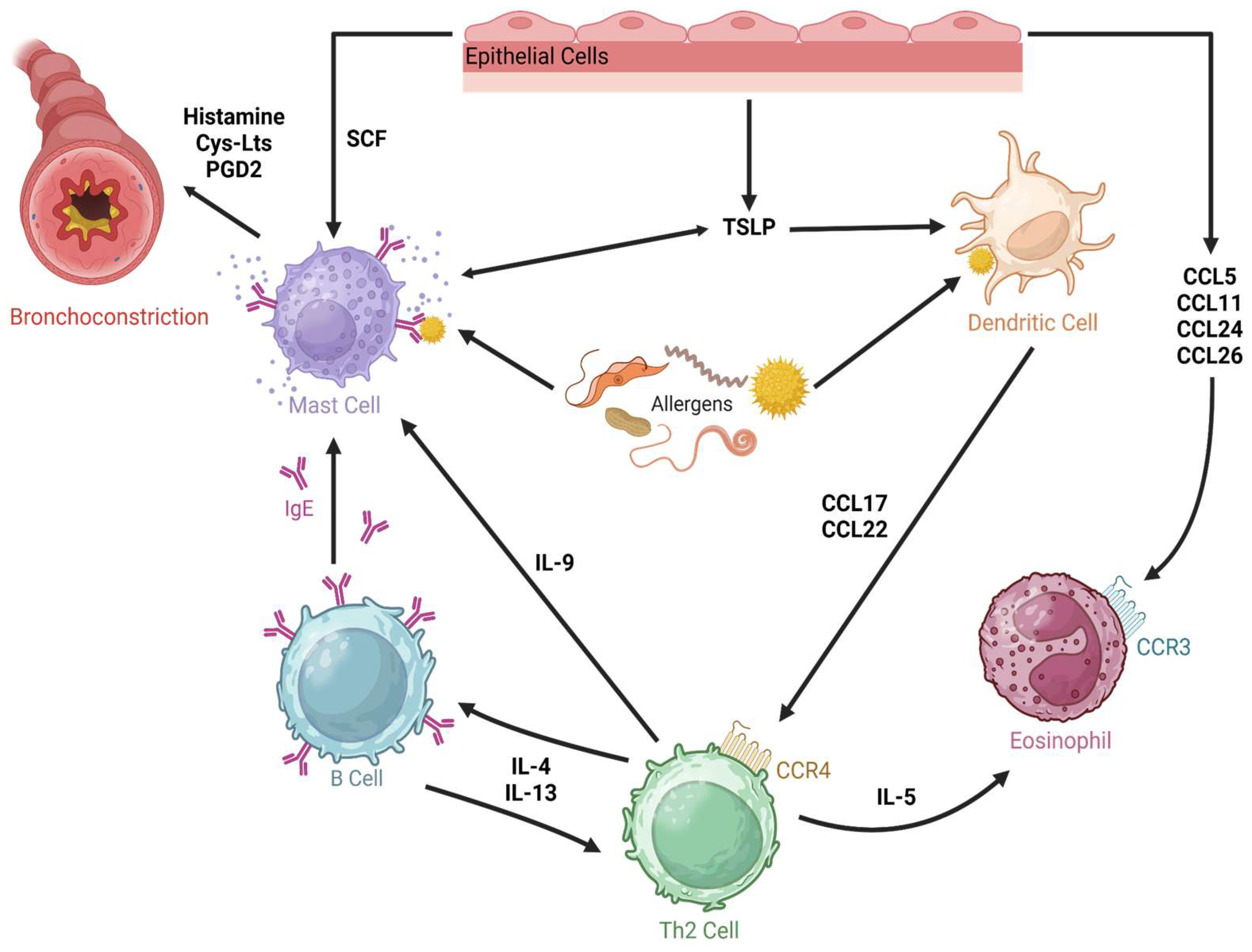
3.2. Asthma
3.3. Pulmonary Fibrosis
3.4. Cystic Fibrosis Bronchiectasis
3.5. COVID-19-Related Respiratory Disease
3.6. Pneumonia
3.7. Lung Cancer
3.8. Tuberculosis
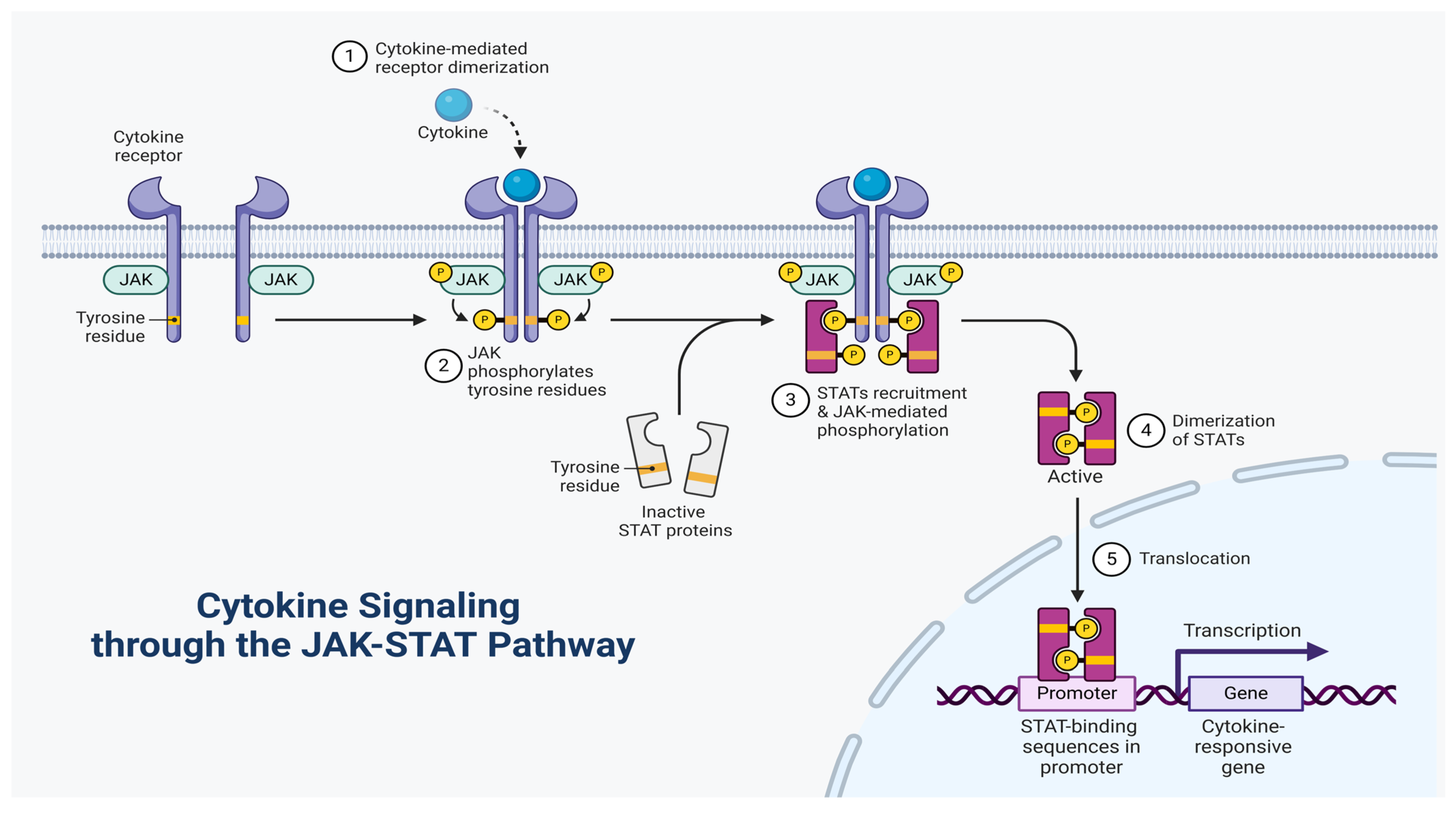
4. Characteristics of Cytokine Receptor Interactions
5. Inflammatory Signaling Involved in Respiratory Diseases
5.1. MAPKs
5.2. Protein Kinase C
5.3. P13K/Akt
5.4. Src Family Kinases
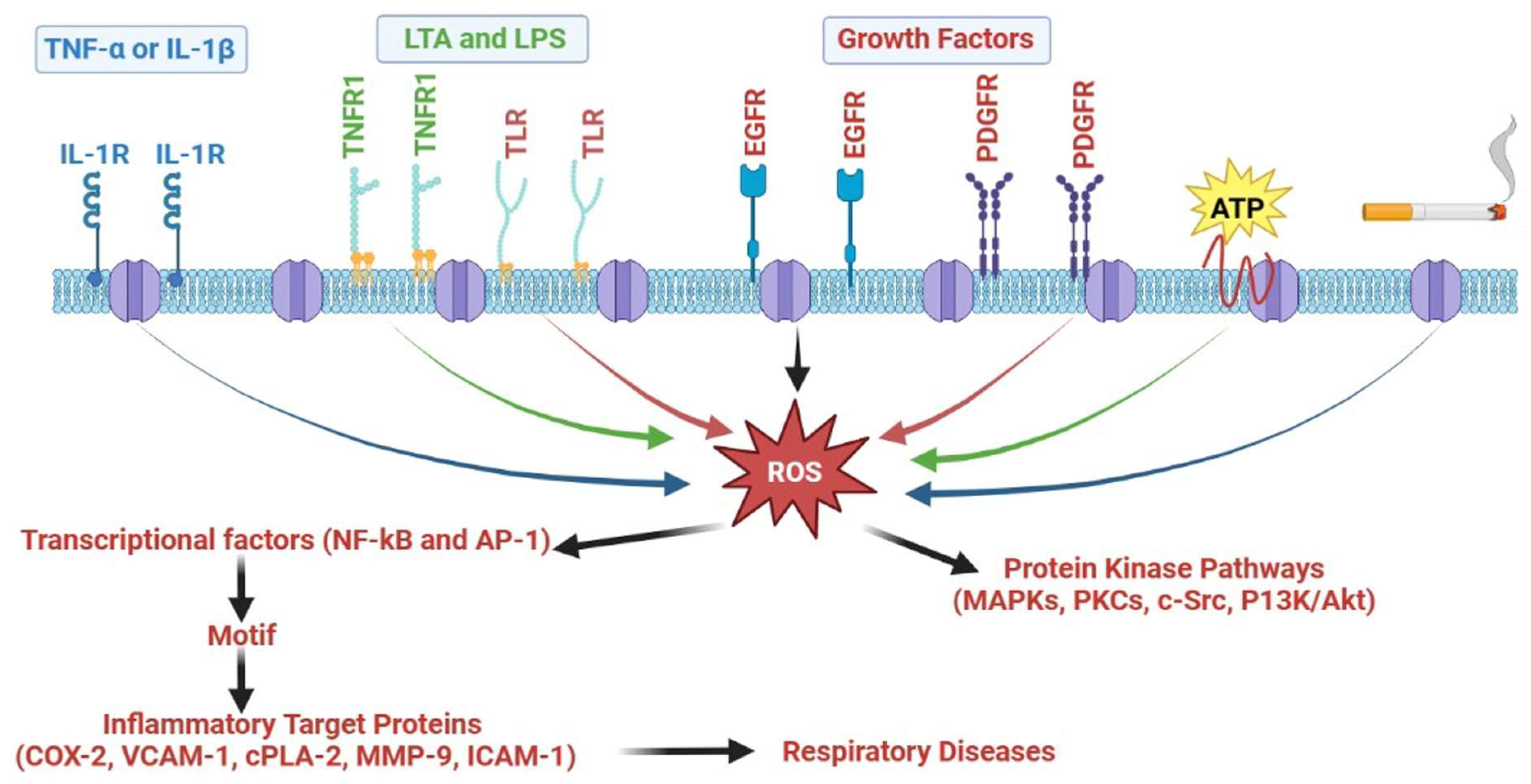
5.5. ROS/NOX
5.6. Activator Protein-1
5.7. Growth Factor Tyrosine Kinase Receptor
5.8. NF-κB
6. Current Cytokine-Targeted Therapeutic Implications
6.1. Future Cytokine-Targeted Therapeutics for Respiratory Diseases
6.1.1. Anti-IL-1β
6.1.2. Anti-IL-6
6.1.3. Anti-IL-8
6.1.4. Anti-IL-4
6.1.5. Anti-IL-5
6.1.6. Anti-IL-9
6.1.7. Anti-IL-13
6.1.8. Anti-IL-17 and Anti-IL-23
6.1.9. Anti-TSLP, Anti-IL-25, and Anti-IL-33
6.1.10. Anti-IL-27
6.1.11. Anti-GM-CSF
6.2. Safety of Anti-Cytokine Therapies
6.3. Ongoing and Completed Clinical Trials for the Management of Respiratory Diseases Based on Cytokine Therapy
6.4. Future Therapeutic Options and Challenges
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agusti, A.; Vogelmeier, C.F.; Halpin, D.M. Tackling the global burden of lung disease through prevention and early diagnosis. Lancet Respir. Med. 2022, 10, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, C.-W.; Fu, Y.-Y.; Li, Y.-Z.; Chen, L.; Zhang, Q.-W.; Chen, Y.-F. Global, regional, and national burden of chronic respiratory diseases and associated risk factors, 1990–2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2023, 10, 1066804. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, H.; Ma, S.; Chen, W.; Sun, L.; Zou, Z. Estimating the global and regional burden of lower respiratory infections attributable to leading pathogens and the protective effectiveness of immunization programs. Int. J. Infect. Dis. 2024, 149, 107268. [Google Scholar] [CrossRef] [PubMed]
- Das, M. WHO urges immediate action to tackle non-communicable diseases. Lancet Oncol. 2022, 23, 1361. [Google Scholar] [CrossRef]
- Denton, E.; O’Hehir, R.E.; Hew, M. The changing global prevalence of asthma and atopic dermatitis. Allergy 2023, 78, 2079. [Google Scholar] [CrossRef]
- Lee, M.O.; Sivasankar, S.; Pokrajac, N.; Smith, C.; Lumba-Brown, A. Emergency department treatment of asthma in children: A review. J. Am. Coll. Emerg. Physicians Open 2020, 1, 1552–1561. [Google Scholar] [CrossRef]
- García-Marcos, L.; Chiang, C.-Y.; Asher, M.I.; Marks, G.B.; El Sony, A.; Masekela, R.; Bissell, K.; Ellwood, E.; Ellwood, P.; Pearce, N. Asthma management and control in children, adolescents, and adults in 25 countries: A Global Asthma Network Phase I cross-sectional study. Lancet Glob. Health 2023, 11, e218–e228. [Google Scholar] [CrossRef]
- Yasaratne, D.; Idrose, N.S.; Dharmage, S.C. Asthma in developing countries in the Asia-Pacific Region (APR). Respirology 2023, 28, 992–1004. [Google Scholar] [CrossRef]
- Cecchi, L.; Annesi-Maesano, I.; d’Amato, G. News on climate change, air pollution, and allergic triggers of asthma. J. Investig. Allergol. Clin. Immunol. 2018, 28, 91–97. [Google Scholar]
- Kaya, A.B.; Çamur, K.C.; Çetin, H.M.; Kaya, S.B.; Erdoğanaras, F. The effects of urban areas, neighborhood and housing on urban health: A systematic review and meta-analysis on asthma. Cities 2023, 143, 104583. [Google Scholar] [CrossRef]
- Mortimer, K.; Kurtulus, S.; Yorgancıoğlu, A.; Romero-Tapia, S.d.J.; Singh, N.; Ahmed, R.; Boladuadua, S.; Masekela, R. Living with Asthma in Low-and Middle-Income Countries in the Six WHO Regions. NEJM Evid. 2023, 3, EVIDpp2300292. [Google Scholar] [CrossRef] [PubMed]
- Balan, I.; Mahmood, S.N.; Jaiswal, R.; Pleshkova, Y.; Manivannan, D.; Negit, S.; Shah, V.; Desai, P.; Akula, N.V.; Nawaz, M.U. Prevalence of active and passive smoking among asthma and asthma-associated emergency admissions: A nationwide prevalence survey study. J. Investig. Med. 2023, 71, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Tiotiu, A.; Ioan, I.; Wirth, N.; Romero-Fernandez, R.; González-Barcala, F.-J. The impact of tobacco smoking on adult asthma outcomes. Int. J. Environ. Res. Public Health 2021, 18, 992. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Folz, R.J.; Ali, U. Environmental factors and their impact on airway diseases: Exploring air pollution, indoor and outdoor allergens, and climate change. Curr. Pulmonol. Rep. 2023, 12, 162–170. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Q. Understanding the Global Cancer Statistics 2018: Implications for cancer control. Sci. China Life Sci. 2021, 64, 1017–1020. [Google Scholar] [CrossRef]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. Współczesna Onkol. 2021, 25, 45–52. [Google Scholar] [CrossRef]
- Kruk, M.E.; Gage, A.D.; Arsenault, C.; Jordan, K.; Leslie, H.H.; Roder-DeWan, S.; Adeyi, O.; Barker, P.; Daelmans, B.; Doubova, S.V. High-quality health systems in the Sustainable Development Goals era: Time for a revolution. Lancet Glob. Health 2018, 6, e1196–e1252. [Google Scholar] [CrossRef]
- Echeverria-Londono, S.; Li, X.; Toor, J.; de Villiers, M.J.; Nayagam, S.; Hallett, T.B.; Abbas, K.; Jit, M.; Klepac, P.; Jean, K. How can the public health impact of vaccination be estimated? BMC Public Health 2021, 21, 2049. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Moghaddam, S.S.; Ghamari, S.-H.; Rad, E.M.; Rezaei, N.; Shobeiri, P.; Aali, A.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abdelmasseh, M. Global burden of chronic respiratory diseases and risk factors, 1990–2019: An update from the Global Burden of Disease Study 2019. EClinicalMedicine 2023, 59, 101936. [Google Scholar] [CrossRef]
- Branchett, W.J.; Lloyd, C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019, 12, 589–600. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2008, 2, 1–11. [Google Scholar] [PubMed]
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.; Regan, K.; Dorward, D.; Rossi, A. Key Mechanisms Governing Resolution of Lung Inflammation. Semin. Immunopathol. 2016, 38, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Antonelli, M.; Kushner, I. It’s time to redefine inflammation. FASEB J. 2017, 31, 1787–1791. [Google Scholar] [CrossRef]
- Chen, C.-M.; Lu, H.-C.; Tung, Y.-T.; Chen, W. Antiplatelet therapy for acute respiratory distress syndrome. Biomedicines 2020, 8, 230. [Google Scholar] [CrossRef]
- Atamas, S.P.; Chapoval, S.P.; Keegan, A.D. Cytokines in chronic respiratory diseases. F1000 Biol. Rep. 2013, 5, 3. [Google Scholar] [CrossRef]
- Bissonnette, E.Y.; Lauzon-Joset, J.-F.; Debley, J.S.; Ziegler, S.F. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front. Immunol. 2020, 11, 583042. [Google Scholar] [CrossRef]
- Fan, W.; Gui, B.; Zhou, X.; Li, L.; Chen, H. A narrative review on lung injury: Mechanisms, biomarkers, and monitoring. Crit. Care 2024, 28, 352. [Google Scholar] [CrossRef]
- Holt, P.G.; Strickland, D.H.; Wikström, M.E.; Jahnsen, F.L. Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 2008, 8, 142–152. [Google Scholar] [CrossRef]
- Manirambona, E.; Okesanya, O.J.; Olaleke, N.O.; Oso, T.A.; Lucero-Prisno, D.E., III. Evolution and implications of SARS-CoV-2 variants in the post-pandemic era. Discov. Public Health 2024, 21, 16. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Swenson, K.E.; Swenson, E.R. Pathophysiology of acute respiratory distress syndrome and COVID-19 lung injury. Crit. Care Clin. 2021, 37, 749–776. [Google Scholar] [CrossRef] [PubMed]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Vanderwall, A.G.; Milligan, E.D. Cytokines in pain: Harnessing endogenous anti-inflammatory signaling for improved pain management. Front. Immunol. 2019, 10, 3009. [Google Scholar] [CrossRef]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef]
- Zhang, J.-M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Mishra, A.K. Role of cytokines in the control of inflammation: A review. Int. J. Ther. Innov. 2023, 1, 5–8. [Google Scholar]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Harvanová, G.; Duranková, S.; Bernasovská, J. The role of cytokines and chemokines in the inflammatory response. Alergol. Pol. Pol. J. Allergol. 2023, 10, 210–219. [Google Scholar] [CrossRef]
- Ferreira, V.L.; Borba, H.H.; Bonetti, A.d.F.; Leonart, L.; Pontarolo, R. Cytokines and interferons: Types and functions. Autoantibodies Cytokines 2018, 13, 1–25. [Google Scholar]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721. e770. [Google Scholar] [PubMed]
- Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and therapeutic applications of tumor necrosis factor-α (TNF-α) in major depressive disorder: A systematic review. Int. J. Mol. Sci. 2016, 17, 733. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Watanabe, R.; Ito, H.; Fujii, T.; Okuma, K.; Oku, T.; Hirayama, Y.; Ohmura, K.; Murata, K.; Murakami, K. Dynamics of type I and type II interferon signature determines responsiveness to anti-TNF therapy in rheumatoid arthritis. Front. Immunol. 2022, 13, 901437. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Utomo, B.S.R.; Hatta, M.; Sirait, R.H.; Pratiwi, S.; Massi, M.N. The role of cytokine interleukin-2, transcription factor of FoxP3 in the immunological regulation of allergic rhinitis. Int. J. Otolaryngol. Head Neck Surg. 2018, 7, 7–19. [Google Scholar] [CrossRef]
- Dalskov, L.; Gad, H.H.; Hartmann, R. Viral recognition and the antiviral interferon response. EMBO J. 2023, 42, e112907. [Google Scholar] [CrossRef]
- Moreau, T.R.; Bondet, V.; Rodero, M.P.; Duffy, D. Heterogeneity and functions of the 13 IFN-α subtypes–lucky for some? Eur. J. Immunol. 2023, 53, 2250307. [Google Scholar] [CrossRef]
- Strayer, D.R.; Carter, W.A.; Stouch, B.C.; Stittelaar, K.J.; Thoolen, R.J.; Osterhaus, A.D.; Mitchell, W.M. Protection from pulmonary tissue damage associated with infection of cynomolgus macaques by highly pathogenic avian influenza virus (H5N1) by low dose natural human IFN-alpha administered to the buccal mucosa. Antiviral Res. 2014, 110, 175–180. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Metcalf, D. The colony-stimulating factors and cancer. Nat. Rev. Cancer 2010, 10, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Cook, A.D.; Tak, P.P. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat. Rev. Drug Discov. 2017, 16, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.J.; Hayward, J.A.; Huang, C.E.; Huma, Z.; Sanchez, J. Mechanisms of regulation of the chemokine-receptor network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef]
- Alfaro, C.; Sanmamed, M.F.; Rodríguez-Ruiz, M.E.; Teijeira, Á.; Oñate, C.; González, Á.; Ponz, M.; Schalper, K.A.; Pérez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar]
- Yue, X.; Wu, L.; Hu, W. The regulation of leukemia inhibitory factor. Cancer Cell Microenviron. 2015, 2, e877. [Google Scholar]
- Mozaffarian, A.; Brewer, A.W.; Trueblood, E.S.; Luzina, I.G.; Todd, N.W.; Atamas, S.P.; Arnett, H.A. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J. Immunol. 2008, 181, 7243–7253. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef]
- Li, L.; Xue, M.; Fu, F.; Yin, L.; Feng, L.; Liu, P. IFN-lambda 3 mediates antiviral protection against porcine epidemic diarrhea virus by inducing a distinct antiviral transcript profile in porcine intestinal epithelia. Front. Immunol. 2019, 10, 2394. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Lim, J.-S. Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch. Pharmacal Res. 2016, 39, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Cockx, M.; Gouwy, M.; Van Damme, J.; Struyf, S. Chemoattractants and cytokines in primary ciliary dyskinesia and cystic fibrosis: Key players in chronic respiratory diseases. Cell. Mol. Immunol. 2018, 15, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Investig. 2016, 126, 2394–2403. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, B.; et al. Expression and Cellular Provenance of Thymic Stromal Lymphopoietin and Chemokines in Patients with Severe Asthma and Chronic Obstructive Pulmonary Disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef]
- Gao, P.; Gibson, P.G.; Baines, K.J.; Yang, I.A.; Upham, J.W.; Reynolds, P.N.; Hodge, S.; James, A.L.; Jenkins, C.; Peters, M.J. Anti-inflammatory deficiencies in neutrophilic asthma: Reduced galectin-3 and IL-1RA/IL-1β. Respir. Res. 2015, 16, 5. [Google Scholar] [CrossRef]
- Robinson, D.; Humbert, M.; Buhl, R.; Cruz, A.A.; Inoue, H.; Korom, S.; Hanania, N.A.; Nair, P. Revisiting T ype 2-high and T ype 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clin. Exp. Allergy 2017, 47, 161–175. [Google Scholar] [CrossRef]
- Lai, T.; Tian, B.; Cao, C.; Hu, Y.; Zhou, J.; Wang, Y.; Wu, Y.; Li, Z.; Xu, X.; Zhang, M. HDAC2 suppresses IL17A-mediated airway remodeling in human and experimental modeling of COPD. Chest 2018, 153, 863–875. [Google Scholar] [CrossRef]
- Kang, M.-J.; Choi, J.-M.; Kim, B.H.; Lee, C.-M.; Cho, W.-K.; Choe, G.; Kim, D.-H.; Lee, C.G.; Elias, J.A. IL-18 induces emphysema and airway and vascular remodeling via IFN-γ, IL-17A, and IL-13. Am. J. Respir. Crit. Care Med. 2012, 185, 1205–1217. [Google Scholar] [CrossRef]
- Bernhagen, J. A new cytokine target for chronic obstructive pulmonary disease? EBioMedicine 2021, 69, 103479. [Google Scholar] [CrossRef]
- Song, S.; Liu, B.; Habibie, H.; van den Bor, J.; Smit, M.J.; Gosens, R.; Wu, X.; Brandsma, C.-A.; Cool, R.H.; Haisma, H.J. D-dopachrome tautomerase contributes to lung epithelial repair via atypical chemokine receptor 3-dependent Akt signaling. EBioMedicine 2021, 68, 103479. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.-M.; Zhao, C.-N.; Leng, J.; Leng, R.-X.; Ye, D.-Q.; Zheng, S.G.; Pan, H.-F. Interleukin-13: A promising therapeutic target for autoimmune disease. Cytokine Growth Factor Rev. 2019, 45, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Grabner, A.; Baumlin, N.; Yanucil, C.; Helton, S.; Grosche, A.; Sailland, J.; Geraghty, P.; Viera, L.; Russell, D.W. Fibroblast growth factor 23 and Klotho contribute to airway inflammation. Eur. Respir. J. 2018, 52, 1800236. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- Harun, S.N.; Holford, N.H.; Grimwood, K.; Wainwright, C.E.; Hennig, S. Pseudomonas aeruginosa eradication therapy and risk of acquiring Aspergillus in young children with cystic fibrosis. Thorax 2019, 74, 740–748. [Google Scholar] [CrossRef]
- Muselet-Charlier, C.; Roque, T.; Boncoeur, E.; Chadelat, K.; Clement, A.; Jacquot, J.; Tabary, O. Enhanced IL-1β-induced IL-8 production in cystic fibrosis lung epithelial cells is dependent of both mitogen-activated protein kinases and NF-κB signaling. Biochem. Biophys. Res. Commun. 2007, 357, 402–407. [Google Scholar] [CrossRef]
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic fibrosis lung disease modifiers and their relevance in the new era of precision medicine. Genes 2021, 12, 562. [Google Scholar] [CrossRef]
- Abolfathi, H.; Sheikhpour, M.; Shahraeini, S.S.; Khatami, S.; Nojoumi, S.A. Studies in lung cancer cytokine proteomics: A review. Expert Rev. Proteom. 2021, 18, 49–64. [Google Scholar] [CrossRef]
- Chanda, C. Role of inflammatory cytokines during lung cancer progression: A review. Res. J. Pharm. Technol. 2018, 11, 5163–5165. [Google Scholar] [CrossRef]
- Kellum, J.A.; Kong, L.; Fink, M.P.; Weissfeld, L.A.; Yealy, D.M.; Pinsky, M.R.; Fine, J.; Krichevsky, A.; Delude, R.L.; Angus, D.C. Understanding the inflammatory cytokine response in pneumonia and sepsis: Results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch. Intern. Med. 2007, 167, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Leme, R.; Zing, N.; Murad, N.; Adami, F.; Hinnig, P.; Feder, D.; Chagas, A.; Fonseca, F. IL-6 and TNF-α serum levels are associated with early death in community-acquired pneumonia patients. Braz. J. Med. Biol. Res. 2015, 48, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Dukhinova, M.; Kokinos, E.; Kuchur, P.; Komissarov, A.; Shtro, A. Macrophage-derived cytokines in pneumonia: Linking cellular immunology and genetics. Cytokine Growth Factor Rev. 2021, 59, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-W.; Lin, F.-C.; Tsai, H.-C.; Chang, S.-C. The importance of pro-inflammatory and anti-inflammatory cytokines in Pneumocystis jirovecii pneumonia. Med. Mycol. 2013, 51, 704–712. [Google Scholar] [CrossRef]
- Bagnato, G.L.; Irrera, N.; Pizzino, G.; Santoro, D.; Roberts, W.N.; Bagnato, G.; Pallio, G.; Vaccaro, M.; Squadrito, F.; Saitta, A. Dual αvβ3 and αvβ5 blockade attenuates fibrotic and vascular alterations in a murine model of systemic sclerosis. Clin. Sci. 2018, 132, 231–242. [Google Scholar] [CrossRef]
- Etna, M.P.; Giacomini, E.; Severa, M.; Coccia, E.M. Pro- and anti-inflammatory cytokines in tuberculosis: A two-edged sword in TB pathogenesis. Semin. Immunol. 2014, 6, 543–551. [Google Scholar] [CrossRef]
- Defnet, A.E.; Shah, S.D.; Huang, W.; Shapiro, P.; Deshpande, D.A.; Kane, M.A. Dysregulated retinoic acid signaling in airway smooth muscle cells in asthma. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e22016. [Google Scholar] [CrossRef]
- Gubernatorova, E.O.; Namakanova, O.A.; Gorshkova, E.A.; Medvedovskaya, A.D.; Nedospasov, S.A.; Drutskaya, M.S. Novel anti-cytokine strategies for prevention and treatment of respiratory allergic diseases. Front. Immunol. 2021, 12, 601842. [Google Scholar] [CrossRef]
- Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung fibrosis and fibrosis in the lungs: Is it all about myofibroblasts? Biomedicines 2022, 10, 1423. [Google Scholar] [CrossRef]
- Torrisi, S.E.; Ley, B.; Kreuter, M.; Wijsenbeek, M.; Vittinghoff, E.; Collard, H.R.; Vancheri, C. The added value of comorbidities in predicting survival in idiopathic pulmonary fibrosis: A multicentre observational study. Eur. Respir. J. 2019, 53, 1801587. [Google Scholar] [CrossRef]
- Renema, P.; Hardy, K.S.; Housley, N.; Dunbar, G.; Annamdevula, N.; Britain, A.; Spadafora, D.; Leavesley, S.; Rich, T.; Audia, J.P. cAMP signaling primes lung endothelial cells to activate caspase-1 during Pseudomonas aeruginosa infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1074–L1083. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Alom, S.; Shakya, A.; Ghosh, S.K.; Singh, U.P.; Bhat, H.R. Implication of in silico studies in the search for novel inhibitors against SARS-CoV-2. Arch. Der Pharm. 2022, 355, 2100360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Karimi, A.; Hashemian, S.M.R. Cytokine storm in COVID-19 and the treatment simulacrum. Biomed. Biotechnol. Res. J. 2020, 4, S41–S48. [Google Scholar]
- Zhang, C.; Wu, Z.; Li, J.-W.; Zhao, H.; Wang, G.-Q. Cytokine release syndrome in severe COVID-19: Interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; Saik, I.E.I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 virus use multiple receptors to enter host cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, K.; Wang, D.; Yue, X.; Song, D.; Zhu, Y.; Wu, J. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-κB. Virology 2007, 365, 324–335. [Google Scholar] [CrossRef]
- Ebihara, N.; Matsuda, A.; Nakamura, S.; Matsuda, H.; Murakami, A. Role of the IL-6 classic-and trans-signaling pathways in corneal sterile inflammation and wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8549–8557. [Google Scholar] [CrossRef] [PubMed]
- Kihel, A.; Hammi, I.; Darif, D.; Lemrani, M.; Riyad, M.; Guessous, F.; Akarid, K. The different faces of the NLRP3 inflammasome in cutaneous Leishmaniasis: A review. Cytokine 2021, 147, 155248. [Google Scholar] [CrossRef] [PubMed]
- Orlov, M.; Wander, P.L.; Morrell, E.D.; Mikacenic, C.; Wurfel, M.M. A case for targeting Th17 cells and IL-17A in SARS-CoV-2 infections. J. Immunol. 2020, 205, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Feldman, C. The global burden of community-acquired pneumonia in adults, encompassing invasive pneumococcal disease and the prevalence of its associated cardiovascular events, with a focus on pneumolysin and macrolide antibiotics in pathogenesis and therapy. Int. J. Mol. Sci. 2023, 24, 11038. [Google Scholar] [CrossRef]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J. Three unique interstitial macrophages in the murine lung at steady state. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef]
- Schyns, J.; Bai, Q.; Ruscitti, C.; Radermecker, C.; De Schepper, S.; Chakarov, S.; Farnir, F.; Pirottin, D.; Ginhoux, F.; Boeckxstaens, G. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun. 2019, 10, 3964. [Google Scholar] [CrossRef]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Rich, H.E.; Alcorn, J.F. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. 2018, 11, 199–208. [Google Scholar] [CrossRef]
- Costantini, A.; Viola, N.; Berretta, A.; Galeazzi, R.; Matacchione, G.; Sabbatinelli, J.; Storci, G.; De Matteis, S.; Butini, L.; Rippo, M.R. Age-related M1/M2 phenotype changes in circulating monocytes from healthy/unhealthy individuals. Aging 2018, 10, 1268. [Google Scholar] [CrossRef]
- Fernandez-Botran, R.; Uriarte, S.M.; Arnold, F.W.; Rodriguez-Hernandez, L.; Rane, M.J.; Peyrani, P.; Wiemken, T.; Kelley, R.; Uppatla, S.; Cavallazzi, R.; et al. Contrasting Inflammatory Responses in Severe and Non-severe Community-acquired Pneumonia. Inflammation 2014, 37, 1158–1166. [Google Scholar] [CrossRef]
- Paats, M.S.; Bergen, I.M.; Hanselaar, W.E.; van Zoelen, E.C.G.; Hoogsteden, H.C.; Hendriks, R.W.; Van der Eerden, M.M. Local and systemic cytokine profiles in nonsevere and severe community-acquired pneumonia. Eur. Respir. J. 2013, 41, 1378–1385. [Google Scholar] [CrossRef]
- Kragsbjerg, P.; Jones, I.; Vikerfors, T.; Holmberg, H. Diagnostic value of blood cytokine concentrations in acute pneumonia. Thorax 1995, 50, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Lee, H.-J.; Chang, J.-E. Inflammatory cytokine: An attractive target for cancer treatment. Biomedicines 2022, 10, 2116. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Brenner, D.R.; Fanidi, A.; Grankvist, K.; Muller, D.C.; Brennan, P.; Manjer, J.; Byrnes, G.; Hodge, A.; Severi, G.; Giles, G.G. Inflammatory cytokines and lung cancer risk in 3 prospective studies. Am. J. Epidemiol. 2017, 185, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.-S.; Liu, L.; Qin, Y.-W. IL-6 and TNF-α promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Lin, Q.; Xue, L.; Tian, T.; Zhang, B.; Guo, L.; Lin, G.; Chen, Z.; Fan, K.; Gu, X. Prognostic value of serum IL-17 and VEGF levels in small cell lung cancer. Int. J. Biol. Markers 2015, 30, 359–363. [Google Scholar] [CrossRef]
- Timperi, E.; Focaccetti, C.; Gallerano, D.; Panetta, M.; Spada, S.; Gallo, E.; Visca, P.; Venuta, F.; Diso, D.; Prelaj, A. IL-18 receptor marks functional CD8+ T cells in non-small cell lung cancer. Oncoimmunology 2017, 6, e1328337. [Google Scholar] [CrossRef]
- Lee, H.-M.; Yuk, J.-M.; Shin, D.-M.; Jo, E.-K. Dectin-1 is Inducible and Plays an Essential Role for Mycobacteria-Induced Innate Immune Responses in Airway Epithelial Cells. J. Clin. Immunol. 2009, 6, 795–805. [Google Scholar] [CrossRef]
- Roy, S.; Sharma, S.; Sharma, M.; Aggarwal, R.; Bose, M. Induction of nitric oxide release from the human alveolar epithelial cell line A549: An in vitro correlate of innate immune response to Mycobacterium tuberculosis. Immunology 2004, 3, 471–480. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Jiao, J.; Huang, Q. Mycobacterium tuberculosis infection induces IL-10 gene expression by disturbing histone deacetylase 6 and histonedeacetylase 11 equilibrium in macrophages. Tuberculosis 2018, 108, 118–123. [Google Scholar] [CrossRef]
- Olsen, A.; Chen, Y.; Ji, Q.; Zhu, G.; De Silva, A.D.; Vilchèze, C.; Weisbrod, T.; Li, W.; Xu, J.; Larsen, M.; et al. Targeting Mycobacterium tuberculosis Tumor Necrosis Factor Alpha-Downregulating Genes for the Development of Antituberculous Vaccines. mBio 2016, 7, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Spangler, J.B.; Moraga, I.; Mendoza, J.L.; Garcia, K.C. Insights into cytokine–receptor interactions from cytokine engineering. Annu. Rev. Immunol. 2015, 33, 139–167. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.; Ueda, G.; Kim, A.R.; Jude, K.M.; Fallas, J.A.; Guo, Y.; Hafer, M.; Miao, Y.; Saxton, R.A.; Piehler, J. Topological control of cytokine receptor signaling induces differential effects in hematopoiesis. Science 2019, 364, eaav7532. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and specificity: Insights from the interleukin 6 family of cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef]
- Hercus, T.R.; Dhagat, U.; Kan, W.L.; Broughton, S.E.; Nero, T.L.; Perugini, M.; Sandow, J.J.; D’Andrea, R.J.; Ekert, P.G.; Hughes, T. Signalling by the βc family of cytokines. Cytokine Growth Factor Rev. 2013, 24, 189–201. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Kossiakoff, A.A.; de Vos, A.M. Structural basis for cytokine hormone-receptor recognition and receptor activation. Adv. Protein Chem. 1998, 52, 67–108. [Google Scholar]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural basis of IL-1 family cytokine signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Scheide-Noeth, J.-P.; Rosen, M.; Baumstark, D.; Dietz, H.; Mueller, T.D. Structural basis of interleukin-5 inhibition by the small cyclic peptide AF17121. J. Mol. Biol. 2019, 431, 714–731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Gao, J.; Xue, Y.; Qin, Y.; Li, X.; Sun, Z.; Xie, H.; Chang, M.; Nie, P.; Zou, J. Identification and expression analysis of IL-4/13 receptors in grass carp Ctenopharyngodon idella. Fish Shellfish. Immunol. 2019, 87, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.R. The role of structure in the biology of interferon signaling. Front. Immunol. 2020, 11, 606489. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Cochran, J.R. Targeting ligand–receptor interactions for development of cancer therapeutics. Curr. Opin. Chem. Biol. 2017, 38, 62–69. [Google Scholar] [CrossRef]
- Deshpande, D.A.; Guedes, A.G.; Graeff, R.; Dogan, S.; Subramanian, S.; Walseth, T.F.; Kannan, M.S. CD38/cADPR signaling pathway in airway disease: Regulatory mechanisms. Mediat. Inflamm. 2018, 2018, 8942042. [Google Scholar] [CrossRef]
- Santana, F.P.; da Silva, R.C.; Ponci, V.; Pinheiro, A.J.; Olivo, C.R.; Caperuto, L.C.; Arantes-Costa, F.M.; Claudio, S.R.; Ribeiro, D.A.; Tibério, I.F. Dehydrodieugenol improved lung inflammation in an asthma model by inhibiting the STAT3/SOCS3 and MAPK pathways. Biochem. Pharmacol. 2020, 180, 114175. [Google Scholar] [CrossRef]
- Pelaia, C.; Vatrella, A.; Gallelli, L.; Lombardo, N.; Sciacqua, A.; Savino, R.; Pelaia, G. Role of p38 mitogen-activated protein kinase in asthma and COPD: Pathogenic aspects and potential targeted therapies. Drug Des. Dev. Ther. 2021, 1275–1284. [Google Scholar] [CrossRef]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef]
- Verma, A.K.; Singh, M.F.; Awasthi, A. Role of p38 MAP Kinase Inhibitor (SB239063) and Vitamin B 12 against Neuroinflammation. Int. J. Pharm. Investig. 2021, 11, 94–98. [Google Scholar] [CrossRef]
- Che, L.; Yu, C.; Chen, G.; Lin, J.; Xie, Z.; Xia, T.; Luo, W.; Cai, X.; Liu, S. The inflammatory response induced by RELMβ upregulates IL-8 and IL-1β expression in bronchial epithelial cells in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2021, 2503–2513. [Google Scholar] [CrossRef]
- Popmihajlov, Z.; Sutherland, D.J.; Horan, G.S.; Ghosh, A.; Lynch, D.A.; Noble, P.W.; Richeldi, L.; Reiss, T.F.; Greenberg, S. CC-90001, a c-Jun N-terminal kinase (JNK) inhibitor, in patients with pulmonary fibrosis: Design of a phase 2, randomised, placebo-controlled trial. BMJ Open Respir. Res. 2022, 9, e001060. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.; Borchard, G.; Peptide Structure Optimisation. UnrTablavelling the Modulation of Tight Junctions: Molecular Mechanisms and Permeation Enhancement. 2022. Available online: https://m2.mtmt.hu/api/citation/32564792?&format=xml (accessed on 21 February 2025).
- Mehta, M.; Dhanjal, D.S.; Satija, S.; Wadhwa, R.; Paudel, K.R.; Chellappan, D.K.; Mohammad, S.; Haghi, M.; Hansbro, P.M.; Dua, K. Advancing of cellular signaling pathways in respiratory diseases using nanocarrier based drug delivery systems. Curr. Pharm. Des. 2020, 26, 5380–5392. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Li, J.; Zhou, L.; Iasvoyskaia, S.; Corbit, K.C.; Soh, J.-W.; Weinstein, I.B.; Brasier, A.R.; Lin, A.; Hershenson, M.B. Regulation of airway epithelial cell NF-κB-dependent gene expression by protein kinase Cδ. J. Immunol. 2003, 170, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Nakano, R.; Kitanaka, T.; Namba, S.; Kitanaka, N.; Sugiya, H. Protein kinase Cε regulates nuclear translocation of extracellular signal-regulated kinase, which contributes to bradykinin-induced cyclooxygenase-2 expression. Sci. Rep. 2018, 8, 8535. [Google Scholar] [CrossRef]
- Wu, T.; Han, C.; Shelhamer, J.H. Involvement of p38 and p42/44 MAP kinases and protein kinase C in the interferon-γ and interleukin-1α-induced phosphorylation of 85-kDa cytosolic phospholipase A2 in primary human bronchial epithelial cells. Cytokine 2004, 25, 11–20. [Google Scholar] [CrossRef]
- Lien, C.-F.; Chen, S.-J.; Tsai, M.-C.; Lin, C.-S. Potential role of protein kinase C in the pathophysiology of diabetes-associated atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef]
- Lin, C.-H.; Cheng, H.-W.; Ma, H.-P.; Wu, C.-H.; Hong, C.-Y.; Chen, B.-C. Thrombin induces NF-κB activation and IL-8/CXCL8 expression in lung epithelial cells by a Rac1-dependent PI3K/Akt pathway. J. Biol. Chem. 2011, 286, 10483–10494. [Google Scholar] [CrossRef]
- Lee, I.T.; Lin, C.C.; Wu, Y.C.; Yang, C.M. TNF-α induces matrix metalloproteinase-9 expression in A549 cells: Role of TNFR1/TRAF2/PKCα-dependent signaling pathways. J. Cell. Physiol. 2010, 224, 454–464. [Google Scholar] [CrossRef]
- Basu, B.; Ghosh, S.; Das, S.; Das, A. Implications of Phosphoinositide 3-Kinase (PI3K) Signalling in Cellular and Molecular Mechanisms of Respiratory Diseases. In Targeting Cellular Signalling Pathways in Lung Diseases; Springer: Berlin/Heidelberg, Germany, 2021; pp. 601–623. [Google Scholar]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sin. B 2022, 12, 18–32. [Google Scholar] [CrossRef]
- Khezri, M.R.; Varzandeh, R.; Ghasemnejad-Berenji, M. The probable role and therapeutic potential of the PI3K/AKT signaling pathway in SARS-CoV-2 induced coagulopathy. Cell. Mol. Biol. Lett. 2022, 27, 6. [Google Scholar] [CrossRef]
- Shimizu, T.; Saito, T.; Aoki-Saito, H.; Okada, S.; Ikeda, H.; Nakakura, T.; Fukuda, H.; Arai, S.; Fujiwara, K.; Nakajima, Y. Resolvin E3 ameliorates high-fat diet-induced insulin resistance via the phosphatidylinositol-3-kinase/Akt signaling pathway in adipocytes. FASEB J. 2022, 36, e22188. [Google Scholar] [CrossRef] [PubMed]
- Traina, G.; Bolzacchini, E.; Bonini, M.; Contini, D.; Mantecca, P.; Caimmi, S.M.E.; Licari, A. Role of air pollutants mediated oxidative stress in respiratory diseases. Pediatr. Allergy Immunol. 2022, 33, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. 2020, 127, 110183. [Google Scholar] [CrossRef] [PubMed]
- Agraval, H.; Sharma, J.R.; Prakash, N.; Yadav, U.C. Fisetin suppresses cigarette smoke extract-induced epithelial to mesenchymal transition of airway epithelial cells through regulating COX-2/MMPs/β-catenin pathway. Chem. Biol. Interact. 2022, 351, 109771. [Google Scholar] [CrossRef]
- Severgnini, M.; Takahashi, S.; Tu, P.; Perides, G.; Homer, R.J.; Jhung, J.W.; Bhavsar, D.; Cochran, B.H.; Simon, A.R. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2005, 171, 858–867. [Google Scholar] [CrossRef]
- van der Vliet, A. Nox enzymes in allergic airway inflammation. Biochim. Biophys. Acta (BBA) General. Subj. 2011, 1810, 1035–1044. [Google Scholar] [CrossRef]
- Albano, G.D.; Gagliardo, R.P.; Montalbano, A.M.; Profita, M. Overview of the mechanisms of oxidative stress: Impact in inflammation of the airway diseases. Antioxidants 2022, 11, 2237. [Google Scholar] [CrossRef]
- Mumby, S.; Adcock, I.M. Recent evidence from omic analysis for redox signalling and mitochondrial oxidative stress in COPD. J. Inflamm. 2022, 19, 10. [Google Scholar] [CrossRef]
- Kyo, M.; Zhu, Z.; Nanishi, M.; Shibata, R.; Ooka, T.; Freishtat, R.J.; Mansbach, J.M.; Camargo Jr, C.A.; Hasegawa, K. Association of nasopharyngeal and serum glutathione metabolism with bronchiolitis severity and asthma risk: A prospective multicenter cohort study. Metabolites 2022, 12, 674. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Janssen-Heininger, Y.M.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Asp. Med. 2018, 63, 59–69. [Google Scholar] [CrossRef]
- Gao, Z.; Ye, J. Inhibition of transcriptional activity of c-JUN by SIRT1. Biochem. Biophys. Res. Commun. 2008, 376, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, H.-Z.; Liu, J.-J.; Jia, Y.-Y.; Zhang, Z.-Q.; Yang, R.-F.; Zhang, Y.; Xu, J.; Wei, Y.-S.; Liu, D.-P. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J. Biol. Chem. 2010, 285, 7097–7110. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Abubakar-Waziri, H.; Lakhdar, R.; Raby, K.; Dixey, P.; Adcock, I.M.; Mumby, S.; Bhavsar, P.K.; Chung, K.F. Molecular mechanisms of oxidative stress in asthma. Mol. Asp. Med. 2022, 85, 101026. [Google Scholar] [CrossRef] [PubMed]
- Kohri, K.; Ueki, I.; Shim, J.; Burgel, P.; Oh, Y.; Tam, D.; Dao-Pick, T.; Nadel, J. Pseudomonas aeruginosa induces MUC5AC production via epidermal growth factor receptor. Eur. Respir. J. 2002, 20, 1263–1270. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lee, K.S.; Park, S.J.; Park, H.S.; Lim, J.S.; Park, K.H.; Im, M.J.; Choi, I.W.; Lee, H.K.; Kim, U.H. Blockade of airway hyperresponsiveness and inflammation in a murine model of asthma by a prodrug of cysteine, L-2-oxothiazolidine-4-carboxylic acid. FASEB J. 2004, 18, 1917–1919. [Google Scholar] [CrossRef]
- Oh, J.H.; Park, E.J.; Park, J.-W.; Lee, J.; Lee, S.H.; Kwon, T.K. A novel cyclin-dependent kinase inhibitor down-regulates tumor necrosis factor-α (TNF-α)-induced expression of cell adhesion molecules by inhibition of NF-κB activation in human pulmonary epithelial cells. Int. Immunopharmacol. 2010, 10, 572–579. [Google Scholar] [CrossRef]
- Lee, I.-T.; Luo, S.-F.; Lee, C.-W.; Wang, S.-W.; Lin, C.-C.; Chang, C.-C.; Chen, Y.-L.; Chau, L.-Y.; Yang, C.-M. Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef]
- Lin, H.-C.; Tu, Y.-F.; Hwang, G.-J.; Huang, H. From precision education to precision medicine. Educ. Technol. Soc. 2021, 24, 123–137. [Google Scholar]
- Coverstone, A.M.; Seibold, M.A.; Peters, M.C. Diagnosis and management of T2-high asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 442–450. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Lipworth, B.; Brattsand, R. Benefit: Risk profile of budesonide in obstructive airways disease. Drugs 2019, 79, 1757–1775. [Google Scholar] [CrossRef]
- Proboszcz, M.; Goryca, K.; Nejman-Gryz, P.; Przybyłowski, T.; Górska, K.; Krenke, R.; Paplińska-Goryca, M. Phenotypic variations of mild-to-moderate obstructive pulmonary diseases according to airway inflammation and clinical features. J. Inflamm. Res. 2021, 2793–2806. [Google Scholar] [CrossRef] [PubMed]
- Breiteneder, H.; Peng, Y.Q.; Agache, I.; Diamant, Z.; Eiwegger, T.; Fokkens, W.J.; Traidl-Hoffmann, C.; Nadeau, K.; O’Hehir, R.E.; O’Mahony, L. Biomarkers for diagnosis and prediction of therapy responses in allergic diseases and asthma. Allergy 2020, 75, 3039–3068. [Google Scholar] [CrossRef] [PubMed]
- Parnes, J.R.; Molfino, N.A.; Colice, G.; Martin, U.; Corren, J.; Menzies-Gow, A. Targeting TSLP in asthma. J. Asthma Allergy 2022, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Allam, V.S.R.R.; Pavlidis, S.; Liu, G.; Kermani, N.Z.; Simpson, J.; To, J.; Donnelly, S.; Guo, Y.-K.; Hansbro, P.M.; Phipps, S. Macrophage migration inhibitory factor promotes glucocorticoid resistance of neutrophilic inflammation in a murine model of severe asthma. Thorax 2023, 78, 661–673. [Google Scholar] [CrossRef]
- Lunding, L.P.; Skouras, D.B.; Vock, C.; Dinarello, C.A.; Wegmann, M. The NLRP3 Inflammasome Inhibitor OLT1177® Ameliorates Experimental Allergic Asthma in Mice. J. Immunol. 2022, 208, 109.102. [Google Scholar] [CrossRef]
- Uysal, P. Novel Applications of Biomarkers in Chronic Obstructive Pulmonary Disease. In Biomarkers in Medicine; Bentham Science Publishers: Sharjah, United Arab Emirates, 2022; p. 425. [Google Scholar]
- Gevaert, P.; Han, J.K.; Smith, S.G.; Sousa, A.R.; Howarth, P.H.; Yancey, S.W.; Chan, R.; Bachert, C. The Roles of Eosinophils and Interleukin-5 in the Pathophysiology of Chronic Rhinosinusitis with Nasal Polyps. In International Forum of Allergy & Rhinology; Willey: Hoboken, NJ, USA, 2022; pp. 1413–1423. [Google Scholar]
- Hameed, R.M.; Abood, H.A.A.N.; Ahmed, M.M. Interleukin-5 and Interleukin-5 Receptor Polymorphism in Asthma; IntechOpen: London, UK, 2022. [Google Scholar]
- Pelaia, C.; Paoletti, G.; Puggioni, F.; Racca, F.; Pelaia, G.; Canonica, G.W.; Heffler, E. Interleukin-5 in the pathophysiology of severe asthma. Front. Physiol. 2019, 10, 1514. [Google Scholar] [CrossRef]
- Kay, A.B.; Hamid, Q.; Robinson, D.S.; Bentley, A.M.; Tsicopoulos, A.; Ying, S.; Moqbel, R.; Corrigan, C.J.; Durham, S.R. Asthma, Eosinophils, and Interleukin-5. In Eosinophils in Allergy and Inflammation; CRC Press: Boca Raton, FL, USA, 2019; pp. 395–406. [Google Scholar]
- Hearn, A.P.; Kent, B.D.; Jackson, D.J. Biologic treatment options for severe asthma. Curr. Opin. Immunol. 2020, 66, 151–160. [Google Scholar] [CrossRef]
- Shrimanker, R.; Keene, O.; Hynes, G.; Wenzel, S.; Yancey, S.; Pavord, I.D. Prognostic and predictive value of blood eosinophil count, fractional exhaled nitric oxide, and their combination in severe asthma: A post hoc analysis. Am. J. Respir. Crit. Care Med. 2019, 200, 1308–1312. [Google Scholar] [CrossRef]
- FitzGerald, J.M.; Bleecker, E.R.; Nair, P.; Korn, S.; Ohta, K.; Lommatzsch, M.; Ferguson, G.T.; Busse, W.W.; Barker, P.; Sproule, S.; et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 388, 2128–2141. [Google Scholar] [CrossRef]
- Brightling, C.E.; Bleecker, E.R.; Panettieri, R.A.; Bafadhel, M.; She, D.; Ward, C.K.; Xu, X.; Birrell, C.; van der Merwe, R. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: A randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir. Med. 2014, 2, 891–901. [Google Scholar] [CrossRef]
- Wu, A.Y.; Sur, S.; Grant, J.A.; Tripple, J.W. Interleukin-4/interleukin-13 versus interleukin-5: A comparison of molecular targets in biologic therapy for the treatment of severe asthma. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.-y.; Li, Y.; Xue, G.-h.; Li, K.-r.; Zheng, Y.-F.; Zhang, Z.-q.; Jiang, Q.; Liu, Y.-y.; Zhou, X.-z.; Cao, C. Requirement of Gαi1 and Gαi3 in interleukin-4-induced signaling, macrophage M2 polarization and allergic asthma response. Theranostics 2021, 11, 4894. [Google Scholar] [CrossRef] [PubMed]
- Husna, S.M.N.; Shukri, N.M.; Ashari, N.S.M.; Wong, K.K. IL-4/IL-13 axis as therapeutic targets in allergic rhinitis and asthma. PeerJ 2022, 10, e13444. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Ford, L.; Pearlman, D.; Spector, S.; Sher, L.; Skobieranda, F.; Wang, L.; Kirkesseli, S.; Rocklin, R.; Bock, B. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 2013, 368, 2455–2466. [Google Scholar] [CrossRef]
- Shastri, M.D.; Allam, V.S.R.R.; Shukla, S.D.; Jha, N.K.; Paudel, K.R.; Peterson, G.M.; Patel, R.P.; Hansbro, P.M.; Chellappan, D.K.; Dua, K. Interleukin-13: A pivotal target against influenza-induced exacerbation of chronic lung diseases. Life Sci. 2021, 283, 119871. [Google Scholar] [CrossRef]
- Scott, G.; Asrat, S.; Allinne, J.; Lim, W.K.; Nagashima, K.; Birchard, D.; Srivatsan, S.; Ajithdoss, D.K.; Oyejide, A.; Ben, L.-H. IL-4 and IL-13, not eosinophils, drive type 2 airway inflammation, remodeling and lung function decline. Cytokine 2023, 162, 156091. [Google Scholar] [CrossRef]
- Wechsler, M.E. Current and emerging biologic therapies for asthma and COPD. Respir. Care 2018, 63, 699–707. [Google Scholar] [CrossRef]
- Osei, E.T.; Brandsma, C.-A.; Timens, W.; Heijink, I.H.; Hackett, T.-L. Current perspectives on the role of interleukin-1 signalling in the pathogenesis of asthma and COPD. Eur. Respir. J. 2020, 55, 1900563. [Google Scholar] [CrossRef]
- Behzadi, P.; Sameer, A.S.; Nissar, S.; Banday, M.Z.; Gajdács, M.; García-Perdomo, H.A.; Akhtar, K.; Pinheiro, M.; Magnusson, P.; Sarshar, M. The Interleukin-1 (IL-1) superfamily cytokines and their single nucleotide polymorphisms (SNPs). J. Immunol. Res. 2022, 2022, 2054431. [Google Scholar] [CrossRef]
- Ma, Y.; Thornton, S.; Boivin, G.P.; Hirsh, D.; Hirsch, R.; Hirsch, E. Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum. 1998, 41, 1798–1805. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Zhang, R.-G.; Chen, F.-Y.; Qiu, Z.-E.; Chen, L.; Huang, Z.-X.; Huang, J.; Zhu, Y.-X.; Zhao, L.; Zhou, W.-L. Cellular mechanism underlying the facilitation of contractile response induced by tumor necrosis factor-α in mouse tracheal smooth muscle. Am. J. Pathol. 2022, 192, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Y.; Zhang, X.; Shi, M.; Meng, X.; Li, D. Clinical Efficacy of Qizhi Yifei Granules on Patients with High Peripheral Blood Eosinophil-Type Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Curr. Top. Nutraceutical Res. 2022, 20, 298. [Google Scholar] [CrossRef]
- Ghosh, A.J.; Hobbs, B.D.; Yun, J.H.; Saferali, A.; Moll, M.; Xu, Z.; Chase, R.P.; Morrow, J.; Ziniti, J.; Sciurba, F. Lung tissue shows divergent gene expression between chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Papaioannou, O.; Sampsonas, F.; Tzouvelekis, A. Infliximab-induced interstitial lung disease. BMJ Case Rep. CP 2021, 14, e245726. [Google Scholar] [CrossRef] [PubMed]
- Velthuis, K.; Jessurun, N.T.; Nguyen, T.D.; Scholl, J.; Jansen, J.R.; van Lint, J.A.; Kosse, L.J.; Ten Klooster, P.M.; Vonkeman, H.E. First-time adverse drug reactions, survival analysis, and the share of adverse drug reactions in treatment discontinuation in real-world rheumatoid arthritis patients: A comparison of first-time treatment with adalimumab and etanercept. Expert Opin. Drug Saf. 2023, 22, 485–492. [Google Scholar] [CrossRef]
- Osipov, V.N.; Khachatryan, D.S.; Balaev, A.N. Biologically active quinazoline-based hydroxamic acids. Med. Chem. Res. 2020, 29, 831–845. [Google Scholar] [CrossRef]
- Henderson, A.G.; Davis, J.M.; Keith, J.D.; Green, M.E.; Oden, A.M.; Rowe, S.M.; Birket, S.E. Static mucus impairs bacterial clearance and allows chronic infection with Pseudomonas aeruginosa in the cystic fibrosis rat. Eur. Respir. J. 2022, 60, 2101032. [Google Scholar] [CrossRef]
- He, Y.; Peng, S.; Xiong, W.; Xu, Y.; Liu, J. Association between Polymorphism of Interleukin-1beta and Interleukin-1 Receptor Antagonist Gene and Asthma Risk: A Meta-Analysis. Sci. World J. 2015, 2015, 685684. [Google Scholar] [CrossRef]
- Rogliani, P.; Calzetta, L.; Ora, J.; Matera, M.G. Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2015, 31, 15–27. [Google Scholar] [CrossRef]
- Croasdell Lucchini, A.; Gachanja, N.N.; Rossi, A.G.; Dorward, D.A.; Lucas, C.D. Epithelial cells and inflammation in pulmonary wound repair. Cells 2021, 10, 339. [Google Scholar] [CrossRef]
- Nieri, D.; Daniele, M.; Lombardi, S.; Bazzan, E.; Santerini, S.; De Cusatis, G.; Vagaggini, B.; Cosio, M.G.; Saetta, M.; Paggiaro, P. Circulating extracellular vesicles are associated with disease severity and interleukin-6 levels in COPD: A Pilot study. J. Clin. Med. 2021, 10, 5014. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Tay, J.; Ton, S.; Agrawal, S.; Gupta, S. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J. Immunol. 2009, 182, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, N.; Freire, A.X.; Bauer, D.C.; Harris, T.B.; Newman, A.B.; Kritchevsky, S.B.; Meibohm, B.; Study, H.A. Predictors of mortality in elderly subjects with obstructive airway disease: The PILE score. Ann. Epidemiol. 2010, 20, 223–232. [Google Scholar] [CrossRef]
- Nixon, L.S.; Yung, B.; Bell, S.C.; Stuart Elborn, J.; Shale, D.J. Circulating immunoreactive interleukin-6 in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1998, 157, 1764–1769. [Google Scholar] [CrossRef]
- Corren, J. New targeted therapies for uncontrolled asthma. J. Allergy Clin. Immunol. Pract. 2019, 7, 1394–1403. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef]
- Voynow, J.A.; Shinbashi, M. Neutrophil elastase and chronic lung disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef]
- Krick, S.; Baumlin, N.; Aller, S.P.; Aguiar, C.; Grabner, A.; Sailland, J.; Mendes, E.; Schmid, A.; Qi, L.; David, N.V. Klotho inhibits interleukin-8 secretion from cystic fibrosis airway epithelia. Sci. Rep. 2017, 7, 14388. [Google Scholar] [CrossRef]
- Lampronti, I.; Manzione, M.G.; Sacchetti, G.; Ferrari, D.; Spisani, S.; Bezzerri, V.; Finotti, A.; Borgatti, M.; Dechecchi, M.C.; Miolo, G. Differential Effects of Angelicin Analogues on NF-κB Activity and IL-8 Gene Expression in Cystic Fibrosis IB3-1 Cells. Mediat. Inflamm. 2017, 2017, 2389487. [Google Scholar] [CrossRef]
- Jaudszus, A.; Arnold, C.; Hentschel, J.; Hünniger, K.; Baier, M.; Mainz, J.G. Increased cytokines in cystic fibrosis patients’ upper airways during a new P. aeruginosa colonization. Pediatr. Pulmonol. 2018, 53, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Ganesan, S.; Comstock, A.T.; Meldrum, C.A.; Mahidhara, R.; Goldsmith, A.M.; Curtis, J.L.; Martinez, F.J.; Hershenson, M.B.; Sajjan, U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Sarir, H.; Mortaz, E.; Janse, W.T.; Givi, M.E.; Nijkamp, F.P.; Folkerts, G. IL-8 production by macrophages is synergistically enhanced when cigarette smoke is combined with TNF-α. Biochem. Pharmacol. 2010, 79, 698–705. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, H.; Zhang, H.; Ma, W.; Wang, F.; Liu, C.; He, S. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 2011, 56, 717–725. [Google Scholar] [CrossRef]
- Huang, A.-X.; Lu, L.-W.; Liu, W.-J.; Huang, M. Plasma inflammatory cytokine IL-4, IL-8, IL-10, and TNF-α levels correlate with pulmonary function in patients with asthma-chronic obstructive pulmonary disease (COPD) overlap syndrome. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2800. [Google Scholar] [CrossRef]
- Mahler, D.A.; Huang, S.; Tabrizi, M.; Bell, G.M. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: A pilot study. Chest 2004, 126, 926–934. [Google Scholar] [CrossRef]
- Lazaar, A.L.; Miller, B.E.; Tabberer, M.; Yonchuk, J.; Leidy, N.; Ambery, C.; Bloomer, J.; Watz, H.; Tal-Singer, R. Effect of the CXCR2 antagonist danirixin on symptoms and health status in COPD. Eur. Respir. J. 2018, 52, 1801020. [Google Scholar] [CrossRef]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef]
- Almikhlafi, M.A.; Haghayeghi, K.; Gardner, A. Endothelin A (ETA) and Endothelin B (ETB) Receptor Subtypes Potentiate Epidermal Growth Factor (EGF)-Mediated Proliferation in Human Asthmatic Bronchial Airway Smooth Muscle. Cureus 2022, 14, e28333. [Google Scholar] [CrossRef]
- Dyneva, M.E.; Aminova, G.E.; Kurbacheva, O.; Ilina, N.I. Dupilumab: New opportunities for the treatment of asthma and chronic rhinosinusitis with nasal polyps. Russ. J. Allergy 2021, 18, 18–31. [Google Scholar] [CrossRef]
- Ijaz, B.; Shabbir, A.; Shahzad, M.; Mobashar, A.; Sharif, M.; Basheer, M.I.; Tareen, R.B. Amelioration of airway inflammation and pulmonary edema by Teucrium stocksianum via attenuation of pro-inflammatory cytokines and up-regulation of AQP1 and AQP5. Respir. Physiol. Neurobiol. 2021, 284, 103569. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Ashraf, S.; Parvez, M.K.; Al-Dosari, M.S.; Ul-Haq, Z. Structure and ligand-based drug discovery of IL-4 inhibitors via interaction-energy-based learning approaches. J. Biomol. Struct. Dyn. 2022, 40, 6503–6521. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, G.; Canonica, G.W.; Matucci, A.; Paolini, R.; Triggiani, M.; Paggiaro, P. Targeted therapy in severe asthma today: Focus on immunoglobulin E. Drug Des. Dev. Ther. 2017, 11, 1979–1987. [Google Scholar] [CrossRef]
- Doran, E.; Cai, F.; Holweg, C.T.; Wong, K.; Brumm, J.; Arron, J.R. Interleukin-13 in asthma and other eosinophilic disorders. Front. Med. 2017, 4, 139. [Google Scholar] [CrossRef]
- Nwaru, B.I.; Ekerljung, L.; Rådinger, M.; Bjerg, A.; Mincheva, R.; Malmhäll, C.; Axelsson, M.; Wennergren, G.; Lotvall, J.; Lundbäck, B. Cohort profile: The West Sweden Asthma Study (WSAS): A multidisciplinary population-based longitudinal study of asthma, allergy and respiratory conditions in adults. BMJ Open 2019, 9, e027808. [Google Scholar] [CrossRef]
- Maskey, A.; Srivastava, K.; Soffer, G.; Dunkin, D.; Yuan, Q.; Li, X.-M. Induction of severe eosinophilic esophagitis and multi-organ inflammation by airborne allergens is associated with IL-4/IL-13 and CCL11 but not IgE in genetic susceptible mice. J. Inflamm. Res. 2022, 15, 5527–5540. [Google Scholar] [CrossRef]
- Yanagibashi, T.; Satoh, M.; Nagai, Y.; Koike, M.; Takatsu, K. Allergic diseases: From bench to clinic-Contribution of the discovery of interleukin-5. Cytokine 2017, 98, 59–70. [Google Scholar] [CrossRef]
- Farah, C.S.; Badal, T.; Reed, N.; Rogers, P.G.; King, G.G.; Thamrin, C.; Peters, M.J.; Seccombe, L.M. Mepolizumab improves small airway function in severe eosinophilic asthma. Respir. Med. 2019, 148, 49–53. [Google Scholar] [CrossRef]
- Van Toor, J.J.; van der Mark, S.C.; Kappen, J.H.; In’t Veen, J.; Braunstahl, G.J. Mepolizumab add-on therapy in a real world cohort of patients with severe eosinophilic asthma: Response rate, effectiveness, and safety. J. Asthma 2021, 58, 651–658. [Google Scholar] [CrossRef]
- Simpson, J.; Bafadhel, M. Alternatives to induced sputum for identifying inflammatory subtypes of asthma. Respirology 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Aleman Paramo, F.; Kjarsgaard, M.; Salter, B.; Nair, G.; LaVigne, N.; Radford, K.; Sehmi, R.; Nair, P. Weight-adjusted intravenous reslizumab in severe asthma with inadequate response to fixed-dose subcutaneous mepolizumab. Am. J. Respir. Crit. Care Med. 2018, 197, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Vatrella, A.; Maglio, A.; Pelaia, C.; Ciampo, L.; Pelaia, G.; Vitale, C. Eosinophilic inflammation: An appealing target for pharmacologic treatments in severe asthma. Biomedicines 2022, 10, 2181. [Google Scholar] [CrossRef] [PubMed]
- Rayees, S.; Din, I.; Rayees, S.; Din, I. Cytokine-Based Therapies. In Asthma: Pathophysiology, Herbal and Modern Therapeutic Interventions; Springer: Berlin/Heidelberg, Germany, 2021; pp. 27–32. [Google Scholar]
- Gong, F.; Pan, Y.H.; Huang, X.; Zhu, H.Y.; Jiang, D.L. From bench to bedside: Therapeutic potential of interleukin-9 in the treatment of asthma. Exp. Ther. Med. 2017, 13, 389–394. [Google Scholar] [CrossRef]
- Oh, C.K.; Leigh, R.; McLaurin, K.K.; Kim, K.; Hultquist, M.; Molfino, N.A. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir. Res. 2013, 14, 93. [Google Scholar] [CrossRef]
- Pera, T.; Penn, R.B. G Protein-Coupled Receptors in Airway Smooth Muscle Function and Obstructive Lung Disease. In Signal Transduction and Smooth Muscle; CRC Press: Boca Raton, FL, USA, 2018; pp. 205–243. [Google Scholar]
- Iftikhar, I.H.; Schimmel, M.; Bender, W.; Swenson, C.; Amrol, D. Comparative efficacy of anti IL-4, IL-5 and IL-13 drugs for treatment of eosinophilic asthma: A network meta-analysis. Lung 2018, 196, 517–530. [Google Scholar] [CrossRef]
- Jain, M.; Doughty, D.; Clawson, C.; Li, X.; White, N.; Agoram, B.; van der Merwe, R. Tralokinumab pharmacokinetics and tolerability when administered by different subcutaneous injection methods and rates. Int. J. Clin. Pharmacol. Ther. 2017, 55, 606. [Google Scholar] [CrossRef]
- Shaikh, S.B.; Prabhu, A.; Bhandary, Y.P. Interleukin-17A: A potential therapeutic target in chronic lung diseases. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 921–928. [Google Scholar] [CrossRef]
- Van Lommel, A.T.; Bollé, T.; Hellings, P.W. Pulmonary neuroepithelial bodies as hypothetical immunomodulators: Some new findings and a review of the literature. In Airway Chemoreceptors in Vertebrates; Taylor & Francis Group: Abingdon, UK, 2019; pp. 311–330. [Google Scholar]
- Pelaia, C.; Crimi, C.; Vatrella, A.; Busceti, M.T.; Gaudio, A.; Garofalo, E.; Bruni, A.; Terracciano, R.; Pelaia, G. New treatments for asthma: From the pathogenic role of prostaglandin D2 to the therapeutic effects of fevipiprant. Pharmacol. Res. 2020, 155, 104490. [Google Scholar] [CrossRef]
- Moermans, C.; Damas, K.; Guiot, J.; Njock, M.-S.; Corhay, J.-L.; Henket, M.; Schleich, F.; Louis, R. Sputum IL-25, IL-33 and TSLP, IL-23 and IL-36 in airway obstructive diseases. Reduced levels of IL-36 in eosinophilic phenotype. Cytokine 2021, 140, 155421. [Google Scholar] [CrossRef]
- Hatta, M.; Surachmanto, E.E.; Islam, A.A.; Wahid, S. Expression of mRNA IL-17F and sIL-17F in atopic asthma patients. BMC Res. Notes 2017, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.C.; Naluai, Å.T.; Oscarsson, M.; Torkzadeh, S.; Bohman, A.; Bende, M.; Harandi, A.M. Differential expression of angiotensin-converting enzyme 2 in nasal tissue of patients with chronic rhinosinusitis with nasal polyps. medRxiv 2021. [Google Scholar] [CrossRef]
- Naji AL-Hasnawi, A.T.; Jabbar AL-Hasnawi, S.M. Role of interleukin 25 and interleukin 33 as immunological markers in pediatric asthma. Eur. Asian J. Biosci. 2020, 14, 4625–4630. [Google Scholar]
- Bonser, L.R.; Erle, D.J. The airway epithelium in asthma. Adv. Immunol. 2019, 142, 1–34. [Google Scholar]
- Kearley, J.; Buckland, K.F.; Mathie, S.A.; Lloyd, C.M. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2–IL-33 pathway. Am. J. Respir. Crit. Care Med. 2009, 179, 772–781. [Google Scholar] [CrossRef]
- Ádám, D.; Arany, J.; Tóth, K.F.; Tóth, B.I.; Szöllősi, A.G.; Oláh, A. Opioidergic signaling—A neglected, yet potentially important player in atopic dermatitis. Int. J. Mol. Sci. 2022, 23, 4140. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Maltby, S.; Tay, H.L.; Eyers, F.; Foster, P.S.; Yang, M. Identification of IFN-γ and IL-27 as critical regulators of respiratory syncytial virus–induced exacerbation of allergic airways disease in a mouse model. J. Immunol. 2018, 200, 237–247. [Google Scholar] [CrossRef]
- Plichta, J.; Kuna, P.; Panek, M. Biologic drugs in the treatment of chronic inflammatory pulmonary diseases: Recent developments and future perspectives. Front. Immunol. 2023, 14, 1207641. [Google Scholar] [CrossRef]
- Yamashita, N.; Tashimo, H.; Ishida, H.; Kaneko, F.; Nakano, J.; Kato, H.; Hirai, K.; Horiuchi, T.; Ohta, K. Attenuation of airway hyperresponsiveness in a murine asthma model by neutralization of granulocyte–macrophage colony-stimulating factor (GM-CSF). Cell. Immunol. 2002, 219, 92–97. [Google Scholar] [CrossRef]
- Dhagat, U.; Hercus, T.R.; Broughton, S.E.; Nero, T.L.; Cheung Tung Shing, K.S.; Barry, E.F.; Thomson, C.A.; Bryson, S.; Pai, E.F.; McClure, B.J. The mechanism of GM-CSF inhibition by human GM-CSF auto-antibodies suggests novel therapeutic opportunities. MAbs 2018, 10, 1018–1029. [Google Scholar] [CrossRef]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Zangrilli, J.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O’Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Castro, M.; Corren, J.; Maspero, J.; Wang, L.; Zhang, B.; Pirozzi, G.; Sutherland, E.R.; Evans, R.R.; Joish, V.N.; et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: A randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 2016, 388, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Esty, B.; Harb, H.; Bartnikas, L.M.; Charbonnier, L.M.; Massoud, A.H.; LeonAstudillo, C.; Visner, G.; Subramaniam, M.; Phipatanakul, W.; Chatila, T.A. Treatment of Severe Persistent Asthma With IL-6 Receptor Blockade. J. Allergy Clin. Immunol. Pract. 2019, 7, 1639–1642.e4. [Google Scholar] [CrossRef] [PubMed]
- Menzies-Gow, A.; Bafadhel, M.; Busse, W.W. An expert consensus framework for asthma remission as a treatment goal. J. Allergy Clin. Immunol. 2020, 145, 757–765. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Agache, I.O.; Soong, W. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J. Allergy Clin. Immunol. 2021, 148, 790–798. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef]
- Brightling, C.E.; Nair, P.; Cousins, D.J. Risankizumab in severe asthma–A phase 2a, placebo-controlled trial. N. Engl. J. Med. 2021, 385, 1669–1679. [Google Scholar] [CrossRef]
- Alcorn, J.F.; Crowe, C.R.; Kolls, J.K. TH17 cells in asthma and COPD. Annu. Rev. Physiol. 2010, 72, 495–516. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Investig. 2008, 11, 3546–3556. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokines | Key Secreting Cells | Roles During Inflammation | References |
|---|---|---|---|
| IL-1 | Endothelial cells, epithelial cells, macrophages, monocytes, dendritic ells | Induces inflammation, fever, and acute phase response, and accelerates production of neutrophils | [25,41,60] |
| IL-2 | CD4+ and CD8+ T cells | Activates NK cells and cytotoxic T cells and increases production of other cytokines | [25,41,60] |
| IL-3 | CD4+ T cells | Produces growth factor for hematopoietic stem cells | [41,60] |
| IL-4 | CD4+ Th2 cells and mast cells | Promotes differentiation of Th2 cells and aids in growth and survival of mast cells, B cells, and T cells | [41,60] |
| IL-5 | CD4+ Th2 cells | Stimulates growth and progression of eosinophil | [25,41] |
| IL-6 | Endothelial cells, macrophages and T lymphocytes | Induces the liver to create acute-phase inflammatory response mediators | [41,60] |
| IL-7 | Stromal cells of bone marrow | Plays a role in adaptive immunity by promoting the growth and division of thymocytes and pre-B cells | [41,60] |
| IL-8 (CXCL8) | Endothelial cells, macrophages, neutrophils | Plays a role in adaptive immunity, controls lymphocyte migration and neutrophil infiltration, and attracts neutrophils and T lymphocytes | [41,60] |
| IL-10 | Macrophages, Th cells, regulatory T cells, dendritic cells | Reduces inflammation by preventing Th1 cells and the release of IL-12 from activated macrophages and dendritic cells | [41,60] |
| IL-12 | Dendritic cells and macrophages | Increases the cytotoxicity of NK cells in innate immunity and triggers the development of Th1 cells in adaptive immunity | [41,60] |
| Type-I IFN (IFN-α, IFN-β) | Fibroblasts, macrophages, dendritic cells, endothelial cells, epithelial cells | NK cell activation, viral replication inhibition, and increases MHC-I molecule expression on virus-infected cells | [41,44,60,61,62] |
| IFN-γ | CD4+ and CD8+ T lymphocytes and NK cells | Promotes the production of MHC-I and II and the processing and presentation of antigens; activates macrophages in innate immune responses and adaptive cell-mediated immune responses | [41,44,61,62] |
| TNF-α | Macrophages and T cells | Brings about inflammation, causes acute phase reaction and fever, and induces neutrophil and endothelial cell activation and cellular apoptosis | [43,60] |
| TGF-β | T cells | Induces cytotoxicity and phagocytosis | [43,60] |
| Chemokines | Endothelial cells, macrophages and T cells | Leukocyte migration from the blood and tissues is controlled and stimulated by chemokines | [41,53,56] |
| G-CSF | Endothelial cells, fibroblast and macrophages | Encourages the production of growth factors and neutrophil maturation in inflammatory reactions | [41,52,60] |
| M-CSF | Macrophages and T cells | Encourages mononuclear phagocyte maturation and growth factors | [41,52,60] |
| GM-CSF | Endothelial cells, fibroblast, macrophages, and T cells | Activates mature granulocytes and encourages the maturation and expansion of neutrophil, eosinophil, and monocyte cells | [41,52,60] |
| Lung Disease | Cytokines Involved | References |
| Asthma | CCR2-5 agonist, CXCR2-3 agonist, EGF, eotaxin, GM-CSF, IL-1 β, IL-4, IL-5, IL-6, IL-8, IL-9, IL-13, IL-17, IL-25, IL-33, NGF, RANTES, SCF, TGF-β, TNF-α, TSLP, VEGF | [63,65,66,67] |
| COPD | CCR2 agonist, CCR3 agonist, CCR5 agonist, CXCR2 agonist, CXCR3 agonist, EGF, GM-CSF, IL-1β, IL-4, IL-6, IL-8, IL-12, IL-17, IL-18, IL-32, IFN-γ, TGF-β, TNF-α, TSLP | [63,65,68,69,70,71] |
| COVID-19-associated lung disease | IL-1β, IL-8, IL-6, IL-17, TNF-α | [72,73] |
| Cystic fibrosis bronchiectasis | IL-1β | [74,75,76,77,78] |
| Lung cancer | IFN-γ, IL-1β, IL-6, IL-8, IL-10, IL-17, IL-18, IL-22,TGF-β, TNF-α, VEGF | [79,80] |
| Pneumonia | IFN-γ, IL-lβ, IL-6, IL-8, IL-10, IL-33, MCP-1, TNF | [81,82,83,84] |
| Pulmonary fibrosis | CCL17, CCL18/PARC, CTGF, CXCL12/SDF-1, GM-CSF, IL-1, IL-4, IL-10, IL-13, IL-17, MCP-1, oncostatin M, PDGF, TGF- β | [63,65,72,85] |
| Pulmonary tuberculosis | TNF-α, IL-1β, IL-12, IL-18, IL- 23, IL-27, IL-10, IL-6, IL-17, IL-22, IFN- β, IFN- γ, TGF- β | [86] |
| Targeted Cytokine | Intervention Molecule | Type of Antibody | Disease Condition | Clinical Efficacy | Status | Company/Organization | Clinical Trials Associated |
|---|---|---|---|---|---|---|---|
| IL-5 | Mepolizumab | Humanized, IgG1 monoclonal antibody | COPD, high levels of sputum eosinophils, and severe eosinophilic asthma | Reduces moderate and severe exacerbations | Phase III | GlaxoSmithKline | NCT02105961 NCT02555371 NCT01691859 NCT02135692 |
| Severe bilateral chronic rhinosinusitis | Improved nasal polyp size and nasal obstruction | Phase III | GlaxoSmithKline | NCT03085797 | |||
| Severe asthma and comorbid conditions | Controls asthma, improved lung function and health-related quality of life, as well as various comorbid pathologies | Phase II | GlaxoSmithKline | NCT01000506 | |||
| Phase III | GlaxoSmithKline | NCT01691521 | |||||
| Phase III | GlaxoSmithKline | NCT01842607 | |||||
| Phase III | GlaxoSmithKline | NCT02281318 | |||||
| Hypereosinophilic syndrome | Decreased disease flares and blood eosinophil count | Phase III | GlaxoSmithKline | NCT03306043 NCT02836496 | |||
| Benralizumab | Humanized, afucosylated anti-IL-5 receptor α monoclonal antibody | Severe eosinophilic asthma, COPD | Reduced the exacerbations in severe COPD | Phase III | AstraZeneca | NCT01914757 | |
| Phase III | AstraZeneca | NCT01928771 | |||||
| Phase II | MedImmune LLC | NCT01227278 | |||||
| Phase III | AstraZeneca | NCT02155660 | |||||
| Phase III | AstraZeneca | NCT02138916 | |||||
| Chronic rhinosinusitis | Reduced NP score, nasal blockage, and difficulty with sense of smell | Phase III | AstraZeneca | NCT03401229 | |||
| Severe eosinophilic asthma | Improved nasal polyposis and lung function | Phase III | AstraZeneca | NCT03170271 | |||
| Severe, uncontrolled asthma | Found to be safe and effective in long-term studies | Phase III | AstraZeneca | NCT02258542 | |||
| Severe asthma | Clinical remission is achievable and reduced exacerbation rates | Phase III | AstraZeneca | NCT02075255 | |||
| Reslizumab | Humanized monoclonal antibody | Eosinophilic granulomatosis with polyangiitis (EGPA) | Found to be safe and effective in EGPA | Phase II | National Jewish Health | NCT02947945 | |
| Late-onset eosinophilic asthma | Reduction in asthma exacerbations and improved lung function | Phase III | Teva Branded Pharmaceutical Products R&D, Inc. | NCT01287039 NCT01285323 | |||
| IL-4 | Dupilumab | Human anti-interleukin-4 receptor α monoclonal antibody | Uncontrolled Asthma | Lower rates of severe asthma exacerbation, better lung function | Phase III | Sanofi | NCT02414854 |
| Glucocorticoid-dependent severe asthma | Decreased the rate of severe exacerbations and increased FEV1 | Phase III | Sanofi | NCT02528214 | |||
| Chronic rhinosinusitis with nasal polyps | Improved upper and lower airway outcome measures | Phase III | Sanofi | NCT02912468 NCT02898454 | |||
| Persistent asthma | Improved lung function and decreased Th2-associated inflammatory markers | Phase II | Sanofi | NCT01312961 | |||
| IL-13 | Lebrikizumab | Anti-interleukin-13 monoclonal antibody | Uncontrolled asthma in adolescent patients | Reduced asthma exacerbation rates | Phase III | Hoffmann-La-Roche | NCT01875003 |
| Idiopathic pulmonary fibrosis | Benefits lung function | Phase II | Hoffmann-La-Roche | NCT01872689 | |||
| Adults with asthma | Improved lung function with high levels of serum periostin | Phase II | Genentech, Inc. | NCT00930163 | |||
| Tralokinumab | Anti-interleukin-13 monoclonal antibody | Moderate-to-severe asthma | Improved lung function with no improvement in ACQ-6 | Phase IIa | MedImmune LLC | NCT00873860 | |
| TNF-α | Infliximab | Anti-TNF-α antibody | Asthma | Results are not posted | Phase II | Imperial College London | NCT00278083 |
| NCT Number | Title | Conditions | Interventions | Phase | Status | Company/Organization |
|---|---|---|---|---|---|---|
| NCT00001908 | T-Cell Cytokine Changes During IL-4 Receptor Treatment for Asthma | Asthma Hypersensitivity | -- | -- | Completed | National Institute of Allergy and Infectious Diseases |
| NCT00455767 | Safety and Efficacy Study of Depelestat in ARDS Patients | Fibrosis Lung Disease Respiratory Disorders Acute Respiratory Distress Syndrome (ARDS) Pulmonary Fibrosis Inflammation | EPI-hNE4, Placebo | Phase II | Completed | Debiopharm International SA |
| NCT00753103 | Anti-Cytokine Therapy for Vasculitis | Wegener’s Granulomatosis Renal Limited Vasculitis Microscopic Polyangiitis | Infliximab, Cyclophosphamide, Prednisolone, Azathioprine, Mycophenolate Mofetil, Methylprednisolone | Phase II | Completed | University Hospital Birmingham NHS Foundation Trust |
| NCT01253941 | Effects of Mud Bath Therapy in Chronic Obstructive Pulmonary Disease | COPD | Mud Bath Therapy | N/A | Completed | Fondazione Salvatore Maugeri |
| NCT02113072 | Recurrent Wheezing in Infants: Risk Factors and Prevention With Probiotics | Respiratory Tract Diseases, Wheezing | Beclomethasone, Probiotics, Placebo | Phase III | Completed | Universidade Federal de Pernambuco |
| NCT02557958 | Chronic Obstructive Pulmonary Disease Transcription Factor and Cytokine Study | COPD | Azithromycin, Placebo | Early Phase I | Completed | NYU Langone Health |
| NCT03595488 | Dupilumab for Aspirin-exacerbated respiratory disease | Aspirin-Exacerbated Respiratory disease | Dupilumab | Phase II | Completed | Rochester General Hospital |
| NCT04102813 | Deep Diaphragmatic Breathing: Neurobiological and Anti-inflammatory Effects | Unrecognized Condition | Functional Therapy | N/A | Completed | Azienda Ospedaliera Universitaria Policlinico Paolo Giaccone Palermo |
| NCT04374149 | Therapeutic Plasma Exchange Alone or in Combination with Ruxolitinib in COVID-19-Associated CRS | Cytokine Release Syndrome COVID-19 | Therapeutic Plasma Exchange Ruxolitinib | Phase II | Completed | Prisma Health Upstate |
| NCT04445272 | Clinical Trial to Evaluate the Effectiveness and Safety of Tocilizumab for Treating Patients With COVID-19 Pneumonia | COVID-19 | Tocilizumab | Phase II | Completed | Fundacion SEIMC-GESIDA |
| NCT04457349 | Therapeutic Plasma Exchange in Resistant Cytokine Storm of COVID-19 | COVID-19 | Therapeutic Plasma Exchange | N/A | Completed | Alexandria University |
| NCT04485169 | Therapeutic Plasma Exchange for Coronavirus Disease-2019 Triggered Cytokine Release Storm | COVID-19 Cytokine Release Syndrome | Therapeutic Plasma Exchange | N/A | Completed | UNICEF |
| NCT05164692 | Effects of Nasal-spraying LiveSpo Navax in Treatment of Acute Respiratory Infections in Children | Acute Respiratory Tract Infections | LiveSpo Navax, 0.9% NaCl Physiological Saline | N/A | Completed | National Children’s Hospital, Vietnam |
| NCT05378022 | Effects of Nasal-spraying LiveSpo Navax in Treatment of Influenza Virus in Children | Acute Respiratory Tract Infections | Combination Product: LiveSpo Navax, 0.9% NaCl Physiological Saline | N/A | Completed | National Children’s Hospital, Vietnam |
| NCT05562843 | Autologous Cellular Therapy With PRP-PC in Chronic Lung Diseases: An Observational Study LI-004 | COPD ILD | Autologous Cellular Therapy with PRP-PC | N/A | Completed | H-CYTE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavda, V.P.; Bezbaruah, R.; Ahmed, N.; Alom, S.; Bhattacharjee, B.; Nalla, L.V.; Rynjah, D.; Gadanec, L.K.; Apostolopoulos, V. Proinflammatory Cytokines in Chronic Respiratory Diseases and Their Management. Cells 2025, 14, 400. https://doi.org/10.3390/cells14060400
Chavda VP, Bezbaruah R, Ahmed N, Alom S, Bhattacharjee B, Nalla LV, Rynjah D, Gadanec LK, Apostolopoulos V. Proinflammatory Cytokines in Chronic Respiratory Diseases and Their Management. Cells. 2025; 14(6):400. https://doi.org/10.3390/cells14060400
Chicago/Turabian StyleChavda, Vivek P., Rajashri Bezbaruah, Nasima Ahmed, Shahnaz Alom, Bedanta Bhattacharjee, Lakshmi Vineela Nalla, Damanbhalang Rynjah, Laura Kate Gadanec, and Vasso Apostolopoulos. 2025. "Proinflammatory Cytokines in Chronic Respiratory Diseases and Their Management" Cells 14, no. 6: 400. https://doi.org/10.3390/cells14060400
APA StyleChavda, V. P., Bezbaruah, R., Ahmed, N., Alom, S., Bhattacharjee, B., Nalla, L. V., Rynjah, D., Gadanec, L. K., & Apostolopoulos, V. (2025). Proinflammatory Cytokines in Chronic Respiratory Diseases and Their Management. Cells, 14(6), 400. https://doi.org/10.3390/cells14060400