3.2. Activation of AMPK Inhibits Flaviviruses at Viral Entry
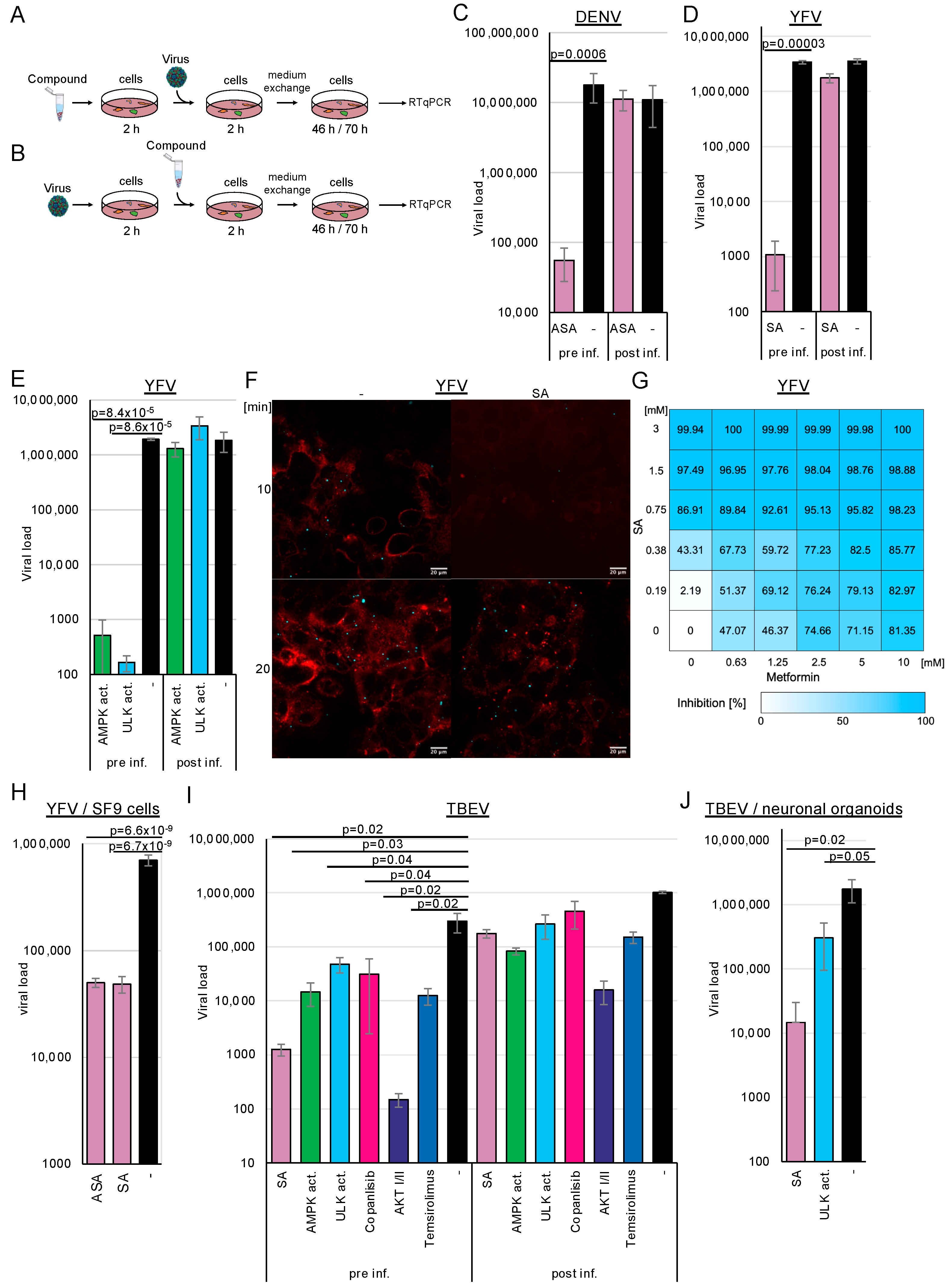
Since other (+)-stranded RNA viruses commonly utilise replicative compartments derived from endosomes, the endoplasmic reticulum, and lysosomes, we analysed whether AMPK activation by ASA/SA inhibits DENV, TBEV, and YFV replication. Huh-7 (DENV, YFV) or human glioma NCE U373 (TBEV) cells were pre-treated with 3 mM ASA/SA, 100 µM AMPK, or ULK1 activators for 2 h prior to infection (
Figure 2A). The AMPK activator does not directly interact with AMPK but inhibits the mitochondrial complex I, reducing cellular ATP levels and subsequently activating AMPK [
32]. The viruses were removed 2 h after infection through a medium exchange. Cell culture supernatants were collected after 2 (YFV, TBEV) or 3 (DENV) days; viral RNAs were isolated and quantified by RTqPCR (
Figure 2C–F). All infection experiments detailed in this manuscript were performed three times in triplicate. The accumulation of TBEV and DENV RNAs was inhibited by more than 2 orders of magnitude (log
10), demonstrating that ASA/SA treatment efficiently suppressed these viruses. Treatment with the AMPK activator inhibited TBEV replication by approximately 1.3 log
10 and YFV by more than 3 log
10, indicating that AMPK activation was sufficient to repress viral replication (
Figure 2E,I,J). The activation of the downstream ULK1 further suppressed YFV and TBEV replication, showing that the inhibition of viral replication was mediated through ULK1.
Metformin, a commonly used antidiabetic drug, has also been shown to activate AMPK [
33,
34]. Given that high doses of SA are necessary for AMPK activation, we aimed to investigate whether a similar activation of AMPK could be achieved through the combination of metformin and SA. We anticipated that both compounds would exhibit additive effects. Cells were treated with increasing concentrations of SA, ranging from 0.185 to 3 mM, and metformin, from 0.625 to 10 mM, and were infected with YFV (
Figure 2G). The EC
50 for SA was calculated as 0.42 mM (0.62 mM on Vero cells) for AMPK activator 17.32 µM (Vero cells: 10.34 µM), while that for metformin was 1.91 mM (Vero cells: 1.61 mM). Concentrations of 1.5 mM SA suppressed YFV replication by 97.49%, whereas 10 mM metformin inhibited viral replication by 81.4%. The analysis of drug synergism using the SynergyFinder software version 3.0 [
35] demonstrated that the compounds acted additively, leading to an inhibition of over 98% with 2.5 mM metformin and 1.5 mM SA, or with 10 mM metformin and 0.75 mM SA. Consequently, the combination of metformin and SA results in either enhanced inhibition of viral replication or requires lower concentrations of the compounds, thereby achieving comparable suppression. This suggests that the decreased concentrations of SA could lead to adverse effects on fever in patients.
Recently, we and others have observed that even direct antivirals can act in a cell-type-specific manner [
31,
36,
37]. Consequently, we aimed to validate our findings regarding TBEV using patient-relevant systems, specifically employing human stem cell-derived 3D neuronal organoid models. The organoids were exposed to 3 mM SA or 100 mM ULK1 activator and were subsequently infected with TBEV. Cell culture supernatants were collected 2 days after infection, and viral RNAs were quantified by RTqPCR (
Figure 2J). SA downregulated TBEV replication by more than 2 log
10, and the ULK1 inhibitor reduced it by 6-fold. These results indicate that the activation of AMPK inhibits TBEV replication even in this model patient-near system. Since mosquitoes transmit YFV, we investigated whether AMPK activation influences viral replication in insect SF9 cells. We observed the antiviral effects of ASA and SA in these cells, providing evidence that the mechanism is conserved (
Figure 2H).
Next, we aimed to determine which replication step was targeted by AMPK activation. Drugs were added either 2 h (
Figure 2A) before (pre-treatment) or 2 h after viral infection (
Figure 2B) to discern whether the compounds inhibited viral replication at the entry or the post-entry steps. For the latter setting, 3 mM ASA, SA and 100 µM AMPK activator (
Figure 2D–I), or 100 µM of the ULK1, were used (
Figure 2E,I). The cells were incubated for 46 h (YFV, TBEV) or 70 h (DENV). Viral RNAs were isolated and quantified by RTqPCR (
Figure 2). We expected a lack of inhibition with AMPK activators, similar to the untreated controls, and wanted to determine whether AMPK activation affected only viral entry since the compounds were added after infection. We observed inhibition of viral replication by more than 2 log
10 when compounds were added 2 h before viral entry. However, treatment with the compounds after infection did not reduce DENV infection, indicating that compound-mediated AMPK activation interfered with DENV entry. Experiments with YFV and TBEV yielded comparable results, suggesting that AMPK activation similarly inhibits viral entry. Remarkably, the pharmacological inhibition of receptor-mediated viral entry by the ULK1 activator suggested that this downstream kinase also regulates flavivirus entry, revealing a previously unrecognised role in the viral infection process.
We generated fluorescently labelled YFV to determine, using an independent assay, that entry is inhibited by AMPK activation. Cells were infected with concentrated YFV (MOI~10). After 1 day, the medium was exchanged for medium containing ATTO 643 DOPE, a compound that stains cell membranes. Cell culture supernatants were harvested after 12 h, and viruses were pelleted by ultracentrifugation, resulting in a coloured pellet. Huh-7 cells were treated with SA for 2 h; then, the medium was replaced with a medium containing a cytoplasmic membrane cell stain (BioTracker 400 blue, Sigma-Aldrich, Taufkirchen, Germany) but no compounds. This dye is taken up by endocytosis. The cells were infected with the stained viruses for 10 or 20 min and fixed with paraformaldehyde. Confocal microscopy revealed that YFV could not be detected in cells treated with SA for 10 min, indicating that the viruses were not endocytosed (
Figure 2F). The membrane dye was not incorporated, suggesting a general block of endocytosis. However, after 20 min of infection without the compound, virus binding was restored, demonstrating that the inhibition was reversible. The results suggest that the AMPK activation blocks one of the first steps of endocytosis.
3.3. AMPK Activation Inhibits Enteroviruses, Alphavirus, and Rubella Virus Entry
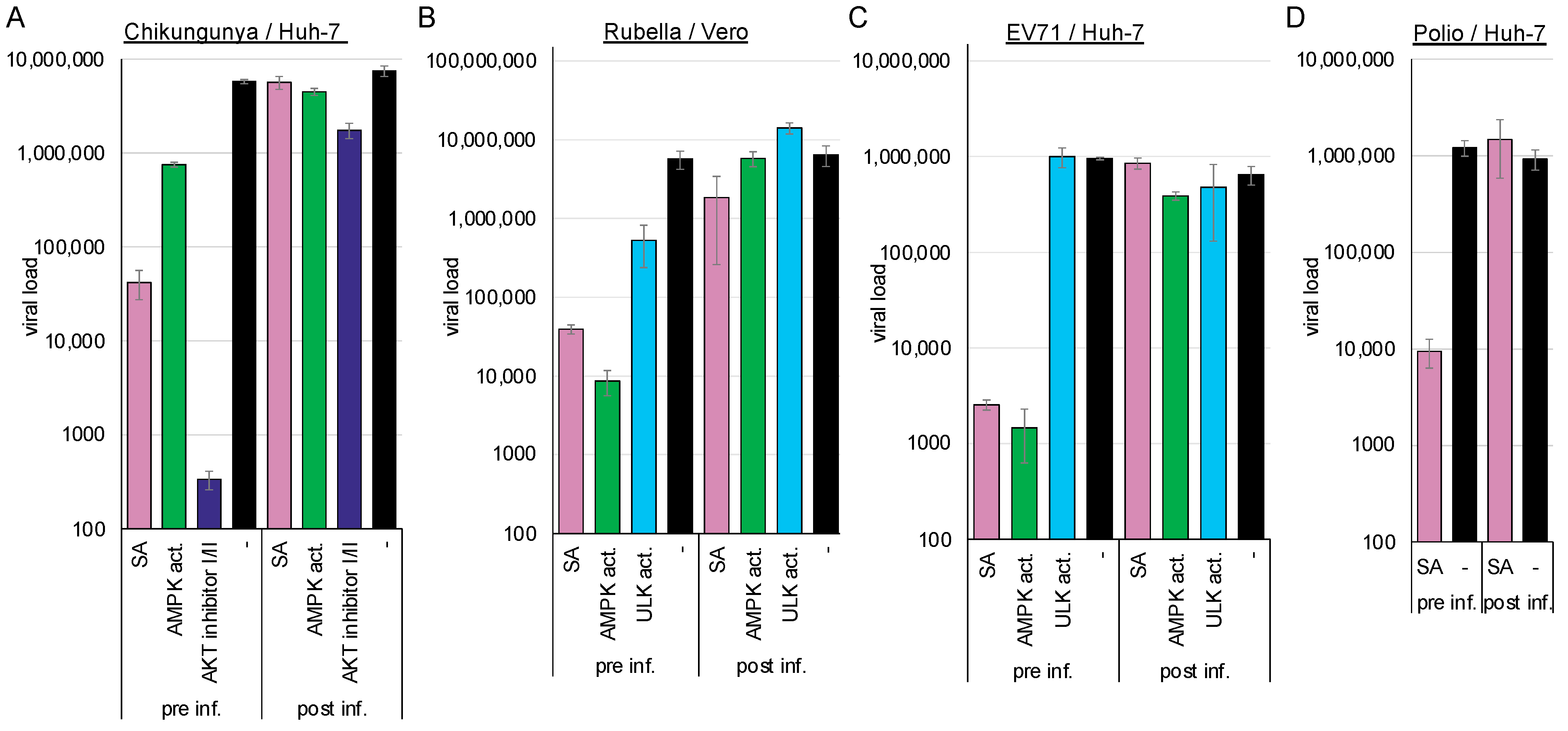
Having observed that flavivirus entry is sensitive to AMPK activation, we sought to analyse whether this phenomenon also applies to other plus-stranded RNA viruses, such as rubella, chikungunya, poliovirus, and enterovirus 71. Like RVFV, the chikungunya virus enters the cell via the Mxra8 receptor. The myelin oligodendrocyte glycoprotein serves as a receptor for the rubella virus, while the scavenger receptor class B member 2, or the P-selectin glycoprotein ligand-1, functions as a receptor for enterovirus 71. Polioviruses utilise CD155 for receptor-mediated endocytosis [
38,
39,
40,
41]. Therefore, we expected that AMPK activation would inhibit early infection steps only if entry is regulated by cellular factors rather than by viral factors.
We compared viral replication in cells pre-treated for 2 h with the compounds or in cells infected 2 h prior to treatment to further analyse whether the activation of AMPK by SA or an AMPK activator suppresses viral entry into the cells. Viral replication was determined by RT-qPCR 2 d post-infection. Activation of AMPK suppressed chikungunya (SA: 2.14 log
10,
p = 1.4 × 10
−5; AMPK activator: log
10 = 0.9,
p = 2.4 × 10
−5) (
Figure 3A), rubella (SA: log
10 = 2.16,
p = 0.03; AMPK activator: log
10 = 2.82,
p = 0.03) (
Figure 3B), EV71 (SA: log
10 = 2.16,
p = 0.03; AMPK activator: log
10 = 2.82,
p = 0.002) (
Figure 3C), and poliovirus (2.11 log
10,
p = 0.002) (
Figure 3D) when cells were preincubated with the compounds. AMPK activation did not significantly reduce viral replication, indicating that viral entry is inhibited, in accordance with our results involving flaviviruses and dextran. The ULK1 activator reduced rubella virus replication when added prior to entry (1.0 log
10,
p = 0.03) but failed to inhibit EV71 replication. This indicates that AMPK induces two distinct antiviral pathways; one is ULK1-dependent and inhibits enveloped viruses, while the other additionally inhibits non-enveloped viruses.
3.4. AMPK Activation Does Not Inhibit SARS-CoV-2 Entry into Calu-3 Cells but Does in Vero Cells
Recently, we demonstrated that ASA and SA inhibit SARS-CoV-2 replication in cell culture and human PCLSs [
23]. Quantifying viral RNA in infected cells at different time points did not support a correlation between the inhibition of viral replication and the interference with viral entry in Calu-3 cell cultures. However, as SARS-CoV-2 can enter cells via direct fusion with the plasma membrane or through endocytosis using the same receptor [
42,
43], we reanalysed the influence of SA on entry using a modified experimental approach with Calu-3 or Vero cells (
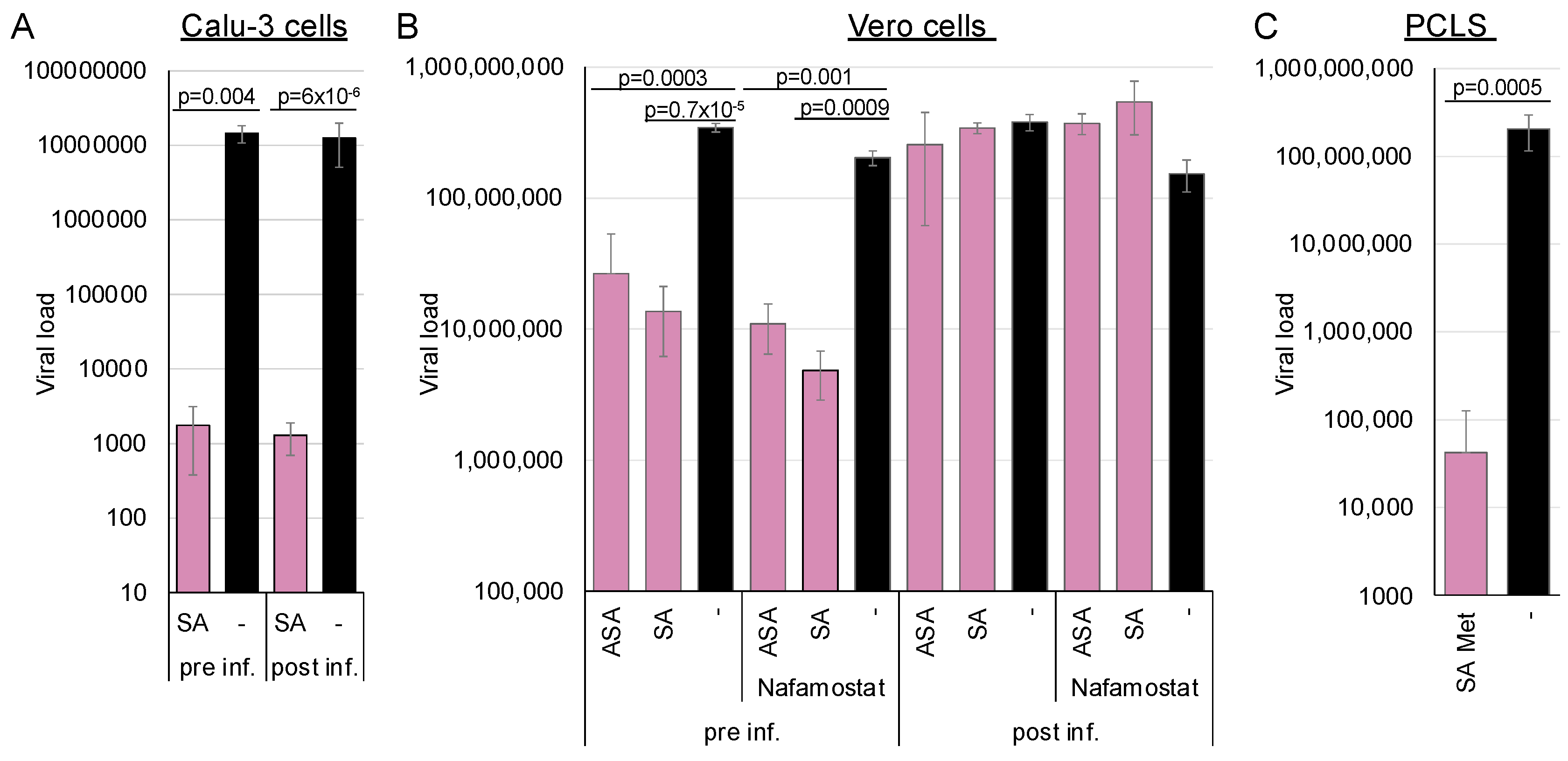
Figure 4A,B). Calu-3 cells express high amounts of the TMPRSS2 protease and, therefore, promote viral entry by fusion at the plasma membrane. Entry into Vero cells is preferentially endocytic, which we reinforced by adding the TMPRSS2 inhibitor nafamostat (
Figure 4B). This experiment enabled the determination of AMPK-mediated influences on the entry pathways since the virus and the receptor remain invariant.
The Calu-3 cells were either preincubated for 2 h before infection with 3 mM SA, or they were infected with SARS-CoV-2, after which 3 mM SA was added 2 h post-infection. In the first case, viral replication should be inhibited if SA targets viral entry, whereas in the second scenario, it should not be affected. The medium was exchanged after 6 h, and viral genomes were quantified after 72 h by RT-qPCR (
Figure 4). Virus replication in Calu-3 was independent of pre- or post-incubation, underlining that AMPK did not target entry via direct fusion at the plasma membrane and confirming our previous findings.
To further analyse whether AMPK activation through a combination of SA and metformin inhibits SARS-CoV-2 in a patient-near system, we treated human PCLSs with 5 mM metformin and 1.5 mM SA (
Figure 4C). The combination effectively suppressed viral replication, demonstrating that AMPK activation inhibits SARS-CoV-2 in a patient-near system.
A similar experiment with Vero cells showed that AMPK activation by ASA or SA before entry inhibited SARS-CoV-2 replication. However, the antiviral effect was completely abolished when viral entry occurred prior to AMPK activation, demonstrating that ASA and SA specifically target endocytic viral entry rather than receptor recognition or binding. Further inhibition of TMPRSS2 by nafamostat slightly decreased viral replication, indicating that TMPRSS2 activity in Vero cells is low. These results suggest that AMPK activation can inhibit SARS-CoV-2 entry in cells expressing low amounts of TMPRSS2 and elucidate the observed correlation between fasting blood glucose and the severity of COVID-19 in patients, even without diagnosed diabetes [
44,
45]. In addition, AMPK activation initiates a second antiviral mechanism, blocking SARS-CoV-2 replication in Calu-3 cells.
3.5. AMPK Activation Inhibits Entry of Negative-Stranded RNA Viruses, Such as Rabies Virus, and Supports Rabies Postexposure Prophylaxis
Since we have demonstrated that the endocytic entry of (+)-stranded RNA viruses is AMPK-dependent, we investigated whether the entry of negative-stranded RNA viruses, specifically the vesicular stomatitis virus and the rabies virus, is influenced by AMPK activation. In particular, if ASA/SA inhibits rabies replication, it could be included in the postexposure prophylaxis alongside antisera to prevent rabies infections. Inhibition and entry analyses were conducted as described above.
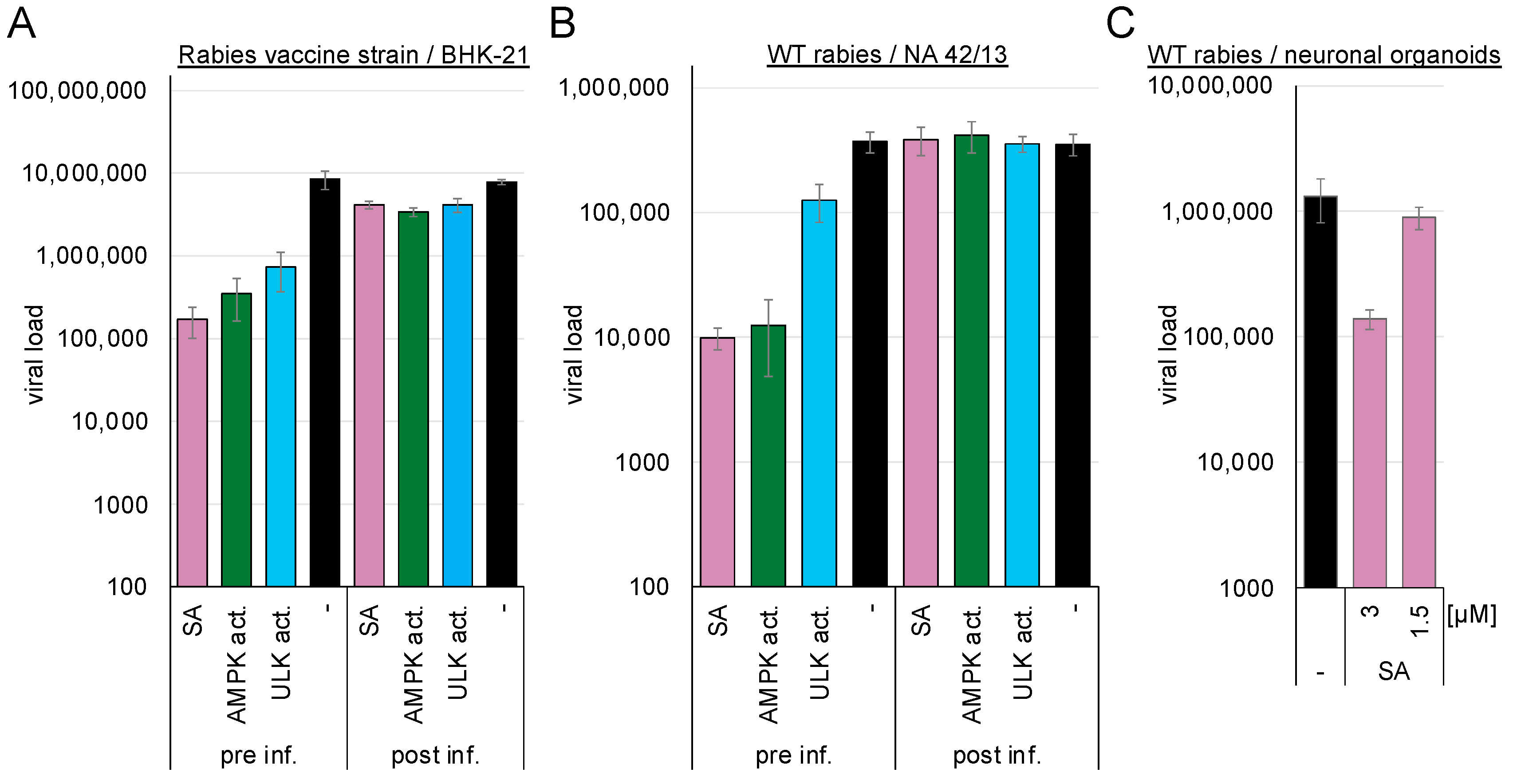
BHK-21 or mouse NA 42/13 cells were either pre-treated with 3 mM ASA/SA, 100 µM AMPK, or ULK1 activators and subsequently infected with the rabies vaccine (BHK-21 cells,
Figure 5A) or the wild-type strain (NA 42/13 cells,
Figure 5B), or they were infected and incubated with the compounds after 2 h. Supernatants were harvested after 75 h, and viral genome copies were determined by RTqPCR. Pre-treatment with SA inhibited replication of the rabies vaccine strain by more than 1.7 log
10 (SA: 1.71 log
10,
p = 0.03). AMPK activation inhibited entry of the vaccine strain by 1.4 log
10 (
p = 0.04) and the wild-type rabies virus by approximately 1.5 log
10 (SA: 1.6 log
10,
p = 0.002, AMPK activator: 1.5 log
10,
p = 0.002) into NA 42/13 cells, while the ULK1 activator reduced entry of the vaccine strain by 1.1 log
10 (
p = 0.046) and the wild-type rabies virus by 0.5 log
10 (
p = 0.013). Thus, activation of AMPK downregulates the entry of the rabies virus.
The AMPK-induced inhibition of viral replication was confirmed in human stem cell-derived 3D neuronal organoid models. The organoids were exposed to 1.5 or 3 mM SA and subsequently infected with wild-type rabies virus. The medium was exchanged after 24 h to remove the non-incorporated virus. Supernatants were collected after 3 d of infection, and viral RNAs were quantified by RTqPCR (
Figure 5C). The introduction of 3 mM SA reduced viral genome amounts by 1 log
10 (
p = 0.05). Similar inhibition of the vesicular stomatitis virus was observed after AMPK activation with SA (
Figure S1).
AMPK activation inhibited rabies wild-type and vaccine strain entry and replication. Thus, it might be beneficial for exposed patients to add an AMPK activator to the approved antisera for postexposure prophylaxis or, if antisera are unavailable, use them to clean the bite wounds.
3.6. AMPK Activation Does Not Influence Receptor Presentation but Downregulates TXNIP, Rab5, and Rab7 by Inactivating eEF2 and Activating the Proteasome
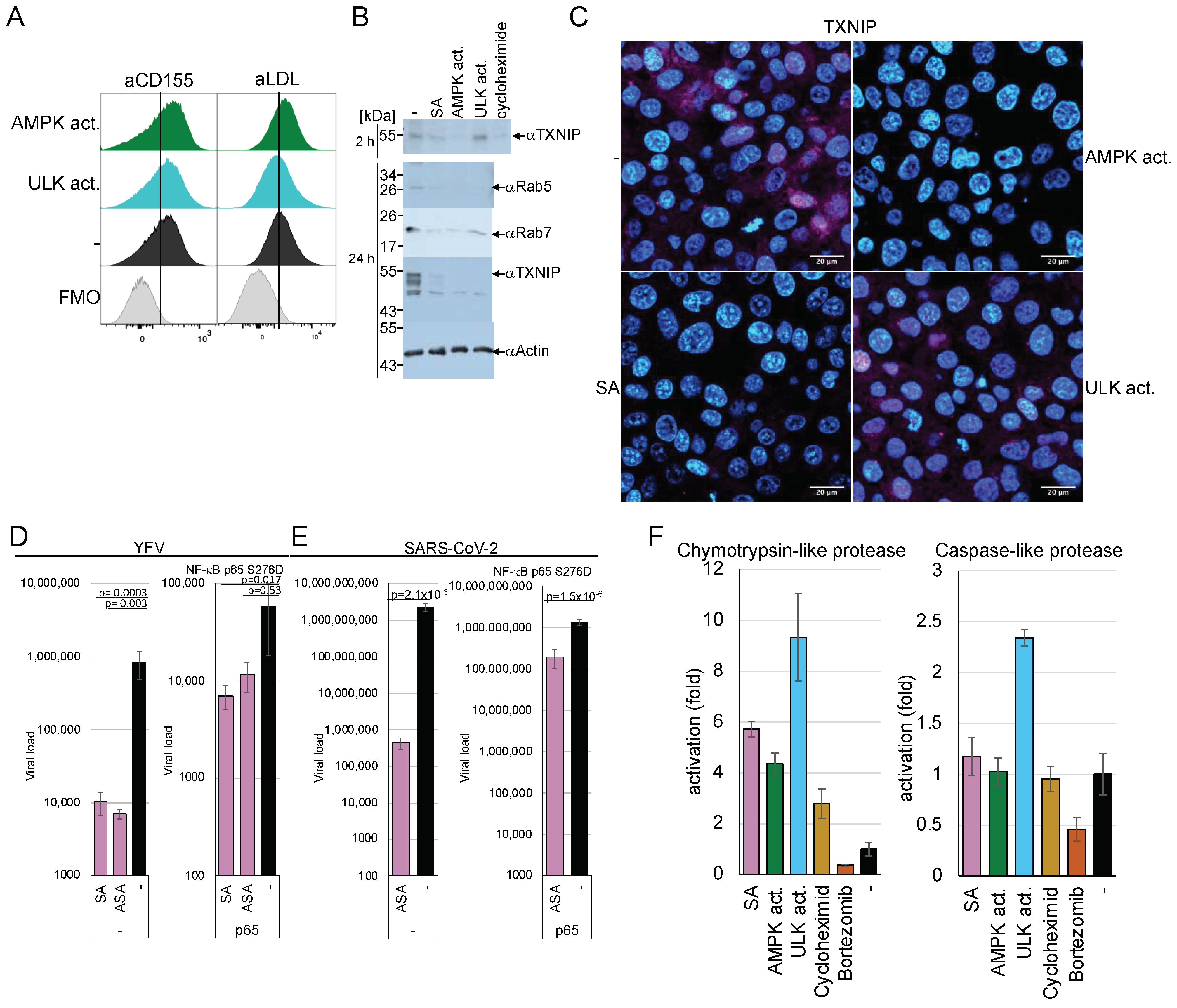
We further investigated the mechanisms involved in the AMPK-mediated downregulation of viral entry. Viruses, such as DENV, TBEV, YFV, chikungunya, rabies, VSV, and SARS-CoV-2 or molecules, such as dextran, utilise specific entry receptors and should be regulated differently. Consequently, AMPK activation is hypothesised to impede viral entry and, more broadly, receptor-mediated endocytosis. First, we determined if AMPK or ULK1 activation changed the receptor presentation at the plasma membrane. We decided to analyse the poliovirus receptor CD155 and the VSV low-density lipoprotein (LDL) entry receptor on MT4 cells by FACS [
46] to address receptor proteins of negative- and non-enveloped, positive-stranded RNA viruses. The LDL receptor supports the entry of Getah, Semliki Forest, and Bebaru viruses in addition to VSV [
47]. MT4 cells were treated with the AMPK or ULK1 activator for 2 h and stained with the respective rabbit-anti-CD155 or mouse-anti-LDL receptor antibodies. FACS analyses showed that the amounts of both receptors did not decrease as a result of AMPK or ULK1 activation (
Figure 6A and
Supplementary Materials Figure S2), indicating that changes in the receptor concentration are not responsible for the observed entry inhibition.
Generally, the α-arrestin TXNIP is known to regulate the endocytosis of GLUT1. TXNIP has a short half-life of about 12 min [
48] and is negatively regulated by AMPK through phosphorylation, which promotes its degradation and causes the inhibition of GLUT1 endocytosis. To investigate whether AMPK-mediated inhibition of viral entry is related to TXNIP degradation. Huh-7 cells were incubated with SA for 24 h or an AMPK activator for 2 h, and TXNIP expression was visualised by Western blotting using specific antibodies (
Figure 6B). The multiple TXNIP forms were probably due to degradation. Activation of AMPK decreased TXNIP amounts after 2 h and 24 h, suggesting that one of the first steps of endocytosis is already inhibited, similar to the inhibition of GLUT1. In contrast, ULK1 activation reduced TXNIP only after 24 h.
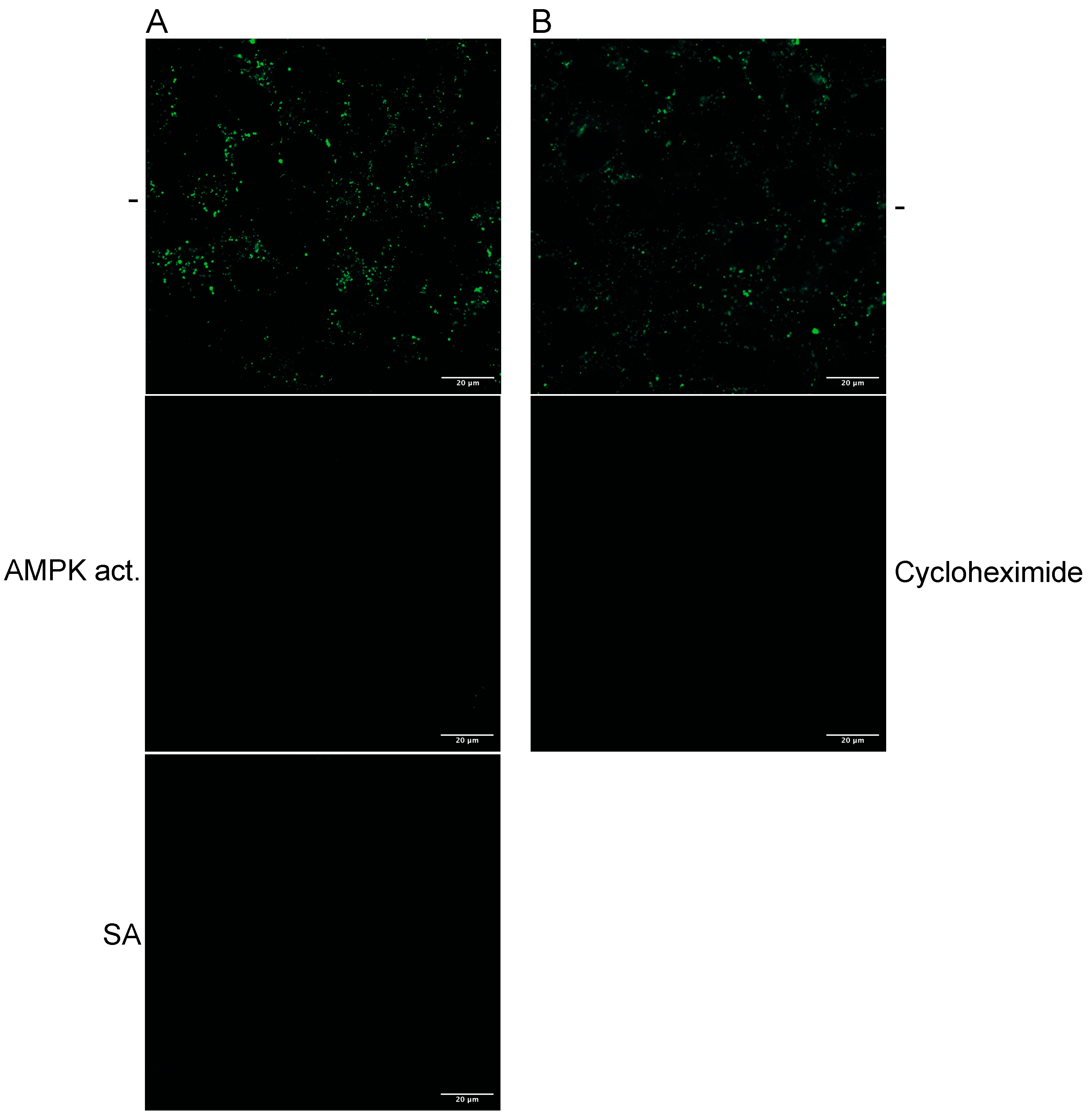
AMPK activates the eEF2 kinase, resulting in the inhibition of translation. The inhibition of translation combined with the short half-life of TXNIP could explain the protein loss due to AMPK activation. The cells were treated with cycloheximide (50 µg/mL) for 2 h to analyse whether the downregulation of TXNIP was due to the inhibition of translation. TXNIP expression was downregulated by inhibition of translation, similar to SA treatment, suggesting that TXNIP is regulated by eEF2 kinase-mediated inactivation (
Figure 6B upper panel). However, ULK1 activation did not significantly decrease TXNIP after 2 h, indicating that the direct phosphorylation by AMPK, which was shown to promote TXNIP degradation by the proteasome [
8], and the inhibition of translation is required for TXNIP reduction. We confirmed the results using immunofluorescence microscopy (
Figure 6C), where TXNIP was reduced by cycloheximide and AMPK activation but not by ULK1 activators. The nuclear localisation of TXNIP has been reported before [
8,
49].
Cycloheximide treatment should block the uptake of dextran if the de novo synthesis of TXNIP is essential for entry. Thus, we treated Huh-7 cells with cycloheximide and added fluorescently labelled dextran. The incorporation was visualised with confocal microscopy (
Figure 1B). SA treatment abolished dextran uptake, while cycloheximide reduced dextran endocytosis compared to the untreated control, indicating that TXNIP enhances dextran uptake.
The p65 subunit of NF-κB downregulates the expression of the eEF2 kinase [
2]. Thus, overexpression of p65 should alleviate the AMPK-mediated inhibition of viral entry if its downregulation is eEF2K-dependent. We constructed a constitutively active p65 S267D mutant expression plasmid. 293T cells were transfected with either the parental vector or the pcDNA-p65 S276D plasmid. The cells were split, treated with SA or ASA, and infected with YFV (
Figure 6D) or SARS-CoV-2 (
Figure 6E). AMPK activation by ASA or SA reduced YFV replication by over 2 orders of magnitude in control cells (
Figure 6D,E). The expression of the constitutively active NF-κB p65 S276D rescues most of the effect of AMPK on viral replication to less than one order of magnitude, indicating that NF-κB restores viral entry. Similar results were obtained for SARS-CoV-2, where AMPK activation inhibited replication by more than 3.6 orders of magnitude. The expression of the constitutively active NF-κB p65 reconstituted viral replication.
In addition to the downregulation of translation, the degradation of TXNIP could contribute to the shutdown of endocytosis through AMPK activation. Therefore, we sought to investigate whether AMPK activation would influence the activity of the proteasome. Huh-7 cells were incubated for 2 h with AMPK and ULK1 activators, cycloheximide, and the proteasome inhibitor bortezomib (100 nM). The activation of AMPK and ULK1 upregulated the cellular chymotrypsin-like protease activity by 5-to-9-fold (
Figure 6F), while only ULK1 activation influenced caspase-like protease activity. TXNIP encodes several predicted highly specific chymotrypsin cleavage sites, while caspase cleavage sites are absent. However, it has been shown that TXNIP is downregulated by caspase activation in T cells. [
50]. The kinase activators did not affect the activity of trypsin-like proteases. The upregulation of proteasome activity could explain the incomplete reconstitution of viral entry by the constitutively active NF-κB p65.
3.7. AMPK Activation Rapidly Downregulates Rab5, Rab7, and TXNIP
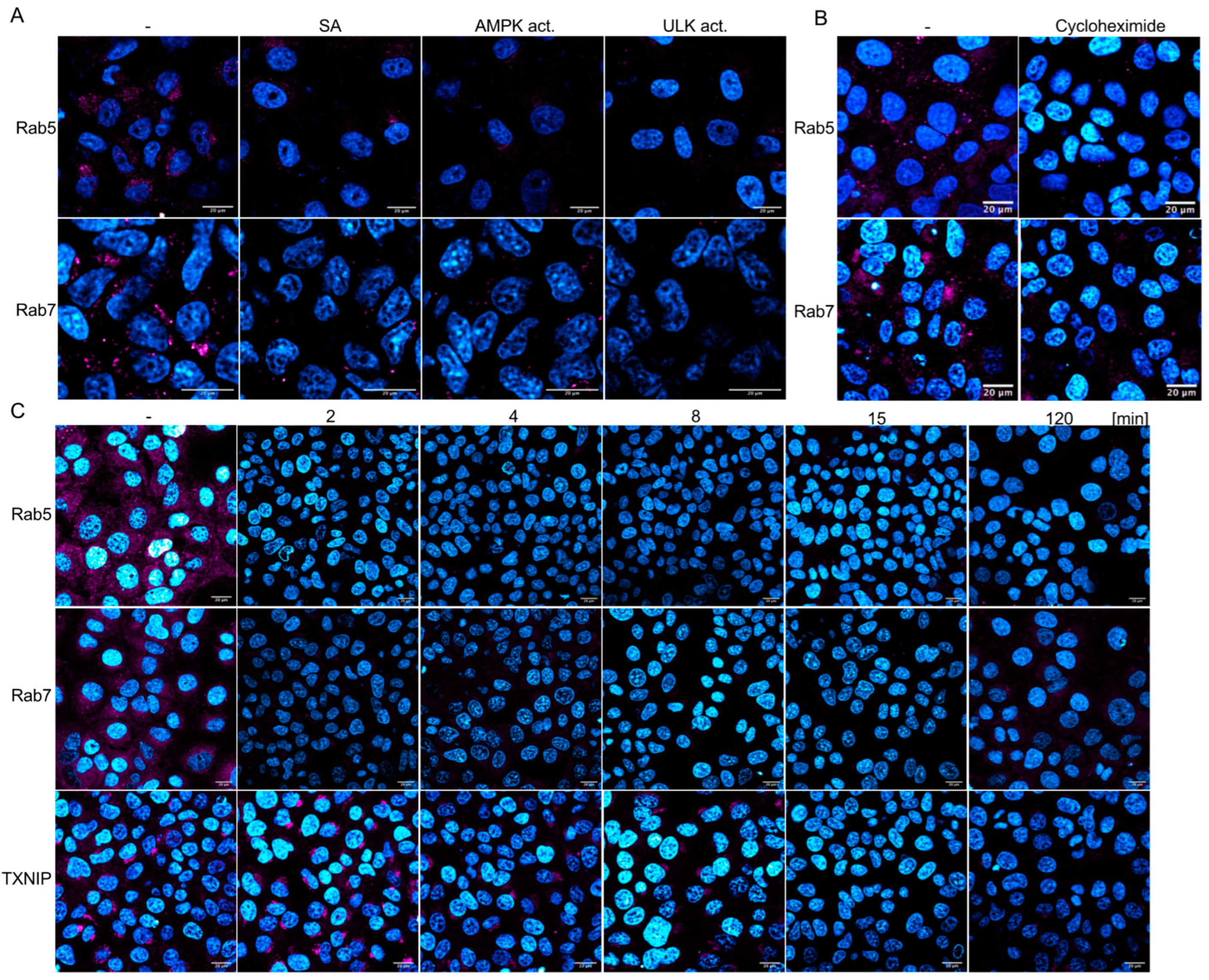
The Ras-related protein Rab5 marks the EE and is enriched in EEs, serving as the primary regulator of the conversion to late endosomes (LEs), where Rab5-GTP recruits Rab7 to the endosome. Thus, EEs are marked by Rab5, whereas during the transition from EEs to LEs, Rab7 replaces Rab5. Analyses of Rab5 and Rab7 amounts by Western blotting revealed that both proteins are downregulated by AMPK and, surprisingly, by ULK1 activation (
Figure 6B), indicating that ULK1 influences the Rab5-positive EEs. These results were confirmed by confocal microscopy using Rab5- and Rab7-specific antibodies (
Figure 7A). Vero cells were treated for 2 h with AMPK or ULK1 activators, then fixed and stained. Confocal microscopy revealed that AMPK activation by SA or the AMPK activator reduced the expression of Rab5 and Rab7. Moreover, ULK1 activation also decreased the number of LEs and EEs. Furthermore, Rab5 and -7 are not only regulated by inhibition of translation but by the activation of the proteasome as well. Thus, the AMPK pathway regulates EEs and LEs, in addition to the Rab4-mediated endosome recycling that has been previously reported [
14]. Considering our results, the upregulation of Rab4 might compensate for the downregulation of Rab5 in EEs and Rab7 in LEs.
Next, we aimed to determine whether Rab5 and Rab7 were downregulated, similar to TXNIP, through AMPK-mediated inhibition of translation or activation of the proteasome. Cells were treated with cycloheximide, and Rab5 and Rab7 were visualised using immunofluorescence (
Figure 7B). The downregulation of translation by cycloheximide resulted in reduced levels of Rab5 and Rab7, similar to that observed during AMPK activation. This reliance on de novo translation is unexpected, given that the half-life of Rab7 has been determined to be 28 h and may also be attributed to the upregulation of proteasomal proteases. Therefore, we examined the time-dependency of SA-mediated AMPK activation on Rab5, Rab7, and TXNIP (
Figure 7C and
Supplementary Materials Figure S3, which includes additional time points). SA decreased Rab5 and Rab7 to undetectable levels after 2 min, while the TXNIP signal remained detectable after 8 min. After 15 min of treatment, neither Rab nor TXNIP signals were observed. However, low levels of Rab7 re-emerged after 2 h. These results substantiate the rapid inhibition of endocytosis and viral entry, indicating that AMPK activation significantly diminishes the half-life of these cellular factors.
The results indicate that AMPK activation inhibits the initial step of endocytosis, as evidenced by the decreased levels of TXNIP in a ULK1-independent way. Nevertheless, both AMPK and ULK1 reduce cellular Rab5 and Rab7 levels within minutes, suggesting a decline in the later stages of endocytosis through translation and proteasome activation (refer to the graphical abstract). In particular, the rapid downregulation of all three endocytosis factors reinforces the observed inhibition of viral entry through drug-induced activation of the AMPK pathway.
3.8. AMPK Activation Modifies the Endo-Lysosomal SARS-CoV-2 Replication Compartment
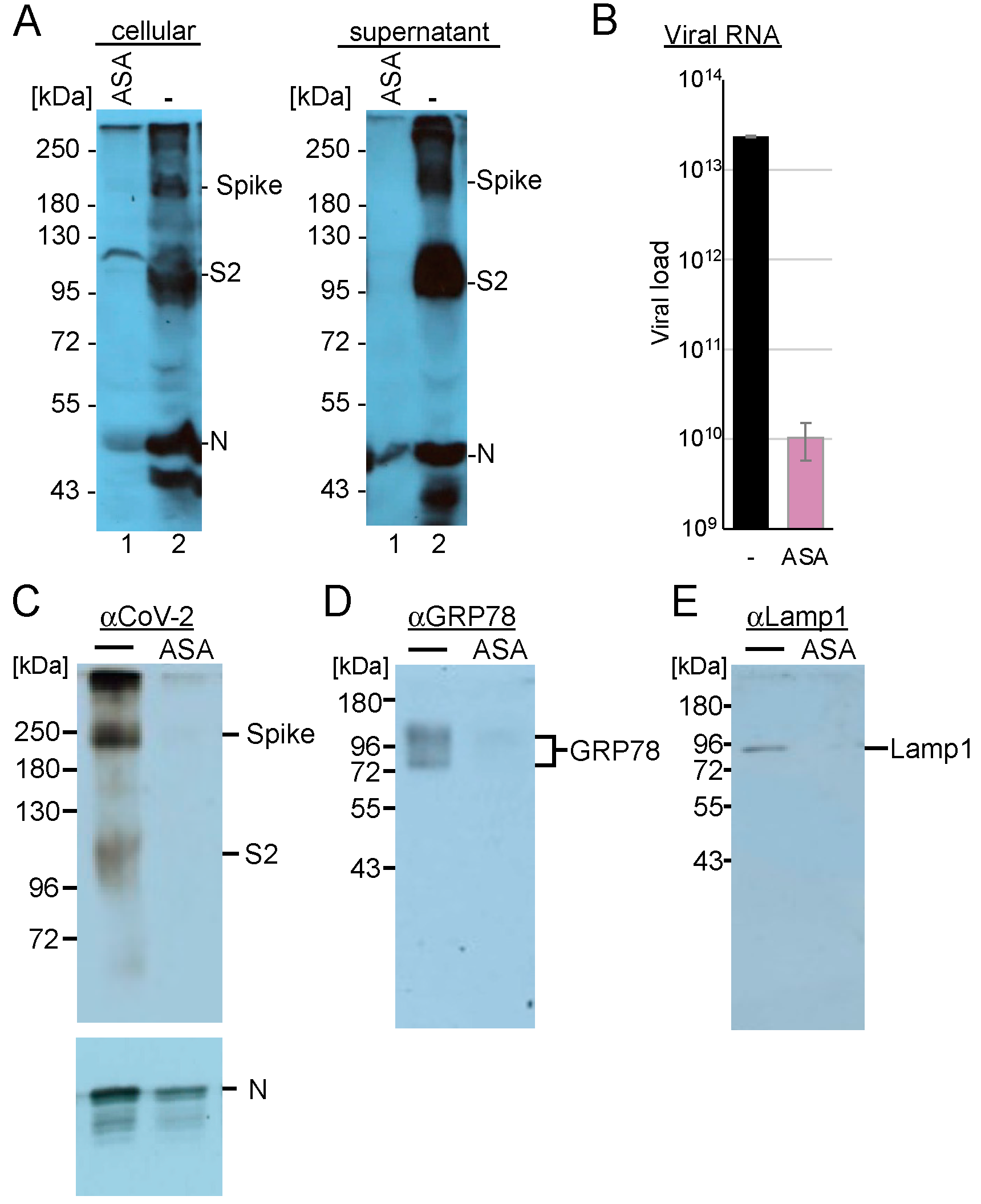
Since Rab7 recruits the V-ATPase required for converting endosomes to lysosomes, we analysed whether AMPK activation inhibits SARS-CoV-2 replication at post-entry steps. First, we determined the influence of AMPK activation on viral gene expression in these cells. Calu-3 cells were incubated with ASA and were infected with SARS-CoV-2. Cells were lysed 3 days after infection, and the amounts of viral N- and spike proteins were detected through Western blotting analysis. Cell culture supernatants were also collected, and the viruses were concentrated by ultracentrifugation through a sucrose cushion. Treatment with AMPK activation reduced the non-processed S protein below the detection level (
Figure 8A), while S and S2 were absent in the supernatant. However, detectable amounts of the N proteins were found in the concentrated viruses.
SARS-CoV-2 replication extensively utilises the lysosomal compartment, and AMPK also regulates lysosomal function as well as the endosomal–lysosomal compartment. Recently, we demonstrated that the serotonin reuptake inhibitor fluoxetine traps SARS-CoV-2 in the lysosomes [
51]. Here, we sought to analyse whether AMPK activation similarly modifies the replication compartment. Calu-3 cells were incubated with ASA and infected with SARS-CoV-2 at an MOI of 10. After 24 h, the cells were lysed, and the lysosomes were isolated. The lysosomal pellets were dissolved in PBS, and viral RNAs and marker proteins were analysed. We determined a reduction of more than 3 log
10 of viral RNAs, with S protein levels being almost undetectable in the cells treated with ASA compared to those in the untreated control (
Figure 8B,C), indicating that AMPK activation inhibited viral transcription and genome replication, resulting in decreased S and N protein levels. Analyses of the endoplasmic reticulum marker GRP78 revealed that the expression of this protein, similar to that of the lysosomal marker protein LAMP1, was reduced in our lysosomal fraction (
Figure 8D,E). These results indicate that AMPK activation with ASA leads to fewer replicative lysosomal compartments, consistent with the observed reductions in Rab5 and Rab7 levels. Our findings suggest that ASA treatment modifies endosomal, lysosomal, and SARS-CoV-2 replication compartments.
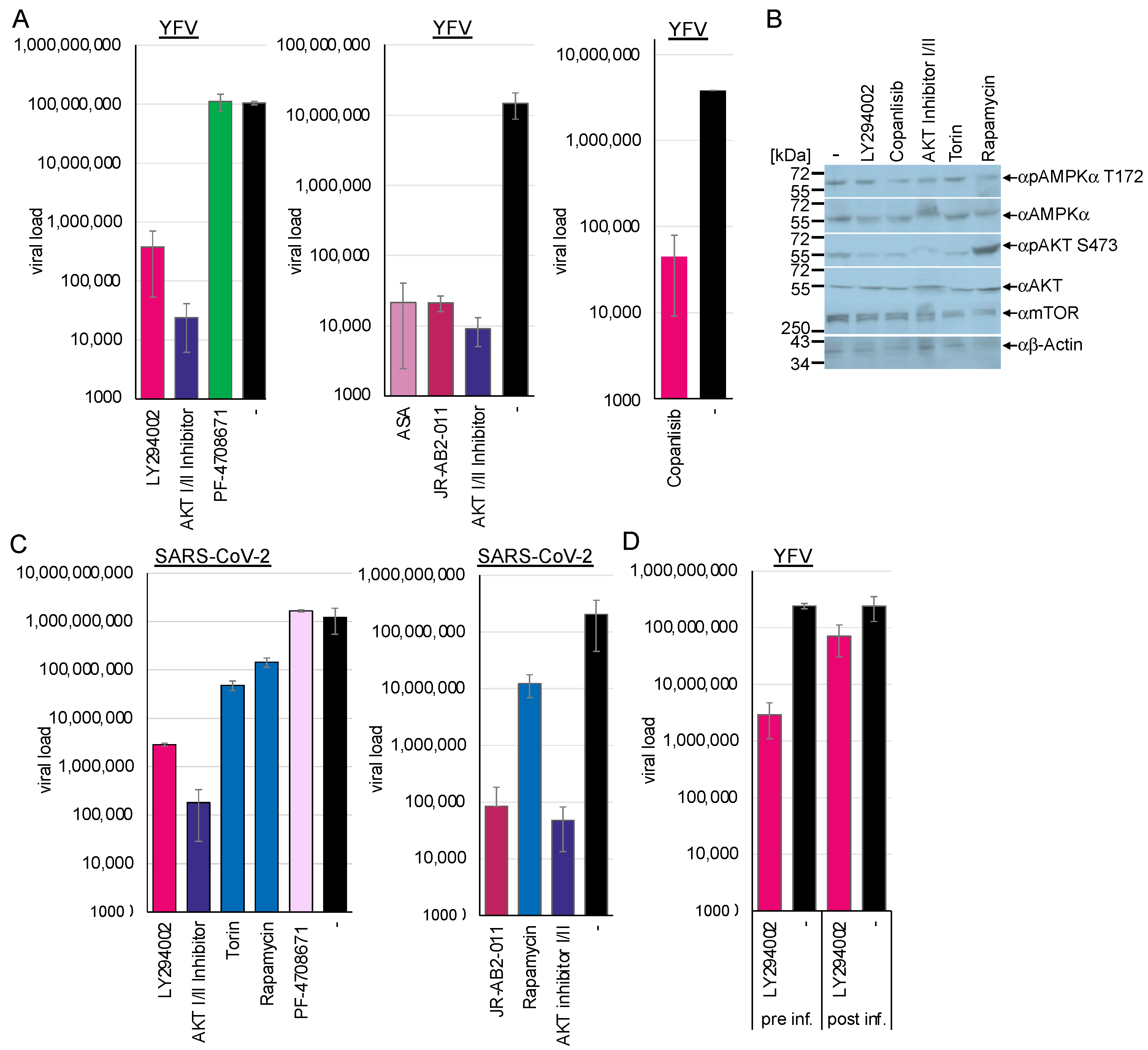
3.9. The PI3K/Akt Kinase Pathway Is Essential for Flavivirus and SARS-CoV-2 Replication
The PI3K/Akt pathway promotes viral endocytosis by activating mTORC1 and silencing AMPK/ULK1. The activation of AMPK antagonises the PI3K/Akt pathway by inhibiting raptor. Thus, we sought to investigate whether the downregulation of the PI3K/Akt pathway influences viral replication by chemically inhibiting several pathway kinases. Huh-7 cells were preincubated with the PI3K inhibitor LY294002 (40 µM) or 300 nM of the novel clinically approved inhibitor PI3K inhibitor copanlisib and were subsequently infected with YFV. Cell culture supernatants were collected 48 h after infection, and viral RNAs were quantified by RTqPCR (
Figure 9A). Western blotting analyses of the treated cells revealed that the PI3K inhibitors reduced Akt serine 473 phosphorylation, while AMPK amounts remained unchanged (
Figure 9B). Inhibition of the PI3K through preincubation with LY294002 reduced viral replication by 2.44 log
10 (
p < 0.0001) and by 1.9 log
10 with copanlisib (
p = 0.0001). This indicated that the regulation of AMPK by the activity of the PI3K/Akt signalling cascade is crucial for viral replication (
Figure 9A). Similarly, treatment of Calu-3 cells with LY294002 inhibited SARS-CoV-2 replication by 2.63 log
10 (
p = 0.002), supporting the importance of PI3K activity (
Figure 9B).
The PI3K is upstream of the Akt kinase, which controls the activation of NF-κB and, in turn, the expression of TXNIP. Thus, we sought to investigate whether the activity of Akt influences SARS-CoV-2 and flavivirus replication. The Akt kinase facilitates the phosphorylation of mTOR. The Akt-I/II inhibitor blocks the activity of Akt kinase I and II and, thus, autophosphorylation. Huh-7 and Calu-3 cells were incubated with 58 µM of Akt-I/II inhibitor and infected with YFV or SARS-CoV-2. Western blotting analyses confirmed reduced Akt phosphorylation (
Figure 9B). Viral replication was determined at 48 h (YFV) or 72 h (SARS-CoV-2) after infection by RT-qPCR (
Figure 9A,C). Treatment with the Akt-I/II inhibitor downregulated viral replication by more than 3 log
10 (YFV: 3.6 log
10,
p < 0.00001; SARS-CoV-2: 3.84 log
10,
p = 0.002). This result indicates that the activity of the Akt kinase is a crucial host factor for viral replication by inhibiting AMPK activation.
If AMPK activation and PI3K/Akt inhibition are two sides of the same coin, the suppression of PI3K or Akt activity should also inhibit viral entry. Thus, we performed an entry assay similar to the experiments above with a PI3K inhibitor. Huh-7 cells were preincubated with LY294002 inhibitor for 2 h and infected or infected and treated with inhibitor 2 h after infection. Viral RNAs were isolated after 48 h and quantified by RTqPCR (
Figure 8D). Inhibition of PI3K before infection resulted in the suppression of flavivirus replication by 1.92 log
10 (
p < 0.0001), indicating that PI3K activity and the PI3K/Akt pathway are essential for flavivirus replication. However, treatment after infection was ineffective (0.5 log
10,
p = 0.009), suggesting that PI3K inhibition suppressed viral entry via increased AMPK activity. The importance of the Akt kinase activity was confirmed in the TBEV and chikungunya entry assays (
Figure 2D and
Figure 3), where preincubation with the AKT I/II inhibitor suppressed viral replication by 3.3 log
10 (TBEV,
p = 0.02) and 4.2 log
10 (chikungunya,
p = 1.3 × 10
−5).
Next, we sought to analyse the downstream effectors of Akt. The activity of the mTOR kinase controls major cellular pathways, such as NF-κB activation [
10], cell metabolism, transcription, protein synthesis, autophagy, and the balance of proteasome activity. Furthermore, mTOR regulates the activity of the lysosomal ATPase, which controls the lysosomal pH [
52]. The mTOR protein exists in two distinct complexes, mTORC1 and mTORC2. The Akt kinase controls the former, while the latter regulates the Akt kinase independently of PI3K activity. First, we investigated the effects of inhibiting mTOR activity with torin-1 and temsirolimus, a novel clinically approved mTOR inhibitor. NCE cells were either preincubated for 2 h or TBEV-infected and treated after 2 h with 10 µM temsirolimus, while Calu-3 cells were treated with 100 nM torin-1 and subsequently infected with SARS-CoV-2. Viral replication was determined using RT-qPCR at 48 h (TBEV) or 72 h (SARS-CoV-2) (
Figure 2D and
Figure 9C) post-infection. Inhibition of both mTORC1 and -2 reduced SARS-CoV-2 replication by 1.4 log
10 (
p = 0.003) (
Figure 8). This demonstrates that mTOR activity is required for viral replication (
Figure 8). Furthermore, the TBEV entry experiments confirmed the inhibition of viral entry (1.4 log
10,
p = 0.026). Experiments with rapamycin were performed to characterise further the mTORC complex, which inhibits SARS-CoV-2. Rapamycin selectively targets mTORC1. Incubation of Calu-3 cells with rapamycin inhibited viral replication (SARS-CoV-2: 0.9 log
10;
p = 0.005), providing evidence that mTORC1 activity is essential for viral replication (
Figure 9C). The connection between mTORC1 and flavivirus, as well as SARS-CoV-2 replication, can be explained by control of AMPK activity and, additionally, in the case of SARS-CoV-2, by the mTOR-mediated acidification of the lysosomes.
Next, we analysed whether mTORC2 is required for SARS-CoV-2 and YFV replication, as mTORC2 activates Akt via serine 473 phosphorylation. Calu-3 and Huh-7 cells were incubated with the specific mTORC2 inhibitor JR-AB2-011 (
Figure 9A,C). The cells were treated with JR-AB2-011 (250 µM) inhibitor and infected with YFV or SARS-CoV-2. Viral genome copies in the supernatants were quantified 48 h and 72 h post-infection by RT-qPCR. JR-AB2-011 suppressed YFV and SARS-CoV-2 replication by approximately 3 log
10 (YFV: 2.84 log
10,
p = 0.0003; SARS-CoV-2: 3.21 log
10,
p = 0.002), indicating that mTORC2 plays a significant role in YFV and SARS-CoV-2 replication, which can lead to Akt activation. Moreover, the activation of AMPK by ASA downregulated the phosphorylation of Akt at serine 473 in either non-infected or SARS-CoV-2-infected Calu-3 cells, mediated by the mTORC2 complex. This provides evidence that AMPK controls not only mTORC1 through phosphorylation of raptor but also mTORC2. This results in the inhibition of viral replication, as we have demonstrated that Akt is essential for SARS-CoV-2 replication.
mTORC1 regulates the translation of specific heat-shock-related mRNAs by controlling the phosphorylation of the ribosomal S6 protein and the translation of 5′TOP-RNAs. Thus, we investigated whether the PI3K signalling cascade inhibits viral replication by downregulating the translation of viral RNA. The S6-kinase inhibitor PF-4708671 specifically suppresses S6 protein phosphorylation. Calu-3 and Huh-7 cells were incubated with 10 µM PF-4708671 and infected with YFV and SARS-CoV-2. Viral RNAs were isolated 48 h and 72 h after infection and quantified by RTqPCR (
Figure 9A,C). PF-4708671 did not influence viral replication, showing that S6 phosphorylation is dispensable for flavivirus or SARS-CoV-2 replication (SARS-CoV-2: −0.13 log
10,
p = 0.17). However, AMPK activation by ASA downregulated S6 phosphorylation in Calu-3 and Huh-7 cells, which is expected as ASA leads to the dephosphorylation of Akt. We have shown that the S6 phosphorylation is not essential for SARS-CoV-2 replication. Thus, the ASA-mediated inhibition of S6 activity should not be responsible for the antiviral effect of ASA.
In summary, we have shown that active PI3K/Akt and mTORC2 pathways and the repression of AMPK/ULK1 are crucial for flavivirus, alphavirus, enterovirus, SARS-CoV-2, and rabies lyssavirus entry and replication. This inhibition of receptor-mediated endocytosis and viral entry was independent of the AMPK activation pathway since ASA/SA, metformin, and the AMPK activator act on different cellular targets. In addition, our results suggest that the regulation of viral endocytosis is ULK1-dependent. Furthermore, the observed downregulation of EEs and LEs supports the reported AMPK-dependent upregulation of Rab4, an endosome recycling factor [
14]. The observed downregulation of LAMP-1 indicates that inhibiting EEs and LEs leads to reduced cellular lysosomes. Our findings suggest that AMPK/ULK1 activation inhibits receptor-mediated endocytosis and changes the number or composition of lysosomes. This central mechanism is a prime target for developing broad-spectrum antivirals. Finally, inhibiting rabies entry might lead to modification of the current postexposure prophylaxis, especially in the absence of antiserum with inexpensive and globally available drugs, such as ASA or metformin.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}