
Defining the Protein Phosphatase 2A (PP2A) Subcomplexes That Regulate FoxO Transcription Factor Localization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning and Cell Culture
2.2. Fluorescence Microscopy
2.3. Chemical Inhibitor Treatments
2.4. Immunoblotting
2.5. Genetic Knockdowns and RT-PCR
2.6. Statistical Analyses and Experimental Replicates
3. Results
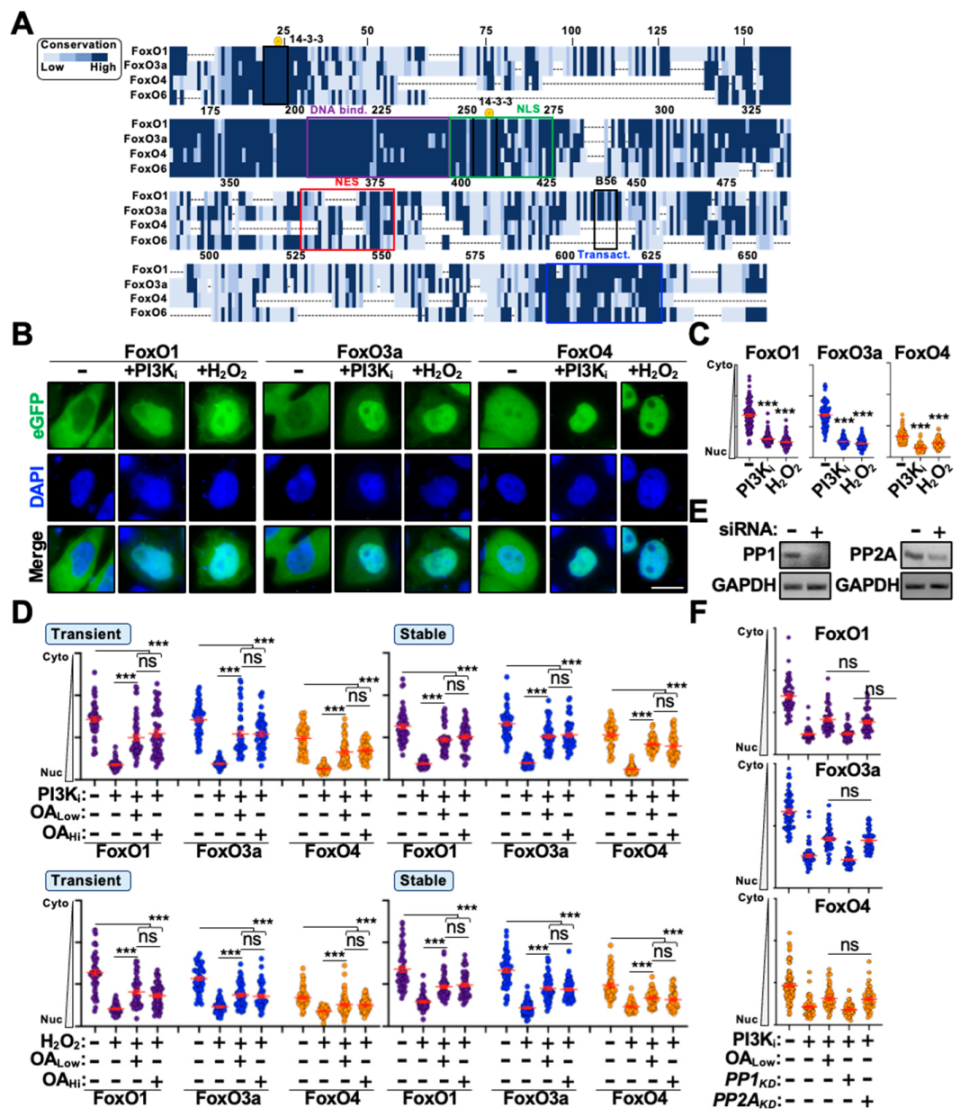
3.1. PP2A Only Partially Regulates FoxO Cytoplasm-to-Nucleus Translocation
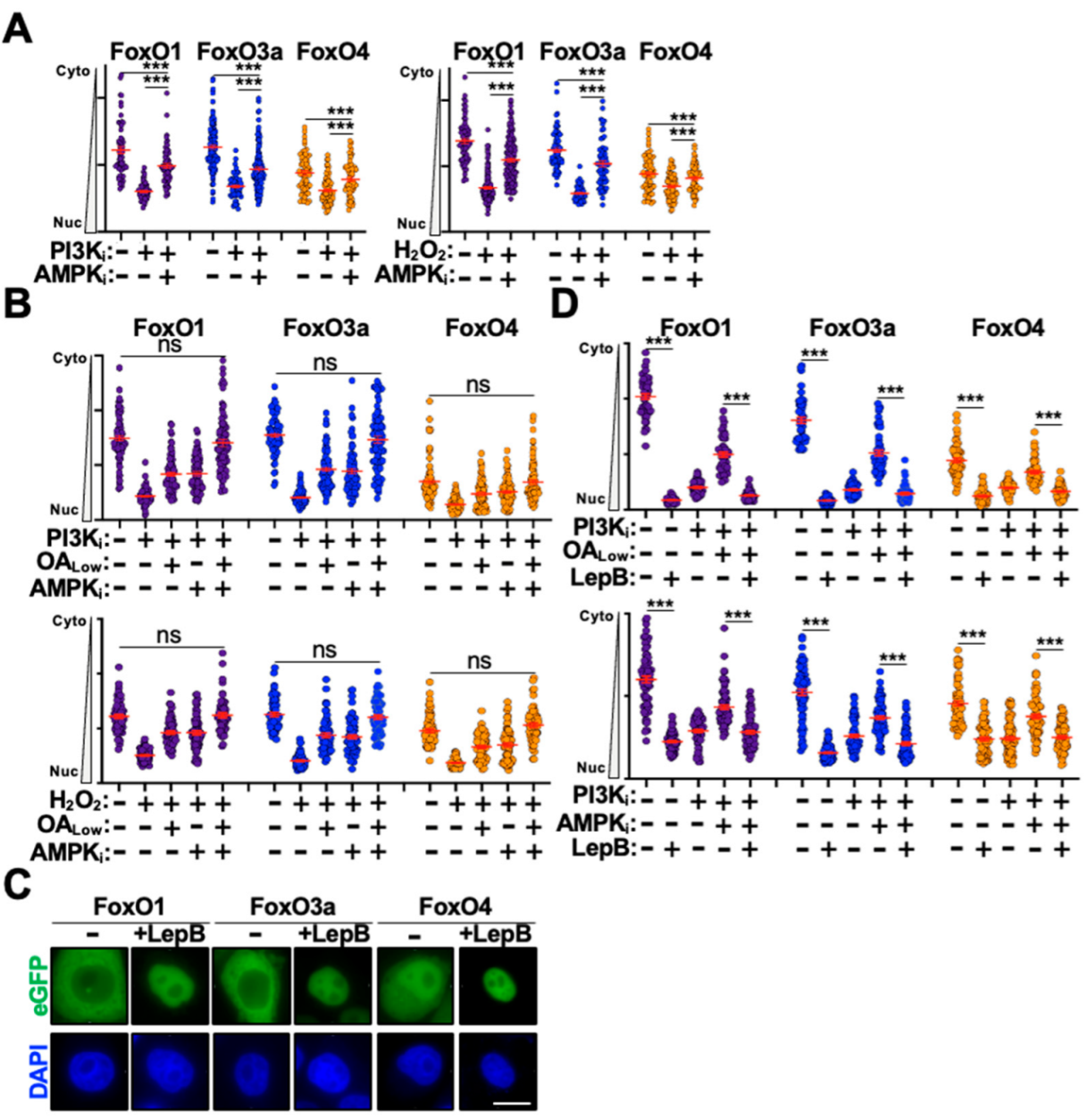
3.2. AMPK and PI3K/AKT Signaling Combine to Regulate FoxO Protein Trafficking
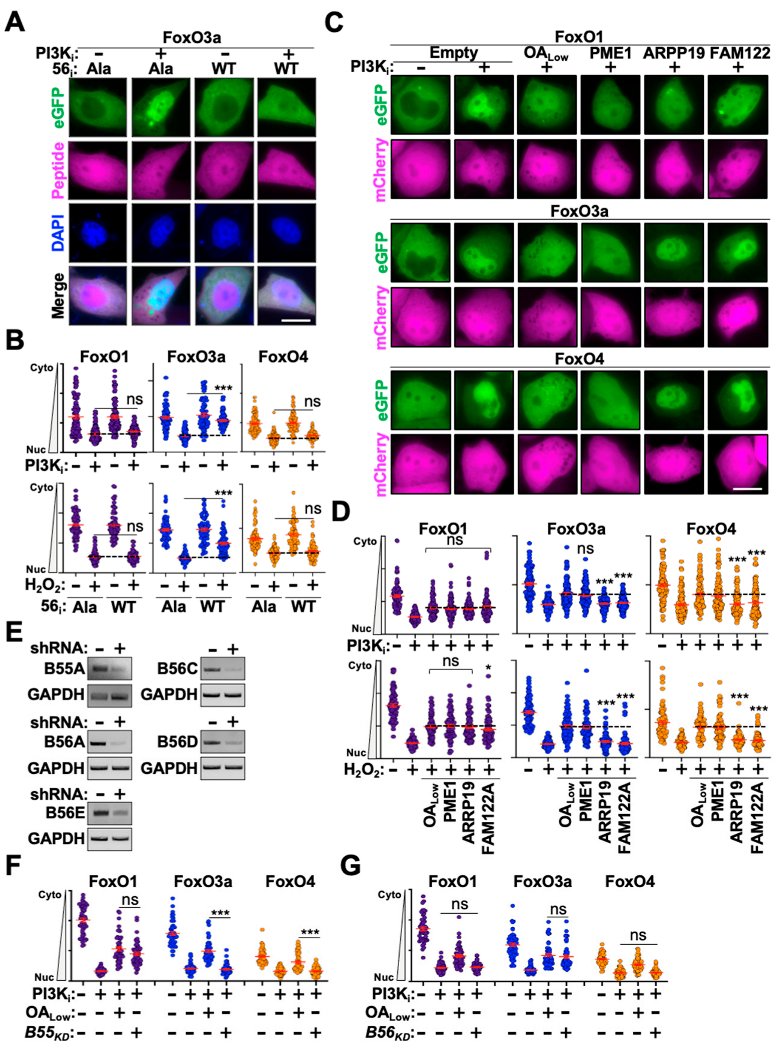
3.3. Defining the PP2A Subcomplexes That Regulate FoxO Nuclear Translocation
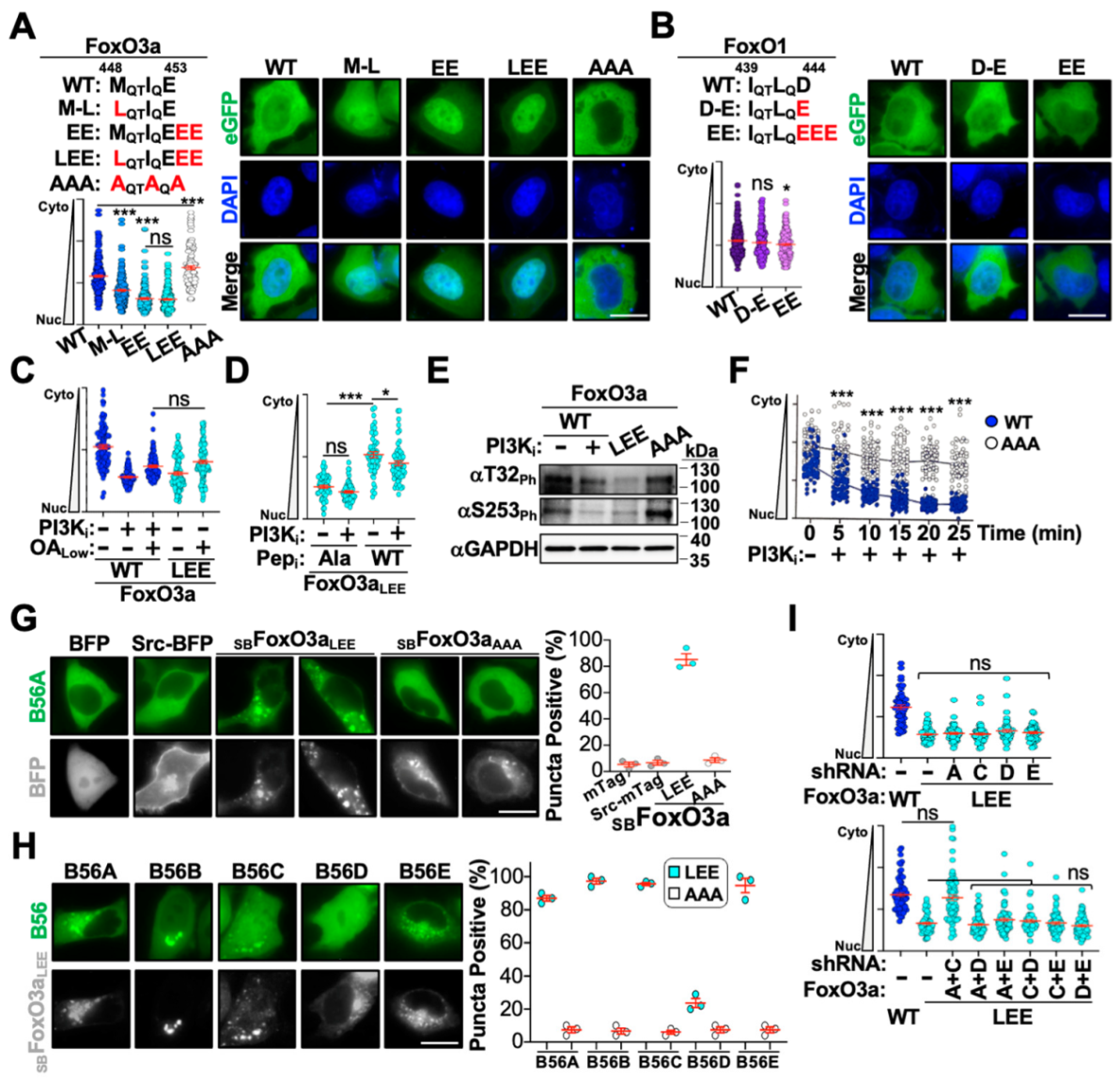
3.4. Altering the FoxO3a B56-Interacting Motif Modulates Nuclear Translocation Kinetics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Anderson, M.J.; Viars, C.S.; Czekay, S.; Cavenee, W.K.; Arden, K.C. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics 1998, 47, 187–199. [Google Scholar] [CrossRef]
- Yang, J.Y.; Hung, M.C. A new fork for clinical application: Targeting forkhead transcription factors in cancer. Clin. Cancer Res. 2009, 15, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Lin, M.; Gu, W.; Su, Z.; Duan, Y.; Song, W.; Liu, H.; Zhang, F. The rules and regulatory mechanisms of FOXO3 on inflammation, metabolism, cell death and aging in hosts. Life Sci. 2023, 328, 121877. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal 2010, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; de Vries-Smits, L.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Wang, M.C.; Bohmann, D.; Jasper, H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 2005, 121, 115–125. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Boccitto, M.; Kalb, R.G. Regulation of Foxo-dependent transcription by post-translational modifications. Curr. Drug Targets 2011, 12, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef]
- Vogt, P.K.; Jiang, H.; Aoki, M. Triple layer control: Phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 2005, 4, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 2011, 1813, 1961–1964. [Google Scholar] [CrossRef]
- van der Horst, A.; de Vries-Smits, A.M.; Brenkman, A.B.; van Triest, M.H.; van den Broek, N.; Colland, F.; Maurice, M.M.; Burgering, B.M. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 2006, 8, 1064–1073. [Google Scholar] [CrossRef]
- Kim, K.H.; Oprescu, S.N.; Snyder, M.M.; Kim, A.; Jia, Z.; Yue, F.; Kuang, S. PRMT5 mediates FoxO1 methylation and subcellular localization to regulate lipophagy in myogenic progenitors. Cell Rep. 2023, 42, 113329. [Google Scholar] [CrossRef]
- Yan, L.; Lavin, V.A.; Moser, L.R.; Cui, Q.; Kanies, C.; Yang, E. PP2A regulates the pro-apoptotic activity of FOXO1. J. Biol. Chem. 2008, 283, 7411–7420. [Google Scholar] [CrossRef]
- Singh, A.; Ye, M.; Bucur, O.; Zhu, S.; Tanya Santos, M.; Rabinovitz, I.; Wei, W.; Gao, D.; Hahn, W.C.; Khosravi-Far, R. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol. Biol. Cell 2010, 21, 1140–1152. [Google Scholar] [CrossRef]
- Jacobs, F.M.; van der Heide, L.P.; Wijchers, P.J.; Burbach, J.P.; Hoekman, M.F.; Smidt, M.P. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J. Biol. Chem. 2003, 278, 35959–35967. [Google Scholar] [CrossRef] [PubMed]
- van der Heide, L.P.; Jacobs, F.M.; Burbach, J.P.; Hoekman, M.F.; Smidt, M.P. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem. J. 2005, 391, 623–629. [Google Scholar] [CrossRef]
- Fowle, H.; Zhao, Z.; Grana, X. PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res. 2019, 144, 55–93. [Google Scholar] [CrossRef]
- Salamango, D.J.; Johnson, M.C. Characterizing the Murine Leukemia Virus Envelope Glycoprotein Membrane-Spanning Domain for Its Roles in Interface Alignment and Fusogenicity. J. Virol. 2015, 89, 12492–12500. [Google Scholar] [CrossRef]
- Salamango, D.J.; Ikeda, T.; Moghadasi, S.A.; Wang, J.; McCann, J.L.; Serebrenik, A.A.; Ebrahimi, D.; Jarvis, M.C.; Brown, W.L.; Harris, R.S. HIV-1 Vif Triggers Cell Cycle Arrest by Degrading Cellular PPP2R5 Phospho-regulators. Cell Rep. 2019, 29, 1057–1065.e4. [Google Scholar] [CrossRef] [PubMed]
- Salamango, D.J.; McCann, J.L.; Demir, O.; Becker, J.T.; Wang, J.; Lingappa, J.R.; Temiz, N.A.; Brown, W.L.; Amaro, R.E.; Harris, R.S. Functional and Structural Insights into a Vif/PPP2R5 Complex Elucidated Using Patient HIV-1 Isolates and Computational Modeling. J. Virol. 2020, 94, e00631-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Liang, H.; Zhao, X.; Liang, L.; Wang, G.; Yang, J.; Jin, Y.; McNutt, M.A.; Yin, Y. Protein Phosphatase 2A (PP2A) Regulates EG5 to Control Mitotic Progression. Sci. Rep. 2017, 7, 1630. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 2011, 14, 593–605. [Google Scholar] [CrossRef]
- Lee, E.J.; Kim, J.M.; Lee, M.K.; Jameson, J.L. Splice variants of the forkhead box protein AFX exhibit dominant negative activity and inhibit AFXalpha-mediated tumor cell apoptosis. PLoS ONE 2008, 3, e2743. [Google Scholar] [CrossRef]
- Sunters, A.; Madureira, P.A.; Pomeranz, K.M.; Aubert, M.; Brosens, J.J.; Cook, S.J.; Burgering, B.M.; Coombes, R.C.; Lam, E.W. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006, 66, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Lasick, K.A.; Jose, E.; Samayoa, A.M.; Shanks, L.; Pond, K.W.; Thorne, C.A.; Paek, A.L. FOXO nuclear shuttling dynamics are stimulus-dependent and correspond with cell fate. Mol. Biol. Cell 2023, 34, ar21. [Google Scholar] [CrossRef]
- Cohen, P.; Holmes, C.F.; Tsukitani, Y. Okadaic acid: A new probe for the study of cellular regulation. Trends Biochem. Sci. 1990, 15, 98–102. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. AMPK exerts dual regulatory effects on the PI3K pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef]
- Kovacic, S.; Soltys, C.L.; Barr, A.J.; Shiojima, I.; Walsh, K.; Dyck, J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003, 278, 39422–39427. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.K.; Liu, H.-Y.; Francisco, J.; Pandya, D.; Donigan, M.; Gallo-Ebert, C.; Giordano, C.; Bata, A.; Nickels, J.T., Jr. Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d. J. Biol. Chem. 2015, 290, 10588–10598. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, X.; Xie, D.; Tang, P.; Li, C.; Zuo, Y.; Jiang, B.; Xue, C. Targeting PP2A activates AMPK signaling to inhibit colorectal cancer cells. Oncotarget 2017, 8, 95810–95823. [Google Scholar] [CrossRef]
- Cairns, J.; Ly, R.C.; Niu, N.; Kalari, K.R.; Carlson, E.E.; Wang, L. CDC25B partners with PP2A to induce AMPK activation and tumor suppression in triple negative breast cancer. NAR Cancer 2020, 2, zcaa039. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar] [CrossRef]
- Coa, L.L.; Abreu, T.F.; Tashima, A.K.; Green, J.; Pascon, R.C.; Vallim, M.A.; Machado-Jr, J. AKT/protein kinase B associates with beta-actin in the nucleus of melanoma cells. Biosci. Rep. 2019, 39, BSR20181312. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y. Neuroprotection signaling of nuclear akt in neuronal cells. Exp. Neurobiol. 2014, 23, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; López-Méndez, B.; Sigurðsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell 2016, 63, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bajaj, R.; Bollen, M.; Peti, W.; Page, R. Expanding the PP2A Interactome by Defining a B56-Specific SLiM. Structure 2016, 24, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Luperchio, A.M.; Jónsson, S.R.; Salamango, D.J. Evolutionary conservation of PP2A antagonism and G2/M cell cycle arrest in Maedi-Visna Virus Vif. Viruses 2022, 14, 1701. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Garvanska, D.H.; Varga, J.K.; Garland, W.; McEwan, B.C.; Hein, J.B.; Weisser, M.B.; Benavides-Puy, I.; Chan, C.B.; Sotelo-Parrilla, P.; et al. Substrate recognition principles for the PP2A-B55 protein phosphatase. Sci. Adv. 2024, 10, eadp5491. [Google Scholar] [CrossRef] [PubMed]
- Cundell, M.J.; Hutter, L.H.; Nunes Bastos, R.; Poser, E.; Holder, J.; Mohammed, S.; Novak, B.; Barr, F.A. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J. Cell Biol. 2016, 214, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Fowle, H.; Zhao, Z.; Xu, Q.; Wasserman, J.S.; Wang, X.; Adeyemi, M.; Feiser, F.; Kurimchak, A.N.; Atar, D.; McEwan, B.C.; et al. PP2A/B55alpha substrate recruitment as defined by the retinoblastoma-related protein p107. eLife 2021, 10, e63181. [Google Scholar] [CrossRef]
- Kruse, T.; Gnosa, S.P.; Nasa, I.; Garvanska, D.H.; Hein, J.B.; Nguyen, H.; Samsoe-Petersen, J.; Lopez-Mendez, B.; Hertz, E.P.T.; Schwarz, J.; et al. Mechanisms of site-specific dephosphorylation and kinase opposition imposed by PP2A regulatory subunits. EMBO J. 2020, 39, e103695. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhu, S.; Fisher, L.A.; Wang, W.; Oakley, G.G.; Li, C.; Peng, A. Protein interactomes of protein phosphatase 2A B55 regulatory subunits reveal B55-mediated regulation of replication protein A under replication stress. Sci. Rep. 2018, 8, 2683. [Google Scholar] [CrossRef]
- Labbe, J.C.; Vigneron, S.; Mechali, F.; Robert, P.; Roque, S.; Genoud, C.; Goguet-Rubio, P.; Barthe, P.; Labesse, G.; Cohen-Gonsaud, M.; et al. The study of the determinants controlling Arpp19 phosphatase-inhibitory activity reveals an Arpp19/PP2A-B55 feedback loop. Nat. Commun. 2021, 12, 3565. [Google Scholar] [CrossRef] [PubMed]
- Padi, S.K.R.; Vos, M.R.; Godek, R.J.; Fuller, J.R.; Kruse, T.; Hein, J.B.; Nilsson, J.; Kelker, M.S.; Page, R.; Peti, W. Cryo-EM structures of PP2A:B55-FAM122A and PP2A:B55-ARPP19. Nature 2024, 625, 195–203. [Google Scholar] [CrossRef]
- Hached, K.; Goguet, P.; Charrasse, S.; Vigneron, S.; Sacristan, M.P.; Lorca, T.; Castro, A. ENSA and ARPP19 differentially control cell cycle progression and development. J. Cell Biol. 2019, 218, 541–558. [Google Scholar] [CrossRef]
- Aakula, A.; Isomursu, A.; Rupp, C.; Erickson, A.; Gupta, N.; Kauko, O.; Shah, P.; Padzik, A.; Pokharel, Y.R.; Kaur, A.; et al. PP2A methylesterase PME-1 suppresses anoikis and is associated with therapy relapse of PTEN-deficient prostate cancers. Mol. Oncol. 2023, 17, 1007–1023. [Google Scholar] [CrossRef]
- Xing, Y.; Li, Z.; Chen, Y.; Stock, J.B.; Jeffrey, P.D.; Shi, Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 2008, 133, 154–163. [Google Scholar] [CrossRef]
- Orea-Soufi, A.; Paik, J.; Braganca, J.; Donlon, T.A.; Willcox, B.J.; Link, W. FOXO transcription factors as therapeutic targets in human diseases. Trends Pharmacol. Sci. 2022, 43, 1070–1084. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Dejean, A.S.; Hedrick, S.M.; Kerdiles, Y.M. Highly specialized role of Forkhead box O transcription factors in the immune system. Antioxid. Redox Signal. 2011, 14, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Zengin, G.; Negrut, N.; Nistor-Cseppento, D.C.; Pavel, F.M.; Corb Aron, R.A.; Bungau, S. Exploring the Genetic Conception of Obesity via the Dual Role of FoxO. Int. J. Mol. Sci. 2021, 22, 3179. [Google Scholar] [CrossRef]
- Wu, C.G.; Balakrishnan, V.K.; Merrill, R.A.; Parihar, P.S.; Konovolov, K.; Chen, Y.C.; Xu, Z.; Wei, H.; Sundaresan, R.; Cui, Q.; et al. B56delta long-disordered arms form a dynamic PP2A regulation interface coupled with global allostery and Jordan’s syndrome mutations. Proc. Natl. Acad. Sci. USA 2024, 121, e2310727120. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- McClinch, K.; Avelar, R.A.; Callejas, D.; Izadmehr, S.; Wiredja, D.; Perl, A.; Sangodkar, J.; Kastrinsky, D.B.; Schlatzer, D.; Cooper, M.; et al. Small-Molecule Activators of Protein Phosphatase 2A for the Treatment of Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 2065–2080. [Google Scholar] [CrossRef]
- Leonard, D.; Huang, W.; Izadmehr, S.; O’Connor, C.M.; Wiredja, D.D.; Wang, Z.; Zaware, N.; Chen, Y.; Schlatzer, D.M.; Kiselar, J.; et al. Selective PP2A Enhancement through Biased Heterotrimer Stabilization. Cell 2020, 181, 688–701.e16. [Google Scholar] [CrossRef] [PubMed]
- Shenolikar, S. A SMAP in the face for cancer. J. Clin. Investig. 2017, 127, 2048–2050. [Google Scholar] [CrossRef]
- Johnson, H.; Narayan, S.; Sharma, A.K. Altering phosphorylation in cancer through PP2A modifiers. Cancer Cell Int. 2024, 24, 11. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef]
- Cucinotta, L.; Filippone, A.; Casili, G.; Lanza, M.; Bova, V.; Capra, A.P.; Giuffrida, R.; Colarossi, C.; Sciacca, D.; Paterniti, I.; et al. The Pivotal Role of Protein Phosphatase 2A (PP2A) in Brain Tumors. Int. J. Mol. Sci. 2022, 23, 15717. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luperchio, A.M.; Salamango, D.J. Defining the Protein Phosphatase 2A (PP2A) Subcomplexes That Regulate FoxO Transcription Factor Localization. Cells 2025, 14, 342. https://doi.org/10.3390/cells14050342
Luperchio AM, Salamango DJ. Defining the Protein Phosphatase 2A (PP2A) Subcomplexes That Regulate FoxO Transcription Factor Localization. Cells. 2025; 14(5):342. https://doi.org/10.3390/cells14050342
Chicago/Turabian StyleLuperchio, Adeline M., and Daniel J. Salamango. 2025. "Defining the Protein Phosphatase 2A (PP2A) Subcomplexes That Regulate FoxO Transcription Factor Localization" Cells 14, no. 5: 342. https://doi.org/10.3390/cells14050342
APA StyleLuperchio, A. M., & Salamango, D. J. (2025). Defining the Protein Phosphatase 2A (PP2A) Subcomplexes That Regulate FoxO Transcription Factor Localization. Cells, 14(5), 342. https://doi.org/10.3390/cells14050342