
Mitochondrial Dysfunction in Neurodegenerative Diseases


Abstract
1. Introduction
2. Mitochondrial Functions and Quality Control
2.1. Mitochondrial Functions
2.2. Mitochondrial Quality Control Mechanisms
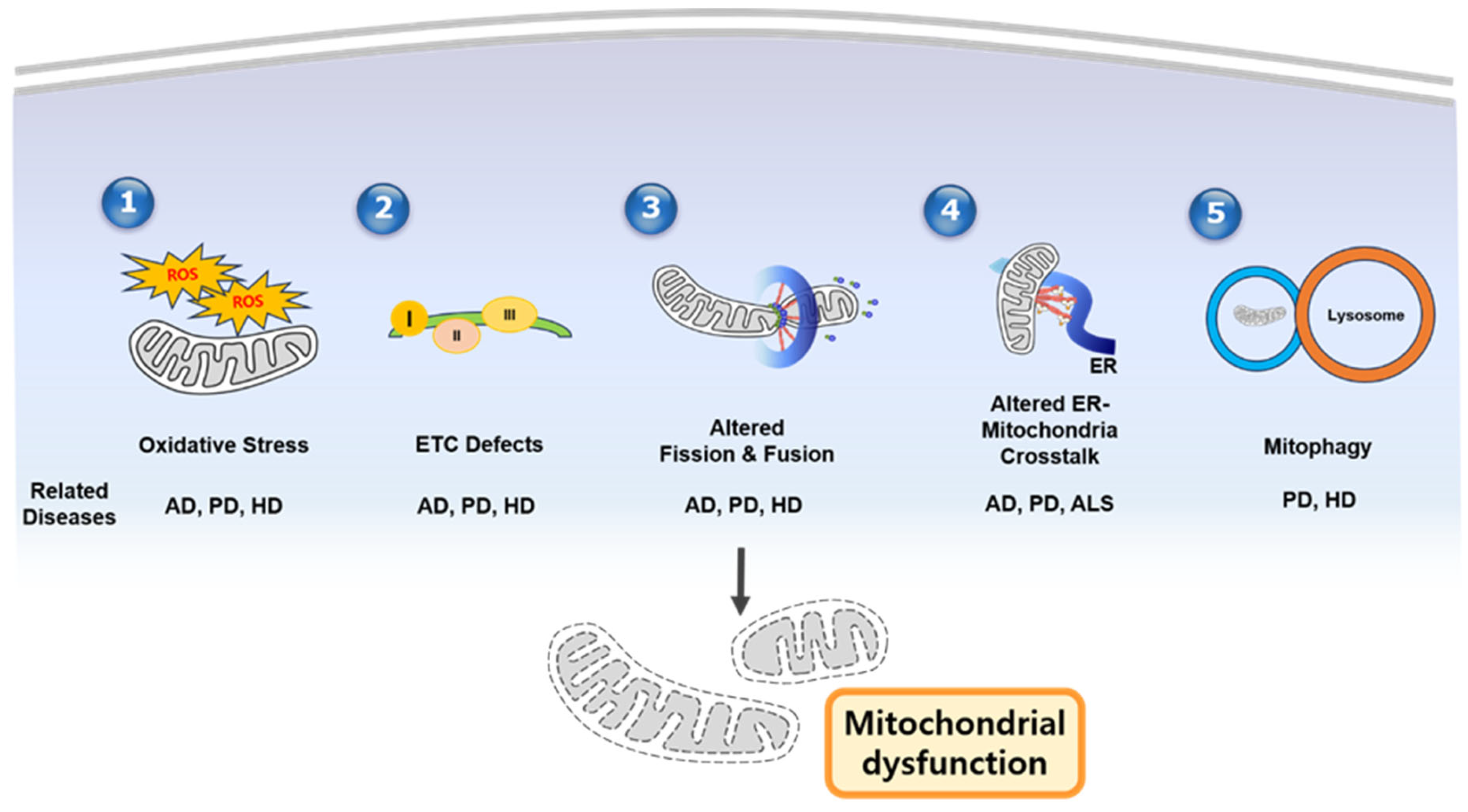
3. Mitochondrial Dysfunction in Neurodegenerative Disease by Mechanism
4. Disease-Specific Mitochondrial Dysfunction
5. Therapeutic Strategies
6. Biomarkers and Future Directions
7. Discussion
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| ALS | amyotrophic lateral sclerosis |
| Aβ | amyloid-β |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| ETC | electron transport chain |
| ROS | reactive oxygen species |
| mtDNA | mitochondrial DNA |
| ER | endoplasmic reticulum |
| MAMs | mitochondria-associated membranes |
| OMM | outer mitochondrial membrane |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane Space |
| OXPHOS | oxidative phosphorylation |
| NADH | nicotinamide adenine dinucleotide |
| FADH2 | flavian adenine dinucleotide |
| ATP | adenosine triphosphate |
| ADP | adenosine diphosphate |
| Mfn1/2 | MItofusin 1/2 |
| OPA1 | optic atrophy 1 |
| Drp1 | dynamin-related protein 1 |
| Fis1 | fission1 |
| Mff | mitochondrial fission factor |
| MiD49 | mitochondrial dynamics protein 49 |
| MCU | mitochondrial calcium uniporter |
| mPTP | mitochondrial permeability transition pore |
| NCLX | Na+/Ca2+ exchanger |
| UPS | Uniquitin-proteasome System |
| MDVs | Mitochondrial-derived Vesicles |
| PINK1 | PTEN-induced Kinase 1 |
| SQSTM1 | Sequestosome 1 |
| NBR1 | neighbor of BRCA1 gene |
| OPTN | optineurin |
| BNIP3 | Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 |
| FUNDC1 | FUN14 domain-containing 1 |
| SOD | Superoxide Dismutase |
| ABAD | amyloid-beta-binding alcohol dehydrogenase |
| TDP-43 | TAR DNA-binding protein-43 |
| DAMPs | damage-associated molecular patterns |
| FUS | fused in sarcoma |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| fAD | familial AD |
| APP | amyloid precursor protein |
| PSEN | presenilin |
| sAD | sporadic AD |
| APOE | apolipoprotein E |
| LRRK2 | leucine-rich repeat kinase 2 |
| SNCA | α-synuclein |
| fALS/sALS | familial/sporadic ALS |
| NAD | nicotinamide adenine dinucleotide |
| SIRT1 | sirtuin 1 |
| NAM | nicotinamide |
| USP30 | ubiquitin-specific Peptidase 30 |
| DCA | dichloroacetate |
| BBB | blood–brain barrier |
| CSF | cerebrospinal fluid |
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| FGF21 | fibroblast growth factor 21 |
| GDF15 | growth differentiation factor 15 |
| PET | Positron Emission Tomography |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| iPSC | induced pluripotent stem cell |
| UPRmt | mitochondrial unfolded protein response |
References
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.U.; Woodling, N.S.; Wang, Q.; Panchal, M.; Liang, X.; Trueba-Saiz, A.; Brown, H.D.; Mhatre, S.D.; Loui, T.; Andreasson, K.I. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Investig. 2015, 125, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.; Mueller, S.; Miller, T.; Miller, C.C. There’s something wrong with my MAM; the ER–mitochondria axis and neurodegeneration. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Lamming, D.W. The mechanistic target of rapamycin: The grand conductor of metabolism and aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef]
- Negida, A.; Elfil, M.; Attia, A.S.; Farahat, R.; El-Nashar, A.; Asaad, S.; Attia, A.S.; El-Remessy, M.; Khafagy, M.E.; Attia, A.S.; et al. Coenzyme Q10 for patients with Parkinson’s disease: A systematic review and meta-analysis of randomized controlled trials. CNS Neurol. Disord. Drug Targets 2023, 22, 919–933. [Google Scholar]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Li, Y.; Wang, S.; Wang, X. Targeting mitochondrial dysfunction and mitophagy for neurodegenerative diseases. Sig. Transduct. Target. Ther. 2024, 9, 124. [Google Scholar]
- Liu, G.; Wang, M.; Lv, X.; Guan, Y.; Li, J.; Xie, J. Identification of mitochondria-related gene biomarkers associated with immune infiltration in acute myocardial infarction. iScience 2024, 27, 110275. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.; Zhu, C.; Farrell, J.J.; Khurshid, Z.; Alzheimer’s Disease Sequencing Project; Alzheimer’s Disease Neuroimaging Initiative; Martin, E.R.; Pericak-Vance, M.A.; Wang, L.S.; Bush, W.S.; et al. Blood-derived mitochondrial DNA copy number is associated with Alzheimer disease, Alzheimer-related biomarkers and serum metabolites. Alzheimer’s Res. Ther. 2024, 16, 234. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Zorov, D.B.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Carteri, R.B. Mitochondria as a Therapeutic Target: Focusing on Traumatic Brain Injury. J. Integr. Neurosci. 2025, 24, 25292. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Complex IV-The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.; Arroum, T.; Wan, J.; Pavelich, L.; Bell, J.; Morse, P.T.; Lee, I.; Grossman, L.I.; Sanderson, T.H.; Malek, M.H.; et al. Regulation of mitochondrial oxidative phosphorylation through tight control of cytochrome c oxidase in health and disease—Implications for ischemia/reperfusion injury, inflammatory diseases, diabetes, and cancer. Redox Biol. 2024, 78, 103426. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tethering protein. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Tabara, L.C.; Segawa, M.; Prudent, J. Molecular mechanisms of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Shutt, T.E. The role of impaired mitochondrial dynamics in MFN2-mediated pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef]
- Kumar, A.; Larrea, D.; Pero, M.E.; Infante, P.; Conenna, M.; Shin, G.J.; Van Elias, V.; Grueber, W.B.; Di Marcotullio, L.; Area-Gomez, E.; et al. MFN2 coordinates mitochondria motility with alpha-tubulin acetylation and this regulation is disrupted in CMT2A. iScience 2024, 27, 109994. [Google Scholar] [CrossRef] [PubMed]
- Zanfardino, P.; Longo, G.; Amati, A.; Morani, F.; Picardi, E.; Girolamo, F.; Pafundi, M.; Cox, S.N.; Manzari, C.; Tullo, A.; et al. Mitofusin 2 mutation drives cell proliferation in Charcot–Marie–Tooth 2A fibroblasts. Hum. Mol. Genet. 2023, 32, 333–350. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, L.; Conde de la Rosa, L.; Torres, S.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial cholesterol: Metabolism and impact on redox biology and disease. Redox Biol. 2023, 61, 102643. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Gray, L.R.; Sultana, M.R.; Rauckhorst, A.J.; Oonthonpan, L.; Tompkins, S.C.; Sharma, A.; Fu, X.; Miao, R.; Pewa, A.D.; Brown, K.S.; et al. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015, 22, 669–681. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The Long History of Iron in the Universe and in Health and Disease. Biochim. Biophys. Acta 2012, 1820, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Perfetto, M. Regulation of heme synthesis by mitochondrial homeostasis proteins. Front. Cell Dev. Biol. 2022, 10, 895521. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007559. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bucci, C.; Marzetti, E. Mitochondrial-derived vesicles: The good, the bad, and the ugly. Int. J. Mol. Sci. 2023, 24, 13835. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Towers, C. Beyond mitophagy: Mitochondrial-derived vesicles can get the job done! Autophagy 2022, 18, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Wodetzki, D.K.; Thorburn, J.; Smith, K.R.; Caino, M.C.; Thorburn, A. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 2021, 56, 2029–2042.e5. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Sig. Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Sabouny, R.; Shutt, T.E. Reciprocal Regulation of Mitochondrial Fission and Fusion. Trends Biochem. Sci. 2020, 45, 564–577. [Google Scholar] [CrossRef]
- Narendra, D.P.; Youle, R.J. Targeting mitochondrial dysfunction: Role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Stern, D.; Gunn Moore, F.J.; Chen, J.X.; Yan, S.S. Inhibition of amyloid-β (Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Zhu, L.; Jia, Q.; Duan, X.; Chen, L.; Wang, X.; Hou, J.; Jiang, G.; Yang, S.; Li, S.; et al. Loss of TDP-43 promotes somatic CAG repeat expansion in Huntington’s disease knock-in mice. Prog. Neurobiol. 2023, 227, 102484. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Cooper, J.M. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s disease patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Beal, M.F. The energetics of Huntington’s disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Zhu, J.; Chu, C.T. Mitochondrial dysfunction in Parkinson’s disease. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S325–S334. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martínez-Guardado, I.; Dalamitros, A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase Drp1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimer’s Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Tsekrekou, M.; Giannakou, M.; Papanikolopoulou, K.; Skretas, G. Protein aggregation and therapeutic strategies in SOD1- and TDP-43- linked ALS. Front. Mol. Biosci. 2024, 11, 1383453. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Demarest, T.G.; Aman, Y.; Figueroa, D.; Morevati, M.; et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef]
- Qin, P.; Sun, Y.; Li, L. Mitochondrial dysfunction in chronic neuroinflammatory diseases. Int. J. Mol. Med. 2024, 53, 68. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Calì, T.; Brini, M. Mitochondria Dysfunction and Neuroinflammation in Neurodegeneration: Who Comes First? Antioxidants 2024, 13, 240. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Sig. Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell. Neurosci. 2013, 55, 26–36. [Google Scholar] [CrossRef]
- Lee, K.S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.K.; Kang, H.Y.; Lu, B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1660. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Azman Ali, R.; Ibrahim, N.F. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tam, B.T.; Sesha-Sai, P.; Diaz-Ramirez, L.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32732. [Google Scholar] [CrossRef] [PubMed]
- Pinho, C.M.; Teixeira, P.F.; Glaser, E. Mitochondrial import and degradation of amyloid-beta peptide. Biochim. Biophys. Acta 2014, 1837, 1069–1074. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jayadev, S.; Lardelli, M.; Newman, M. A Review of the Familial Alzheimer’s Disease Locus PRESENILIN 2 and Its Relationship to PRESENILIN 1. J. Alzheimer’s Dis. 2018, 66, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Xu, H.; Chen, S.; Wang, X.; Yang, J.; Gong, M.; Wei, C.; Tang, Y.; Qu, Q.; Chu, L.; et al. The APOE ε4 exerts differential effects on familial and other subtypes of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1613–1625. [Google Scholar] [CrossRef]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, J.E.; Mahley, R.W.; Huang, Y. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 2018, 360, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Estrada Sánchez, A.M.; Mejía-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Fisher, E.M.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S.I. Regulation of Mitochondrial Transport and Inter-Microtubule Spacing by Tau Phosphorylation at the Sites Hyperphosphorylated in Alzheimer’s Disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Hu, Y.; Wang, Z.-H.; Luo, Y.; Zhang, Y.; Liu, X.-P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.-P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, J.; Götz, M.B.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Pérez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease-repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Sig. Transduct. Target. Ther. 2024, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zheng, Y.; Guo, M.; Ares, I.; Martínez, M.; López-Torres, B.; Martínez-Larrañaga, M.R.; Wang, X.; Anadón, A.; Martínez, M.A. Oxidative stress, the blood-brain barrier and neurodegenerative diseases: The critical beneficial role of dietary antioxidants. Acta Pharm. Sin. B 2023, 13, 3988–4021. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Whitley, B.N.; Engelhart, E.A.; Hoppins, S. Mitochondrial dynamics and their potential as a therapeutic target. Mitochondrion 2019, 49, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. PINK1- and Parkin-mediated local mitophagy in distal neuronal axons. Autophagy 2015, 11, 187–189. [Google Scholar]
- Ghasemi, M.; Brown, R.H. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef]
- Son, S.M.; Jung, E.S.; Shin, H.J.; Byun, J.; Mook-Jung, I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol. Aging 2012, 33, 1006.e11–1006.e23. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Han, R.; He, H.J.; Wang, Z.; Luan, X.Q.; Li, J.; Feng, L.; Chen, S.Y.; Aman, Y.; Xie, C.L. Delineating the Role of Mitophagy Inducers for Alzheimer Disease Patients. Aging Dis. 2021, 12, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Antico, O.; Thompson, P.W.; Hertz, N.T.; Muqit, M.M.; Parton, L.E. Targeting mitophagy in neurodegenerative diseases. Nat. Rev. Drug Discov. 2025; in press. [Google Scholar] [CrossRef] [PubMed]
- Yung, C.; Sha, D.; Li, L.; Chin, L.S. Parkin Protects Against Misfolded SOD1 Toxicity by Promoting Its Aggresome Formation and Autophagic Clearance. Mol. Neurobiol. 2016, 53, 6270–6287. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Wilkin, G.P.; Tarnopolsky, M.A.; Gibala, M.J. A practical model of low-volume high-intensity interval training induces mitochondrial biogenesis in human skeletal muscle: Potential mechanisms. J. Physiol. 2010, 588, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef]
- Andres, R.H.; Ducray, A.D.; Schlattner, U.; Wallimann, T.; Widmer, H.R. Functions and effects of creatine in the central nervous system. Brain Res. Bull. 2008, 76, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.-Y.; DuMouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med. 2001, 344, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Studer, L. Strategies for bringing stem cell–derived dopamine neurons to the clinic—The NYSTEM trial. Prog. Brain Res. 2017, 230, 191–212. [Google Scholar] [PubMed]
- Boskovic, P.; Gao, W.; Kipnis, J. Will cellular immunotherapies end neurodegenerative diseases? Trends Immunol. 2024, 45, 329–337. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Negida, A.; Menshawy, A.; El Ashal, G.; Elfouly, Y.; Hani, Y.; Hegazy, Y.; El Ghonimy, S.; Fouda, S.; Rashad, Y. Coenzyme Q10 for Patients with Parkinson’s Disease: A Systematic Review and Meta-Analysis. CNS Neurol. Disord. Drug Targets 2016, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Therapeutic effects of coenzyme Q10 in neurodegenerative diseases. Methods Enzymol. 2004, 382, 473–487. [Google Scholar]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Coenzyme Q10 and Parkinsonian Syndromes: A Systematic Review. J. Pers. Med. 2022, 12, 975. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef]
- Thal, L.J.; Grundman, M.; Berg, J.; Ernstrom, K.; Margolin, R.; Pfeiffer, E.; Weiner, M.F.; Zamrini, E.; Thomas, R.G. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 2003, 61, 1498–1502. [Google Scholar] [CrossRef]
- Rustin, P.; von Kleist-Retzow, J.C.; Chantrel-Groussard, K.; Sidi, D.; Munnich, A.; Rötig, A. Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: A preliminary study. Lancet 1999, 354, 477–479. [Google Scholar] [CrossRef]
- Buyse, G.; Goemans, N.; van den Hauwe, M.; Meier, T. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12-month, double-blind, randomized-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Jhung, S.; Sano, M.C.; Shungu, D.C.; Millar, W.S.; Hong, X.; Gooch, C.L.; Mao, X.; et al. Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurology 2006, 66, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Barshop, B.A.; Naviaux, R.K.; McGowan, K.A.; Levine, F.; Nyhan, W.L.; Loupis-Geller, A.; Haas, R.H. Chronic treatment of mitochondrial disease patients with dichloroacetate. Mol. Genet. Metab. 2004, 83, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Conte, C.; Fontana, L.; Mittendorfer, B.; Imai, S.; Schechtman, K.B.; Gu, C.; Kunz, I.; Rossi Fanelli, F.; Patterson, B.W.; et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012, 16, 658–664. [Google Scholar] [CrossRef]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Shayota, B.J. Biomarkers of mitochondrial disorders. Neurotherapeutics 2024, 21, e00325. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, 216.e7–216.e10. [Google Scholar] [CrossRef]
- Kurbatova, N.; Garg, M.; Whiley, L.; Chekmeneva, E.; Jiménez, B.; Gómez-Romero, M.; Pearce, J.; Kimhofer, T.; D’hondt, E.; Soininen, H.; et al. Urinary metabolic phenotyping for Alzheimer’s disease. Sci. Rep. 2020, 10, 21745. [Google Scholar] [CrossRef]
- Seol, W.; Kim, H.; Son, I. Urinary Biomarkers for Neurodegenerative Diseases. Exp. Neurobiol. 2020, 29, 325–333. [Google Scholar] [CrossRef]
- Cheslow, L.; Snook, A.E.; Waldman, S.A. Biomarkers for Managing Neurodegenerative Diseases. Biomolecules. 2024, 14, 398. [Google Scholar] [CrossRef]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10 (Suppl. 7), S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Liun, F.; Rokach, J.; Lee, V.Y.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Perluigi, M.; Sultana, R. Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. Eur. J. Pharmacol. 2006, 545, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Taliyan, R.; Chandran, S.K.; Kakoty, V. Therapeutic Approaches to Alzheimer’s Type of Dementia: A Focus on FGF21 Mediated Neuroprotection. Curr. Pharm. Des. 2019, 25, 2555–2568. [Google Scholar] [CrossRef]
- Xue, X.-H.; Tao, L.-L.; Su, D.-Q.; Guo, C.-J.; Liu, H. Diagnostic utility of GDF15 in neurodegenerative diseases: A systematic review and meta-analysis. Brain Behav. 2022, 12, e2502. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.C.; Boada, J.; Rodriguez-Mañas, L.; Barbas, C.; Valero-Paniagua, V.; Martinez-Castelao, A.; Miranda, C.; Formiga, F.; Camafort, M.; The Octabaix Study Group. Association between functional and cognitive impairment and GDF15 in nonagenarians. GeroScience 2021, 43, 985–1001. [Google Scholar]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2018, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Arcuti, S.; Copetti, M.; Alessandria, M.; Savica, R.; Fontana, A.; Liguori, R.; Logroscino, G. Accuracy of clinical diagnosis of dementia with Lewy bodies: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2018, 89, 358–366. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Awuah, W.A.; Ahluwalia, A.; Ghosh, S.; Roy, S.; Tan, J.K.; Adebusoye, F.T.; Ferreira, T.; Bharadwaj, H.R.; Shet, V.; Kundu, M.; et al. The molecular landscape of neurological disorders: Insights from single-cell RNA sequencing in neurology and neurosurgery. Eur. J. Med. Res. 2023, 28, 529. [Google Scholar] [CrossRef]
- Jackson, M.; Marks, L.; May, G.H.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–157. [Google Scholar]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Benek, O.; Korábečný, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]

{kind=link}
| Disease | Central Mitochondrial Defects | Familial (Genetic) Mutations | Sporadic Factors | References |
|---|---|---|---|---|
| AD | Decreased complex IV activity, Aβ-induced ROS elevation, tau-driven transport failures, reduced PGC-1α expression, excessive fission | APP, PSEN1, PSEN2 | APOE ε4, aging, inflammation | [1,9,10,12,81,97,98,99,100,101,102,103,104,105,106] |
| PD | Complex I inhibition, α-synuclein aggregates on mitochondrial membranes, impaired mitophagy (PINK1/Parkin), dopaminergic oxidative stress, possible Drp1/Mfn2 dysregulation | PINK1, Parkin, DJ-1, LRRK2 | Environmental toxins (MPTP, rotenone), α-syn pathology | [2,7,8,14,72,107,108,109,110,111,112,113,114,115] |
| HD | Mutant huntingtin interfering with ETC (II–III), excessive Drp1-driven fission, calcium dysregulation, suppressed mitochondrial biogenesis via PGC-1α | HTT (CAG repeat) | [4,13,73,80,116,117,118] | |
| ALS | Aggregated SOD1 or TDP-43 in mitochondrial compartments, decreased ETC function, severely compromised axonal transport to NMJs, glial-driven inflammatory loops | SOD1, TDP-43, C9orf72 | [5,6,119,120,121,122,123] |
| Strategy Type | Examples/Compounds | Mechanism of Action | Potential Applications and Challenges | References |
|---|---|---|---|---|
| Antioxidants and ETC Stabilizers | Coenzyme Q10, MitoQ, NAD+ precursors | Scavenge ROS; enhance electron flow | Shown partial neuroprotection preclinically; BBB penetration and timing hamper clinical success | [8,16,26,135,136,137,138] |
| Mitochondrial Dynamics Modulators | mdivi-1 (Drp1 inhibitor), Mfn2 upregulators | Correct excessive fission; support fusion | Balancing is crucial; excessive fusion may trap damaged mitochondria | [53,79,80,139,140] |
| Mitophagy Enhancers | Parkin overexpression, resveratrol, NADH, NAM, USP30 inhibitors, PINK1 activators | Selective clearance of depolarized mitochondria | Strong rationale in PD with PINK1/Parkin loss; specificity needed to avoid unintended autophagic overload | [14,62,82,141,142,143,144,145] |
| Gene Therapy | ASOs for mutant huntingtin/SOD1; Lentiviral vectors for Parkin | Silence toxic genes or supplement protective ones | Delivery, stable expression, and immunogenicity remain barriers; especially relevant for monogenic forms | [5,15,146] |
| Lifestyle and Nutritional Interventions | Exercise, ketogenic diet, creatine | Bolster endogenous mitochondrial biogenesis, reduce ROS | Low risk but varied efficacy; synergy with pharmacological agents may yield best outcomes | [147,148,149] |
| Cell and Mitochondrial Transplantation | Stem cell–derived neurons, direct mito transfer | Replace dysfunctional neurons or supply healthy mitochondria | Limited by immune rejection, graft survival, and targeted delivery in chronic neurodegeneration | [150,151,152] |
| Biomarker | Biofluid(s) | Relevance to Neurodegenerative Diseases | Sensitivity/Specificity (If Known) | References |
|---|---|---|---|---|
| mtDNA levels | Blood, CSF, urine | Increased levels may indicate mitochondrial damage | Moderate sensitivity, limited specificity | [167,168,169,170,171] |
| mtDNA mutations | Blood, CSF, urine | Specific mutations may be associated with certain diseases | Variable, depending on the mutation | [167,168,169,170,171] |
| Cytochrome c | Blood, CSF | Marker of mitochondrial damage and apoptosis | Moderate sensitivity | [167,173] |
| F2-isoprostanes | Blood, CSF, urine | Marker of lipid peroxidation | Moderate sensitivity, limited specificity | [167,174,175] |
| Protein carbonyls | Blood, CSF, urine | Marker of protein oxidation | Moderate sensitivity, limited specificity | [137,174,175] |
| 8-OHdG | Blood, CSF, urine | Marker of DNA oxidation | Moderate sensitivity, limited specificity | [167,176,177,178] |
| FGF21 | Blood | Secreted under mitochondrial stress | May track disease progression; requires validation in larger cohort | [179,180] |
| GDF15 | Blood | Upregulated by oxidative and mitochondrial stress | Good sensitivity but moderate specificity; more clinical data needed | [179,181] |
| Urinary metabolites | Urine | Reflect metabolic alterations in neurodegeneration | Under investigation, potential for non-invasive monitoring | [169,170,171] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-M. Mitochondrial Dysfunction in Neurodegenerative Diseases. Cells 2025, 14, 276. https://doi.org/10.3390/cells14040276
Yang H-M. Mitochondrial Dysfunction in Neurodegenerative Diseases. Cells. 2025; 14(4):276. https://doi.org/10.3390/cells14040276
Chicago/Turabian StyleYang, Han-Mo. 2025. "Mitochondrial Dysfunction in Neurodegenerative Diseases" Cells 14, no. 4: 276. https://doi.org/10.3390/cells14040276
APA StyleYang, H.-M. (2025). Mitochondrial Dysfunction in Neurodegenerative Diseases. Cells, 14(4), 276. https://doi.org/10.3390/cells14040276