

Peripheral Immune Profiles in Individuals at Genetic Risk of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
1. Introduction
2. Methods
2.1. Study Participants
2.2. Generation and Processing of Immune Cell and Genome-Wide SNP Data
2.3. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lee, C.; Kodama, L.; Gan, L. Sex Differences in Neurodegeneration: The Role of the Immune System in Humans. Biol. Psychiatry 2022, 91, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lee, C.; Torres, E.R.S.; Carling, G.; Gan, L. Mechanisms of sex differences in Alzheimer’s disease. Neuron 2024, 112, 1208–1221. [Google Scholar] [CrossRef]
- Weiner, H.L. Immune mechanisms and shared immune targets in neurodegenerative diseases. Nat. Rev. Neurol. 2025, 21, 67–85. [Google Scholar] [CrossRef]
- Deecke, L.; Homann, J.; Goldeck, D.; Ohlei, O.; Dobricic, V.; Drewelies, J.; Demuth, I.; Pawelec, G.; Bertram, L.; Lill, C.M.; et al. No increase of CD8+ TEMRA cells in the blood of healthy adults at high genetic risk of Alzheimer’s disease. Alzheimer’s Dement. 2024, 20, 3116–3118. [Google Scholar] [CrossRef]
- Gericke, C.; Kirabali, T.; Flury, R.; Mallone, A.; Rickenbach, C.; Kulic, L.; Tosevski, V.; Hock, C.; Nitsch, R.M.; Treyer, V.; et al. Early β--amyloid accumulation in the brain is associated with peripheral T cell alterations. Alzheimer’s Dement. 2023, 19, 5642–5662. [Google Scholar] [CrossRef]
- Bertram, L.; Böckenhoff, A.; Demuth, I.; Düzel, S.; Eckardt, R.; Li, S.C.; Lindenberger, U.; Pawelec, G.; Siedler, T.; Wagner, G.G.; et al. Cohort Profile: The Berlin Aging Study II (BASE-II). Int. J. Epidemiol. 2014, 43, 703–712. [Google Scholar] [CrossRef]
- Sbierski-Kind, J.; Goldeck, D.; Buchmann, N.; Spranger, J.; Volk, H.-D.; Steinhagen-Thiessen, E.; Pawelec, G.; Demuth, I. T cell phenotypes associated with insulin resistance: Results from the Berlin Aging Study II. Immun. Ageing 2020, 17, 1–11. [Google Scholar]
- Deecke, L.; Goldeck, D.; Ohlei, O.; Homann, J.; Demuth, I.; Bertram, L.; Pawelec, G.; Lill, C.M. Immune cell distributions in the blood of healthy individuals at high genetic risk of Parkinson’s disease. Int. J. Mol. Sci. 2024, 25, 13655. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Zippelius, A.; Kurth, I.; Pittet, M.J.; Touvrey, C.; Iancu, E.M.; Corthesy, P.; Devevre, E.; Speiser, D.E.; Rufer, N. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J. Immunol. 2007, 178, 4112–4119. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Larbi, A.; Derhovanessian, E.; Özcelik, D.; Naumova, E.; Pawelec, G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun. Ageing 2008, 5, 6. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Derhovanessian, E.; Steinhagen-Thiessen, E.; Goldeck, D.; Müller, L.; Pawelec, G. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: Results from the Berlin BASE-II Study. Biogerontology 2015, 16, 631–643. [Google Scholar] [CrossRef]
- Manenti, S.; Orrico, M.; Masciocchi, S.; Mandelli, A.; Finardi, A.; Furlan, R. PD-1/PD-L Axis in Neuroinflammation: New Insights. Front. Neurol. 2022, 13, 877936. [Google Scholar] [CrossRef]
- Deecke, L.; Homann, J.; Goldeck, D.; Luessi, F.; Vandebergh, M.; Ohlei, O.; Toepfer, S.; Zipp, F.; Demuth, I.; Morgan, S.L.; et al. Genome-wide association study reveals different T cell distributions in peripheral blood of healthy individuals at high genetic risk of type 1 diabetes and long COVID. medRxiv, 2024; preprint, under review. [Google Scholar]
- van Rheenen, W.; Van Der Spek, R.A.; Bakker, M.K.; Van Vugt, J.J.; Hop, P.J.; Zwamborn, R.A.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Campisi, L.; Chizari, S.; Ho, J.S.; Gromova, A.; Arnold, F.J.; Mosca, L.; Mei, X.; Fstkchyan, Y.; Torre, D.; Beharry, C.; et al. Clonally expanded CD8 T cells characterize amyotrophic lateral sclerosis-4. Nature 2022, 606, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Bestagno, M.; Burrone, O.; Petrini, M. CD57+ T lymphocytes and functional immune deficiency. J. Leukoc. Biol. 2010, 87, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Maes, M.; Puri, B.K. Could Alzheimer’s Disease Originate in the Periphery and If So How So? Mol. Neurobiol. 2019, 56, 406–434. [Google Scholar] [CrossRef]
- Moretta, L.; Montaldo, E.; Vacca, P.; Del Zotto, G.; Moretta, F.; Merli, P.; Locatelli, F.; Mingar, M.C. Human natural killer cells: Origin, receptors, function, and clinical applications. Int. Arch. Allergy Immunol. 2014, 164, 253–264. [Google Scholar] [CrossRef]
- Jadidi--Niaragh, F.; Shegarfi, H.; Naddafi, F.; Mirshafiey, A. The Role of Natural Killer Cells in Alzheimer’s Disease. Scand. J. Immunol. 2012, 76, 451–456. [Google Scholar] [CrossRef]
- Altmann, A.; Scelsi, M.A.; Shoai, M.; de Silva, E.; Aksman, L.M.; Cash, D.M.; Hardy, J.; Schott, J.M.; Alzheimer’s Disease Neuroimaging Initiative. A comprehensive analysis of methods for assessing polygenic burden on Alzheimer’s disease pathology and risk beyond APOE. Brain Commun. 2020, 2, fcz047. [Google Scholar] [CrossRef]
- Fortea, J.; Pegueroles, J.; Alcolea, D.; Belbin, O.; Dols-Icardo, O.; Vaqué-Alcázar, L.; Videla, L.; Gispert, J.D.; Suárez-Calvet, M.; Johnson, S.C.; et al. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 2024, 30, 1284–1291. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
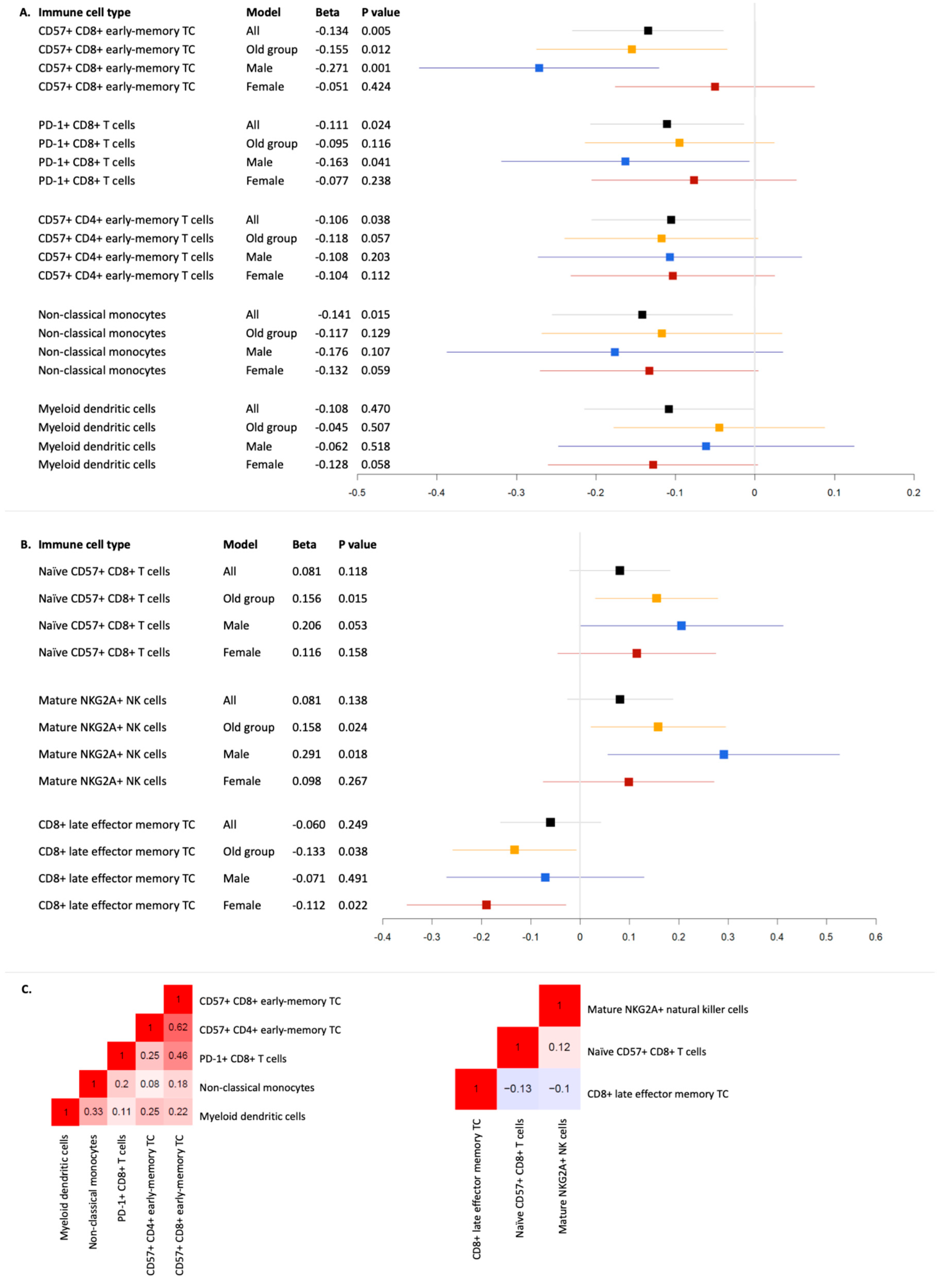
| Immune Cell Type | Model | Beta | ∆R2 | p | |
|---|---|---|---|---|---|
| ALS | CD57+ CD8+ early-memory T cells | All | −0.135 | 0.018 | 0.006 |
| CD57+ CD8+ early-memory T cells | Old group | −0.155 | 0.024 | 0.012 | |
| CD57+ CD8+ early-memory T cells | Female | −0.051 | 0.003 | 0.424 | |
| CD57+ CD8+ early-memory T cells | Male | −0.271 | 0.072 | 0.001 | |
| PD-1+ CD8+ T cells | All | −0.111 | 0.012 | 0.024 | |
| PD-1+ CD8+ T cells | Old group | −0.095 | 0.009 | 0.116 | |
| PD-1+ CD8+ T cells | Female | −0.077 | 0.006 | 0.238 | |
| PD-1+ CD8+ T cells | Male | −0.163 | 0.026 | 0.041 | |
| Non-classical monocytes | All | −0.141 | 0.021 | 0.015 | |
| Non-classical monocytes | Old group | −0.117 | 0.015 | 0.129 | |
| Non-classical monocytes | Female | −0.133 | 0.018 | 0.059 | |
| Non-classical monocytes | Male | −0.176 | 0.031 | 0.107 | |
| CD57+ CD4+ early-memory T cells | All | −0.106 | 0.011 | 0.038 | |
| CD57+ CD4+ early-memory T cells | Old group | −0.118 | 0.014 | 0.057 | |
| CD57+ CD4+ early-memory T cells | Female | −0.104 | 0.011 | 0.112 | |
| CD57+ CD4+ early-memory T cells | Male | −0.108 | 0.011 | 0.203 | |
| Myeloid dendritic cells | All | −0.108 | 0.012 | 0.047 | |
| Myeloid dendritic cells | Old group | −0.045 | 0.002 | 0.507 | |
| Myeloid dendritic cells | Female | −0.128 | 0.017 | 0.058 | |
| Myeloid dendritic cells | Male | −0.062 | 0.004 | 0.518 | |
| AD | Naïve CD57+ CD8+ T cells | All | 0.081 | 0.007 | 0.118 |
| Naïve CD57+ CD8+ T cells | Old group | 0.156 | 0.024 | 0.015 | |
| Naïve CD57+ CD8+ T cells | Female | 0.116 | 0.013 | 0.158 | |
| Naïve CD57+ CD8+ T cells | Male | 0.206 | 0.039 | 0.053 | |
| Mature NKG2A+ natural killer cells | All | 0.081 | 0.007 | 0.138 | |
| Mature NKG2A+ natural killer cells | Old group | 0.158 | 0.024 | 0.024 | |
| Mature NKG2A+ natural killer cells | Female | 0.098 | 0.009 | 0.267 | |
| Mature NKG2A+ natural killer cells | Male | 0.291 | 0.079 | 0.018 | |
| CD8+ late effector memory T cells | All | −0.060 | 0.004 | 0.249 | |
| CD8+ late effector memory T cells | Old group | −0.133 | 0.017 | 0.038 | |
| CD8+ late effector memory T cells | Female | −0.112 | 0.013 | 0.022 | |
| CD8+ late effector memory T cells | Male | −0.071 | 0.005 | 0.491 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deecke, L.; Ohlei, O.; Goldeck, D.; Homann, J.; Toepfer, S.; Demuth, I.; Bertram, L.; Pawelec, G.; Lill, C.M. Peripheral Immune Profiles in Individuals at Genetic Risk of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease. Cells 2025, 14, 250. https://doi.org/10.3390/cells14040250
Deecke L, Ohlei O, Goldeck D, Homann J, Toepfer S, Demuth I, Bertram L, Pawelec G, Lill CM. Peripheral Immune Profiles in Individuals at Genetic Risk of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease. Cells. 2025; 14(4):250. https://doi.org/10.3390/cells14040250
Chicago/Turabian StyleDeecke, Laura, Olena Ohlei, David Goldeck, Jan Homann, Sarah Toepfer, Ilja Demuth, Lars Bertram, Graham Pawelec, and Christina M. Lill. 2025. "Peripheral Immune Profiles in Individuals at Genetic Risk of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease" Cells 14, no. 4: 250. https://doi.org/10.3390/cells14040250
APA StyleDeecke, L., Ohlei, O., Goldeck, D., Homann, J., Toepfer, S., Demuth, I., Bertram, L., Pawelec, G., & Lill, C. M. (2025). Peripheral Immune Profiles in Individuals at Genetic Risk of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease. Cells, 14(4), 250. https://doi.org/10.3390/cells14040250