Single-Cell RNA Sequencing on Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Identified Multi-Ciliary Cells in Breast Cancer

 , , , , ,
, , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.2. Pathological and Molecular Characterization of Breast Carcinomas
2.3. scRNAseq
2.4. Electron Microscopy
2.5. Statistical Analysis
3. Results
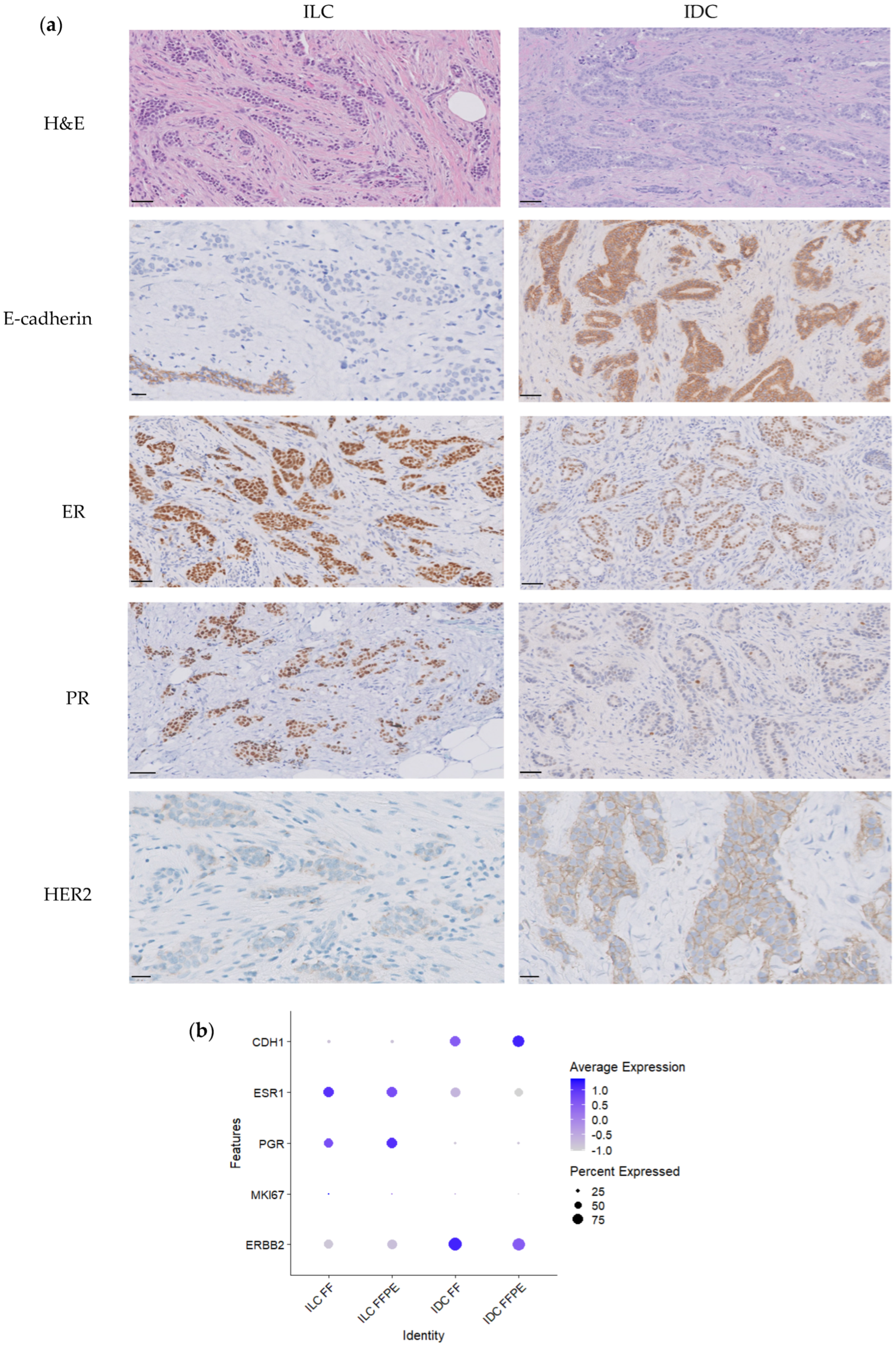
3.1. Clinicopathological and Molecular Features of Tumor Samples
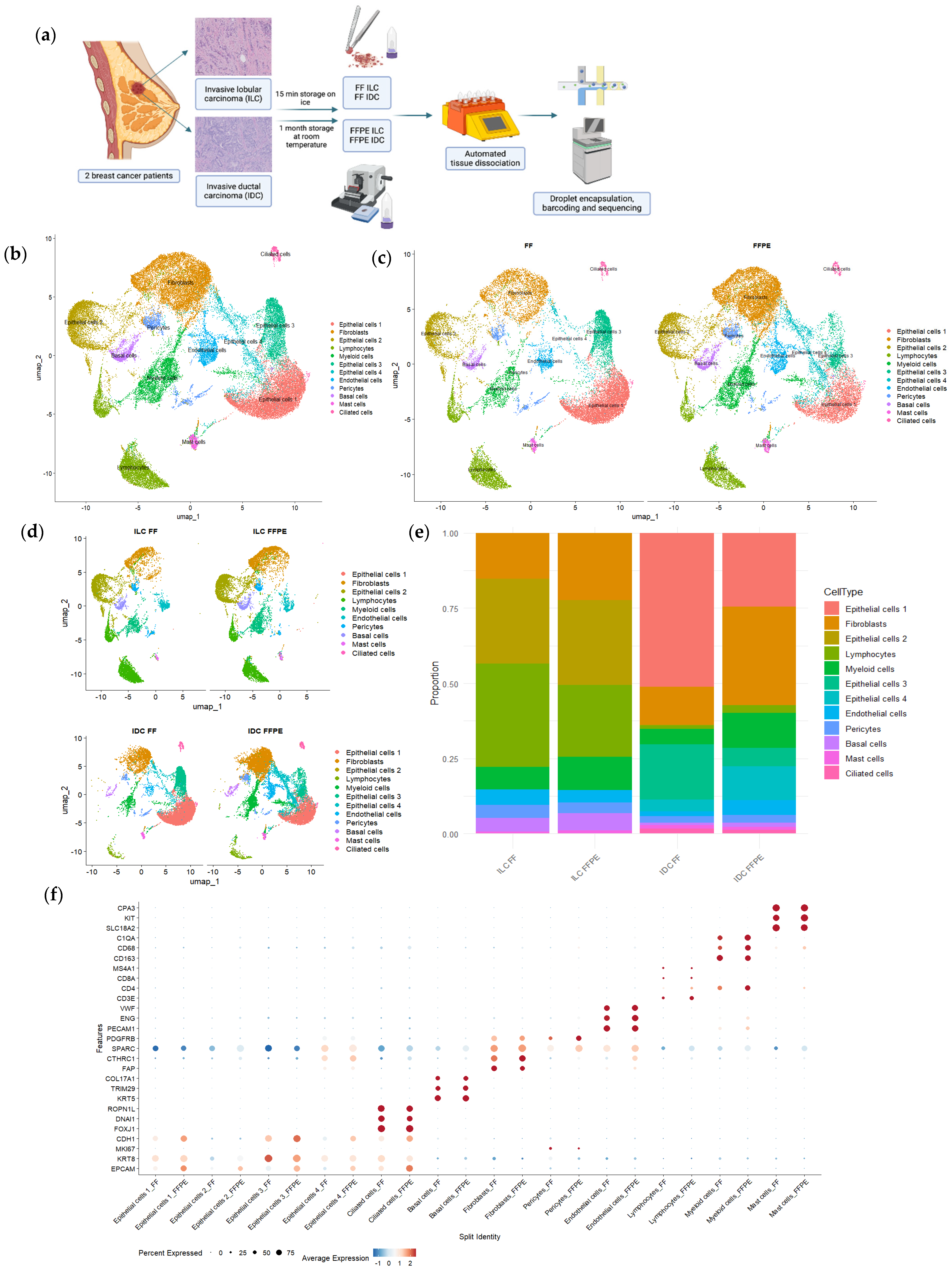
3.2. Assessment of Single-Cell Transcriptome Quality in Fixed Fresh and FFPE Tissue Samples
3.3. Cell Heterogeneity and Gene Expression in Fixed Fresh and FFPE Samples
3.4. scRNAseq on FFPE Captures Immunohistochemical and Immune Features of Tumors
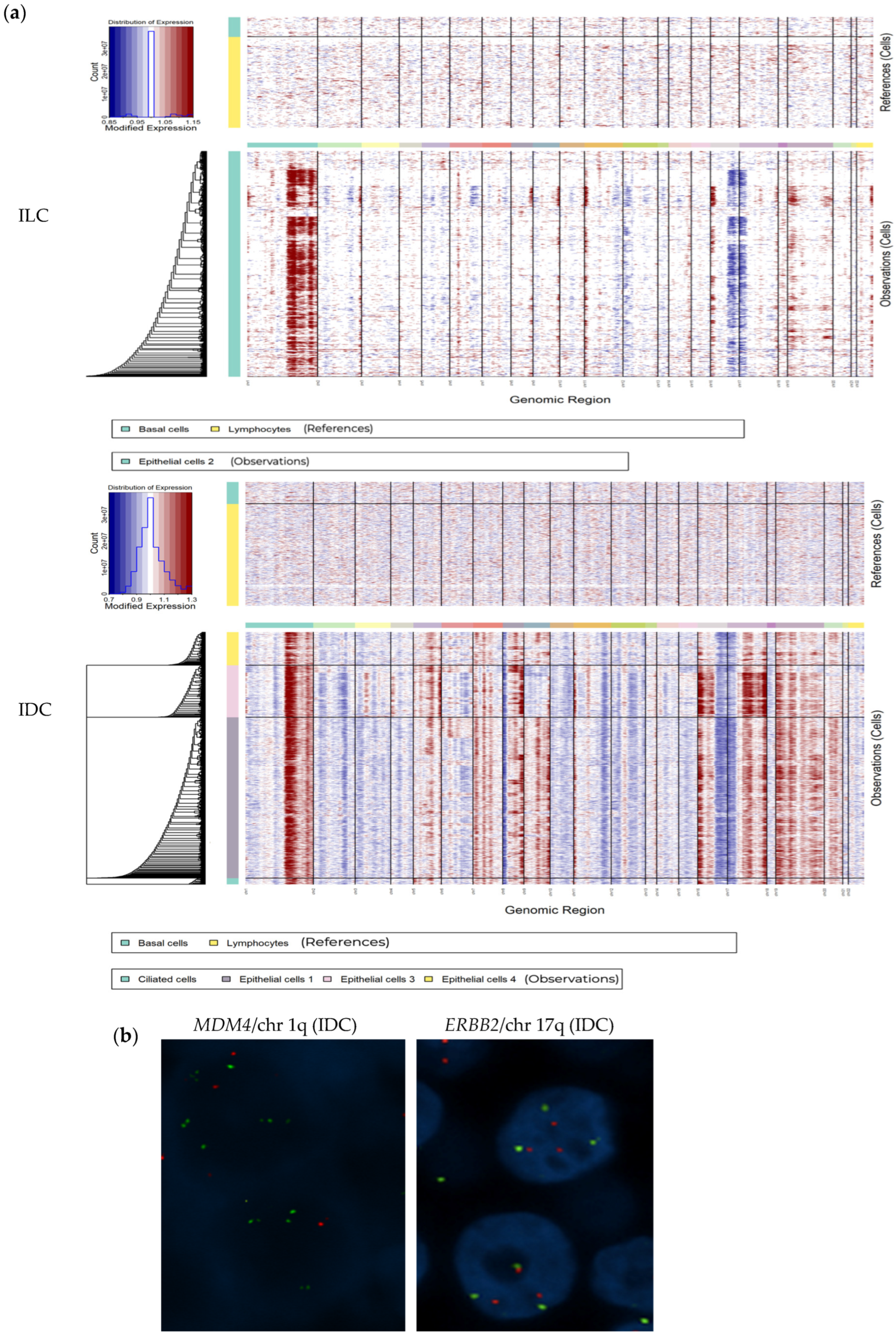
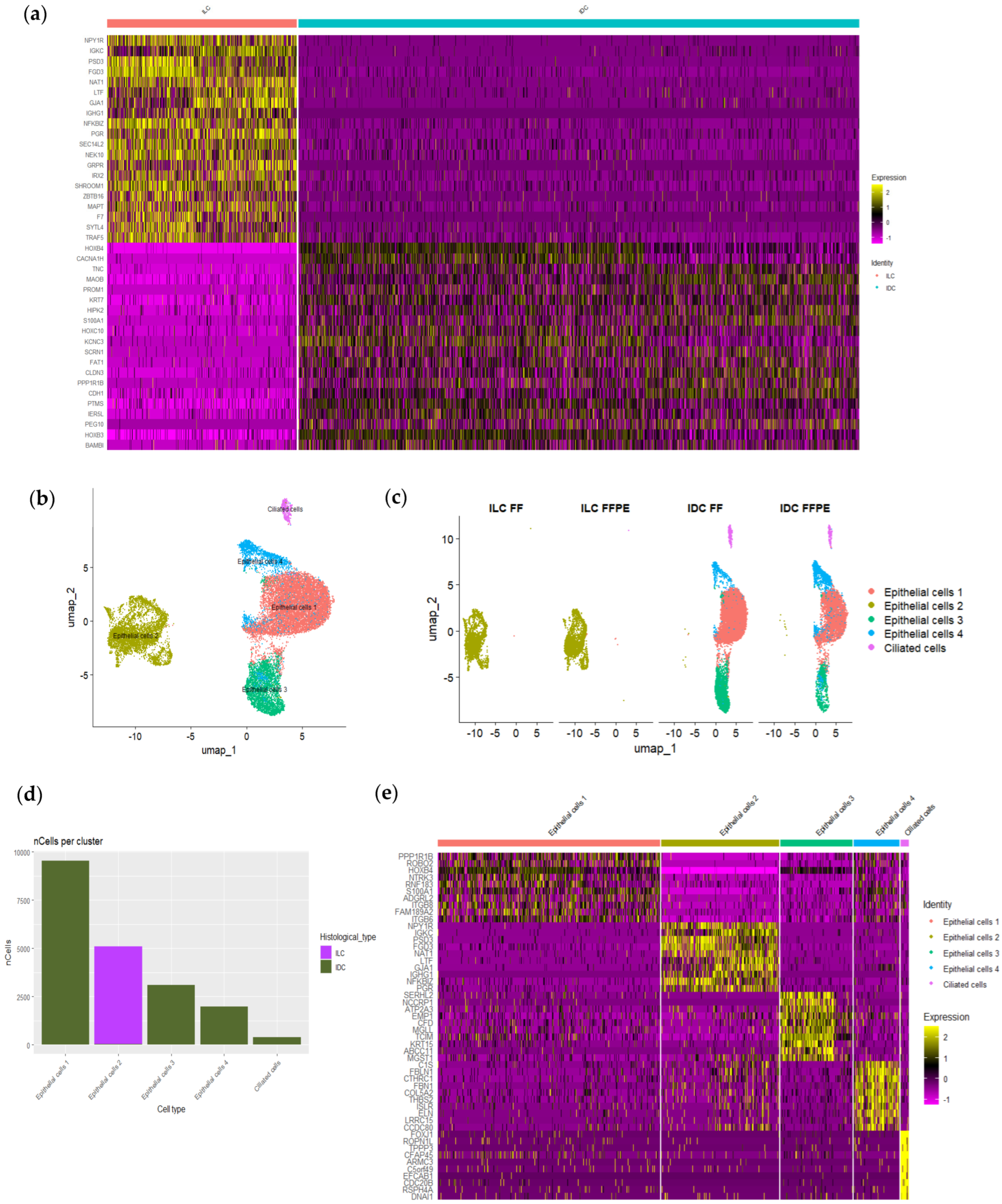
3.5. scRNAseq Identified Epithelial Cells Heterogeneity Among Neoplastic Cells
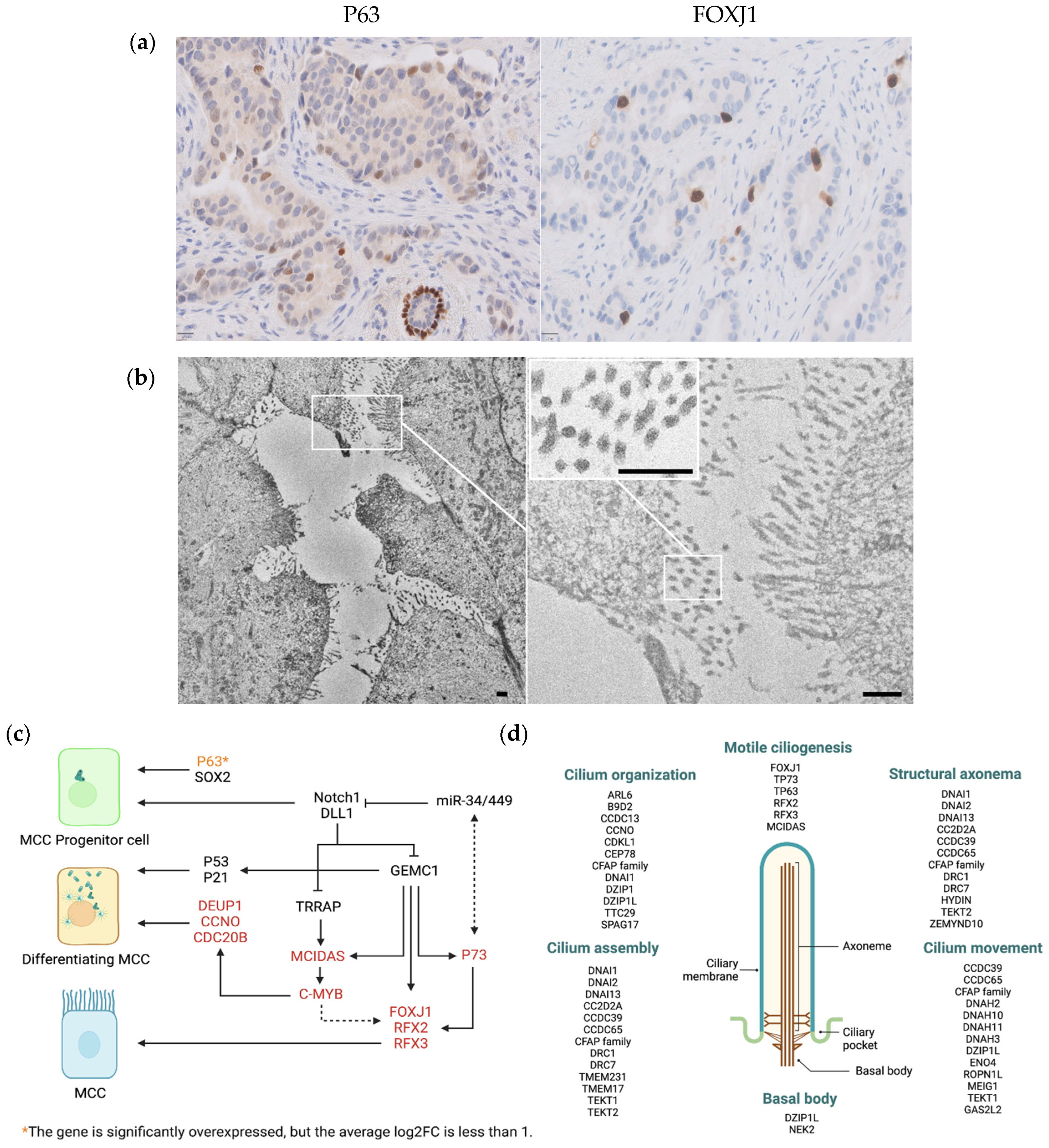
3.6. scRNAseq Identified Neoplastic Epithelial Cells with a Transcriptional Program of Multi-Ciliated Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| CNV | Copy Number Variation |
| FF | Fixed Fresh |
| FFPE | Formalin-Fixed and Paraffin-Embedded |
| FISH | Fluorescence In Situ Hybridization |
| H&E | Hematoxylin and eosin |
| IDC | Invasive Ductal Carcinoma |
| IHC | Immunohistochemistry |
| ILC | Invasive lobular carcinoma |
| MCC | Multi-Ciliated Cell |
| scRNAseq | single-cell RNA sequencing |
| snRNAseq | single-nuclei RNA sequencing |
| TMA | Tisuue microarray |
| WSI | Whole Slide Image |
Appendix A
Appendix A.1. Methods
- Samples
- Processing of fresh tissue samples
- Processing of FFPE tissue samples
- 10× Library preparation and sequencing
- scRNAseq data processing
- Functional enrichment analysis
- Inference of copy number variation (CNV) from scRNAseq
- Trajectory analysis
- Immunohistochemistry and image analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Clon | Supplier |
|---|---|---|
| ER | 6F11 | Leica Biosystems |
| PR | 16 | Leica Biosystems |
| HER2 | 4B5 | Ventana, Roche Diagnostics, Oro Valley, AZ, USA |
| E-cadherin | 36b5 | Leica Biosystems |
| P63 | 7 JUL | Leica Biosystems |
| FOXJ1 | EPR21874 | Abcam, Cambridge, UK |
| CD4 | 4B12 | Leica Biosystems |
| CD8 | 4B11 | Leica Biosystems |
| CD3 | LN10 | Leica Biosystems |
| CD20 | L26 | Leica Biosystems |
| CD68 | 514H12 | Leica Biosystems |
| CD117 (c-KIT) | EP10 | Leica Biosystems |
- Fluorescent In-Situ Hybridization (FISH)
References
- Wang, S.; Sun, S.-T.; Zhang, X.-Y.; Ding, H.-R.; Yuan, Y.; He, J.-J.; Wang, M.-S.; Yang, B.; Li, Y.-B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int. J. Mol. Sci. 2023, 24, 2943. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-Cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, K.; Liu, R.; Song, Y.; Lv, Y.; Bi, P.; Yang, F.; Li, S.; Zhao, J.; Li, X.; et al. Single-cell Transcriptome Sequencing of B-cell Heterogeneity and Tertiary Lymphoid Structure Predicts Breast Cancer Prognosis and Neoadjuvant Therapy Efficacy. Clin. Transl. Med. 2023, 13, e1346. [Google Scholar] [CrossRef]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Tubau Ribera, N.; et al. A Single-cell RNA Expression Atlas of Normal, Preneoplastic and Tumorigenic States in the Human Breast. EMBO J. 2021, 40, e107333. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.D.; Pensa, S.; Steif, A.; Stenning, J.; Kunz, D.J.; Porter, L.J.; Hua, K.; He, P.; Twigger, A.-J.; Siu, A.J.Q.; et al. A Single-Cell Atlas Enables Mapping of Homeostatic Cellular Shifts in the Adult Human Breast. Nat. Genet. 2024, 56, 652–662. [Google Scholar] [CrossRef]
- Nee, K.; Ma, D.; Nguyen, Q.H.; Pein, M.; Pervolarakis, N.; Insua-Rodríguez, J.; Gong, Y.; Hernandez, G.; Alshetaiwi, H.; Williams, J.; et al. Preneoplastic Stromal Cells Promote BRCA1-Mediated Breast Tumorigenesis. Nat. Genet. 2023, 55, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; Li, C.M.-C.; Rosenbluth, J.M.; Selfors, L.M.; Girnius, N.; Lin, J.-R.; Schackmann, R.C.J.; Goh, W.L.; Moore, K.; Shapiro, H.K.; et al. A Human Breast Atlas Integrating Single-Cell Proteomics and Transcriptomics. Dev. Cell 2022, 57, 1400–1420.e7. [Google Scholar] [CrossRef]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Gong, Y.; Pein, M.; Sei, E.; et al. A Spatially Resolved Single Cell Genomic Atlas of the Adult Human Breast. Nature 2023, 620, 181–191. [Google Scholar] [CrossRef]
- Twigger, A.-J.; Engelbrecht, L.K.; Bach, K.; Schultz-Pernice, I.; Pensa, S.; Stenning, J.; Petricca, S.; Scheel, C.H.; Khaled, W.T. Transcriptional Changes in the Mammary Gland during Lactation Revealed by Single Cell Sequencing of Cells from Human Milk. Nat. Commun. 2022, 13, 562. [Google Scholar] [CrossRef]
- Murrow, L.M.; Weber, R.J.; Caruso, J.A.; McGinnis, C.S.; Phong, K.; Gascard, P.; Rabadam, G.; Borowsky, A.D.; Desai, T.A.; Thomson, M.; et al. Mapping Hormone-Regulated Cell-Cell Interaction Networks in the Human Breast at Single-Cell Resolution. Cell Syst. 2022, 13, 644–664.e8. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A Single-Cell and Spatially Resolved Atlas of Human Breast Cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA Sequencing Technologies and Applications: A Brief Overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Trinks, A.; Milek, M.; Beule, D.; Kluge, J.; Florian, S.; Sers, C.; Horst, D.; Morkel, M.; Bischoff, P. Robust Detection of Clinically Relevant Features in Single-Cell RNA Profiles of Patient-Matched Fresh and Formalin-Fixed Paraffin-Embedded (FFPE) Lung Cancer Tissue. Cell. Oncol. 2024, 47, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High Resolution Mapping of the Tumor Microenvironment Using Integrated Single-Cell, Spatial and in Situ Analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef]
- Pizarro, D.; Romero, I.; Pérez-Mies, B.; Redondo, A.; Caniego-Casas, T.; Carretero-Barrio, I.; Cristóbal, E.; Gutiérrez-Pecharromán, A.; Santaballa, A.; D’Angelo, E.; et al. The Prognostic Significance of Tumor-Infiltrating Lymphocytes, PD-L1, BRCA Mutation Status and Tumor Mutational Burden in Early-Stage High-Grade Serous Ovarian Carcinoma—A Study by the Spanish Group for Ovarian Cancer Research (GEICO). Int. J. Mol. Sci. 2023, 24, 11183. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Weigert, M.; Broaddus, C.; Myers, G. Cell Detection with Star-Convex Polygons. arXiv 2018, arXiv:1806.03535. [Google Scholar] [CrossRef]
- González-Martínez, S.; Pizarro, D.; Pérez-Mies, B.; Caniego-Casas, T.; Rodríguez-Peralto, J.L.; Curigliano, G.; Cortés, A.; Gión, M.; Cortés, J.; Palacios, J. Differences in the Molecular Profile between Primary Breast Carcinomas and Their Cutaneous Metastases. Cancers 2022, 14, 1151. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Enríquez, A.; González-Rocha, T.; Burgos, E.; Stolnicu, S.; Mendiola, M.; Nogales, F.F.; Hardisson, D. Transitional Cell Carcinoma of the Endometrium and Endometrial Carcinoma with Transitional Cell Differentiation: A Clinicopathologic Study of 5 Cases and Review of the Literature. Hum. Pathol. 2008, 39, 1606–1613. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Narvaez, D.; Nadal, J.; Nervo, A.; Costanzo, M.V.; Paletta, C.; Petracci, F.E.; Rivero, S.; Ostinelli, A.; Freile, B.; Enrico, D.; et al. The Emerging Role of Tertiary Lymphoid Structures in Breast Cancer: A Narrative Review. Cancers 2024, 16, 396. [Google Scholar] [CrossRef]
- Dinh, H.Q.; Lin, X.; Abbasi, F.; Nameki, R.; Haro, M.; Olingy, C.E.; Chang, H.; Hernandez, L.; Gayther, S.A.; Wright, K.N.; et al. Single-Cell Transcriptomics Identifies Gene Expression Networks Driving Differentiation and Tumorigenesis in the Human Fallopian Tube. Cell Rep. 2021, 35, 108978. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Handfield, L.-F.; Roberts, K.; Nikolakopoulou, K.; Fernando, R.C.; Gardner, L.; Woodhams, B.; Arutyunyan, A.; Polanski, K.; Hoo, R.; et al. Mapping the Temporal and Spatial Dynamics of the Human Endometrium in Vivo and in Vitro. Nat. Genet. 2021, 53, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Q.; Li, Q.; Zhou, J.; Zhao, H. Formation and Function of Multiciliated Cells. J. Cell Biol. 2024, 223, e202307150. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Saavedra Armero, V.; Boudreau, D.K.; Gaudreault, N.; Desmeules, P.; Thériault, S.; Mathieu, P.; Joubert, P.; Bossé, Y. Single-Cell and Single-Nucleus RNA-Sequencing from Paired Normal-Adenocarcinoma Lung Samples Provide Both Common and Discordant Biological Insights. PLoS Genet. 2024, 20, e1011301. [Google Scholar] [CrossRef] [PubMed]
- Tickle, T.I.; Georgescu, C.; Brown, M.; Haas, B. inferCNV of the Trinity CTAT Project. 2019. Available online: https://Github.Com/Broadinstitute/inferCNV (accessed on 1 September 2023).
- Choksi, S.P.; Byrnes, L.E.; Konjikusic, M.J.; Tsai, B.W.H.; Deleon, R.; Lu, Q.; Westlake, C.J.; Reiter, J.F. An Alternative Cell Cycle Coordinates Multiciliated Cell Differentiation. Nature 2024, 630, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, I.; Roy, S.; Chakrabarti, S. Identification of Important Effector Proteins in the FOXJ1 Transcriptional Network Associated With Ciliogenesis and Ciliary Function. Front. Genet. 2019, 10, 23. [Google Scholar] [CrossRef]
- Boutin, C.; Kodjabachian, L. Biology of Multiciliated Cells. Curr. Opin. Genet. Dev. 2019, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.J.P.; Anderson, T.J.; Wells, C.A.; Battersby, S. An Ultrastructural Study of Mucoid Carcinoma of the Breast: Variability of Cytoplasmic Features. Histopathology 1986, 10, 1219–1230. [Google Scholar] [CrossRef]
- Reilova-Velez, J.; Seiler, M.W. Abnormal Cilia in a Breast Carcinoma. An Ultrastructural Study. Arch. Pathol. Lab. Med. 1984, 108, 795–797. [Google Scholar]
- Zhou, X.; Xiao, C.; Han, T.; Qiu, S.; Wang, M.; Chu, J.; Sun, W.; Li, L.; Lin, L. Prognostic Biomarkers Related to Breast Cancer Recurrence Identified Based on Logit Model Analysis. World J. Surg. Oncol. 2020, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Z.; Zhong, Q.; Zhao, L.; Wang, Y.; Chi, H. Identification and Validation of a Novel Prognostic Signature Based on Transcription Factors in Breast Cancer by Bioinformatics Analysis. Gland Surg. 2022, 11, 892–912. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tao, X.; Luo, Y.; Zheng, S.; Lin, N.; Xie, X. A Novel Super-Enhancer-Related Gene Signature Predicts Prognosis and Immune Microenvironment for Breast Cancer. BMC Cancer 2023, 23, 776. [Google Scholar] [CrossRef]
- Weir, A.; Kang, E.-Y.; Meagher, N.S.; Nelson, G.S.; Ghatage, P.; Lee, C.-H.; Riggan, M.J.; Gentry-Maharaj, A.; Ryan, A.; Singh, N.; et al. Increased FOXJ1 Protein Expression Is Associated with Improved Overall Survival in High-Grade Serous Ovarian Carcinoma: An Ovarian Tumor Tissue Analysis Consortium Study. Br. J. Cancer 2023, 128, 137–147. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Campbell, K.R.; Greening, K.; Ho, G.C.; Hopkins, J.; Bui, M.; Douglas, J.M.; Sharlandjieva, V.; Munzur, A.D.; Lai, D.; et al. Single Cell Transcriptomes of Normal Endometrial Derived Organoids Uncover Novel Cell Type Markers and Cryptic Differentiation of Primary Tumours. J. Pathol. 2020, 252, 201–214. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carré, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Setty, M.; Kiseliovas, V.; Levine, J.; Gayoso, A.; Mazutis, L.; Pe’er, D. Characterization of Cell Fate Probabilities in Single-Cell Data with Palantir. Nat. Biotechnol. 2019, 37, 451–460. [Google Scholar] [CrossRef]
- Lin, L.; Sirohi, D.; Coleman, J.F.; Gulbahce, H.E. American Society of Clinical Oncology/College of American Pathologists 2018 Focused Update of Breast Cancer HER2 FISH Testing GuidelinesResults From a National Reference Laboratory. Am. J. Clin. Pathol. 2019, 152, 479–485. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Martínez, S.; Palacios, J.; Carretero-Barrio, I.; Lanza, V.F.; García-Cosío Piqueras, M.; Caniego-Casas, T.; Hardisson, D.; Esteban-Rodríguez, I.; Cortés, J.; Pérez-Mies, B. Single-Cell RNA Sequencing on Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Identified Multi-Ciliary Cells in Breast Cancer. Cells 2025, 14, 197. https://doi.org/10.3390/cells14030197
González-Martínez S, Palacios J, Carretero-Barrio I, Lanza VF, García-Cosío Piqueras M, Caniego-Casas T, Hardisson D, Esteban-Rodríguez I, Cortés J, Pérez-Mies B. Single-Cell RNA Sequencing on Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Identified Multi-Ciliary Cells in Breast Cancer. Cells. 2025; 14(3):197. https://doi.org/10.3390/cells14030197
Chicago/Turabian StyleGonzález-Martínez, Silvia, José Palacios, Irene Carretero-Barrio, Val F. Lanza, Mónica García-Cosío Piqueras, Tamara Caniego-Casas, David Hardisson, Isabel Esteban-Rodríguez, Javier Cortés, and Belén Pérez-Mies. 2025. "Single-Cell RNA Sequencing on Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Identified Multi-Ciliary Cells in Breast Cancer" Cells 14, no. 3: 197. https://doi.org/10.3390/cells14030197
APA StyleGonzález-Martínez, S., Palacios, J., Carretero-Barrio, I., Lanza, V. F., García-Cosío Piqueras, M., Caniego-Casas, T., Hardisson, D., Esteban-Rodríguez, I., Cortés, J., & Pérez-Mies, B. (2025). Single-Cell RNA Sequencing on Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Identified Multi-Ciliary Cells in Breast Cancer. Cells, 14(3), 197. https://doi.org/10.3390/cells14030197