Abstract
Over the past two decades, immune–inflammatory dysregulation has emerged as a central paradigm in the biology of mood disorders. Patients with major depression (MDD) and bipolar disorder (BD) frequently display low-grade systemic inflammation. Elevated C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) identify clinically relevant subgroups of patients characterized by greater severity, cognitive impairment, and poor treatment response. Changes in the gut microbiota and disruptions of the blood–brain barrier (BBB) act as important gateways through which systemic immune activity can influence the brain. At the intracellular level, pattern-recognition receptors activate convergent hubs including NF-κB, JAK/STAT, and MAPK cascades, while the NLRP3 inflammasome integrates mitochondrial dysfunction and oxidative stress with IL-1β release and pyroptosis. These pathways converge on glial dysregulation, impaired BDNF/TrkB signaling, and kynurenine pathway (KP) alterations, fostering excitotoxicity and synaptic deficits. Translational studies demonstrate that elevated CRP and IL-6 predict poor antidepressant outcomes. Anti-inflammatory agents such as infliximab and celecoxib show efficacy in specific subgroups of patients. Emerging multi-omics approaches identify immuno-metabolic biotypes, supporting the rationale for biomarker-guided stratification. These findings define an ‘inflammatory biotype’ of mood disorders and highlight the need for biomarkers and precision-based trials to guide treatment.
1. Introduction
The standard pharmacological treatments for major depressive disorder (MDD) and bipolar disorder (BD) fail to achieve remission in a considerable proportion of patients, many of whom continue to experience residual symptoms, chronic course, and marked functional impairment [1,2]. While the monoaminergic dysfunction model has long provided the dominant explanatory framework, it is now widely recognized as insufficient to account for the marked biological heterogeneity, variability in treatment response, and persistence of cognitive and affective symptoms observed in mood disorders [3,4]. Consequently, attention has shifted toward alternative pathophysiological models capable of integrating systemic and central mechanisms. Over the past two decades, converging lines of evidence have positioned immune–inflammatory dysregulation as a central dimension in the biology of mood disorders [5,6]. Meta-analyses consistently demonstrate low-grade systemic inflammation in both MDD and BD, with elevations in interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and C-reactive protein (CRP) [7,8,9]. Notably, hs-CRP levels above 3 mg/L define a clinically relevant subgroup of patients characterized by heightened symptom severity, psychomotor slowing, cognitive impairment, and reduced responsiveness to antidepressants [9,10,11]. These findings suggest that mood disorders, at least in a biologically distinct subgroup, share features with chronic inflammatory syndromes, rather than representing conditions solely driven by neurotransmitter imbalance [6]. Crucially, inflammatory activity is not confined to peripheral soluble mediators but extends to several peripheral–central interfaces [12,13]. Alterations such as gut microbiota dysbiosis, reduced microbial diversity, depletion of butyrate-producing taxa, and enrichment of pro-inflammatory genera have been linked to altered short-chain fatty acid (SCFA) and indole metabolite production, as well as increased translocation of lipopolysaccharides (LPS) into circulation [14,15,16]. These changes amplify systemic immune activity and provide a mechanistic link between metabolic status and mood dysregulation [15,16]. Similarly, dysfunction of the blood–brain barrier (BBB) constitutes a critical interface: cytokines such as TNF-α and IL-6, together with reactive oxygen species, compromise tight junction integrity, allowing circulating inflammatory mediators to access the central nervous system (CNS) and foster neuroinflammation [12,13]. At the intracellular level, these peripheral signals converge on pattern-recognition receptors (PRRs) and downstream hubs. Toll-like receptor 4 (TLR4) recognizes microbial ligands such as lipopolysaccharides (LPS), while NOD1/2 receptors detect bacterial peptidoglycans, both of which can enter systemic circulation under conditions of bowel barrier dysfunction [14,17]. Engagement of these receptors activates nuclear factor kappa B (NF-κB), Janus kinase/signal transducer and activator of transcription (JAK/STAT), and mitogen-activated protein kinase (MAPK) pathways, thereby sustaining transcriptional programs of immune activation [18]. In parallel, the NLRP3 inflammasome links mitochondrial dysfunction, oxidative stress, and extracellular danger signals to IL-1β/IL-18 release and pyroptosis, establishing a further amplification loop [19,20,21]. These pathways ultimately converge on cellular effectors in the CNS, particularly microglia and astrocytes. Microglia and astrocytes, the primary immune regulators of the brain, become dysregulated under inflammatory conditions: hyperactive microglia release pro-inflammatory cytokines, glutamate, and reactive oxygen species, while impaired astrocytic function reduces trophic support and glutamate clearance [22,23]. Together with mitochondrial dysfunction and redox imbalance, these alterations foster excitotoxicity, energy failure, and progressive synaptic deficits [24,25]. Neurotrophic signaling is likewise disrupted, with cytokine-induced suppression of brain-derived neurotrophic factor (BDNF)/TrkB pathways contributing to dendritic spine loss and impaired synaptic plasticity [26,27]. Interestingly, rapid-acting antidepressants such as ketamine exert part of their therapeutic efficacy through rapid restoration of BDNF-dependent plasticity [28]. A further metabolic dimension is provided by the kynurenine pathway (KP), which represents a major biochemical interface between immune activation and neurotransmission [29,30]. Pro-inflammatory cytokines, particularly interferon-γ, TNF-α, and IL-6, activate indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO), diverting tryptophan metabolism away from serotonin toward kynurenine and its neuroactive metabolites [29,31]. This shift results in increased production of quinolinic acid (QA), a neurotoxic NMDA receptor agonist, and reduced kynurenic acid (KYNA), a neuroprotective NMDA antagonist and α7 nicotinic receptor modulator, thereby contributing to excitotoxicity, impaired cognition, and pathology progression [32,33,34]. From a translational perspective, these mechanistic insights align with clinical evidence. Peripheral biomarker studies, neuroimaging with translocator protein positron emission tomography (TSPO-PET), metabolic assays, gut microbiome analyses, and interventional trials with anti-inflammatory and immunomodulatory agents all converge in supporting the existence of an “inflammatory biotype” of mood disorders [35,36]. Notably, clinical benefits are observed primarily in biomarker-enriched subgroups, underscoring the need for stratified and precision-based approaches [37,38]. Beyond immune–inflammatory dysregulation, mood disorders arise from the interplay of multiple vulnerability domains. Genetic factors contribute substantially, with genome-wide association studies implicating variants in genes regulating immune signaling, synaptic plasticity, and stress-response pathways [39]. Polygenic risk scores suggest that immune-related genetic architectures partly overlap with susceptibility to MDD and BD, reinforcing the biological plausibility of an immunogenetic interface [40]. Stress-related mechanisms represent another critical determinant. Chronic psychosocial stress activates the hypothalamic–pituitary–adrenal (HPA) axis, leading to hypercortisolemia, glucocorticoid receptor resistance, and sustained low-grade inflammation. These alterations potentiate the activation of NF-κB and NLRP3-dependent cascades, linking stress exposure to immune–inflammatory processes and to downstream effects on neuroplasticity and mitochondrial function [41]. Finally, environmental exposures—ranging from early life adversity and trauma to lifestyle factors such as diet, smoking, and physical inactivity—exert long-lasting effects on immune tone and brain health. Early adversity has been associated with persistent microglial priming and heightened inflammatory reactivity, while obesogenic diets and sedentary behavior promote systemic metabolic inflammation, further exacerbating neuropsychiatric vulnerability [6,42]. Collectively, these genetic, stress-related, and environmental dimensions interact with immune–inflammatory pathways, providing a multifactorial framework that captures the complexity and heterogeneity of mood disorders.
In this review, we synthesize findings from the past decade along four thematic domains. The first addresses peripheral triggers and barrier dysfunction, followed by intracellular signaling pathways, downstream final common pathways involving mitochondria, glia, plasticity, and kynurenine metabolism, and inter-individual differences in immune reactivity. The last section focuses on therapeutic implications and stratification approaches. By integrating these perspectives, we aim to outline an immune–inflammatory model of mood disorders that moves beyond descriptive associations toward mechanistic understanding and precision therapeutics.
2. Materials and Methods
We performed an extensive search using the following biomedical databases: Medline using the PubMed interface, Web of Science, and Embase. Search terms were as follows: “major depressive disorder”, “depression”, “bipolar disorder”, “bipolar depression” with immune–inflammatory and neurobiological concepts, including “cytokine”, “C-reactive protein”, “IL-6”, “TNF”, “neuroinflammation”, “translocator protein”, “kynurenine”, “blood–brain barrier”, “microbiome”, “oxidative stress”, “mitochondria”, and “BDNF”. Additional keywords were used for specific domains such as neuroimaging (TSPO-PET), kynurenine metabolites (quinolinic and kynurenic acid), cellular indices (NLR, PLR, MLR, monocyte subsets, T cells), intracellular signaling pathways (NF-κB, JAK/STAT, MAPK), inflammasome biology (NLRP3, IL-1β, IL-18), glial and astrocytic alterations, mitochondrial and redox pathways, synaptic plasticity, gut–brain axis, and BBB markers. No restriction on publication date was applied, and the last search was performed on 26 August 2025. Only articles written in or translated into English were considered. Priority was given to systematic reviews, meta-analyses, randomized controlled trials, large cohort studies, and mechanistic human investigations addressing immune–inflammatory biomarkers in mood disorders. Artificial intelligence was employed exclusively for linguistic polishing of the text. All aspects of the study—conception and design, data collection, analysis, interpretation, and conclusions—were carried out entirely by the authors, who take full responsibility for the integrity and accuracy of the work.
3. Mechanistic Pathways of Immune–Inflammatory Dysregulation in Mood Disorders
3.1. Peripheral Triggers and Barrier Dysfunction
Peripheral triggers include a range of signals generated outside the CNS—such as microbial metabolites, dietary products, and systemic inflammatory mediators—that may contribute to the immune alterations observed in mood disorders [14,15,43]. While their precise role remains debated, a recurrent model suggests that these peripheral factors can shape systemic immune activity and, over time, influence the function of critical interfaces such as the intestinal epithelium and the BBB [13,15]. In this framework, gut dysbiosis and barrier permeability may represent early events, followed by immune activation and ultimately by neuroinflammatory changes that may contribute to the emergence of mood disorders, although the directionality and causality of these processes are not fully established [16,44]. The sequential processes linking peripheral immune activation to central neuroinflammation are schematically depicted in Figure 1.
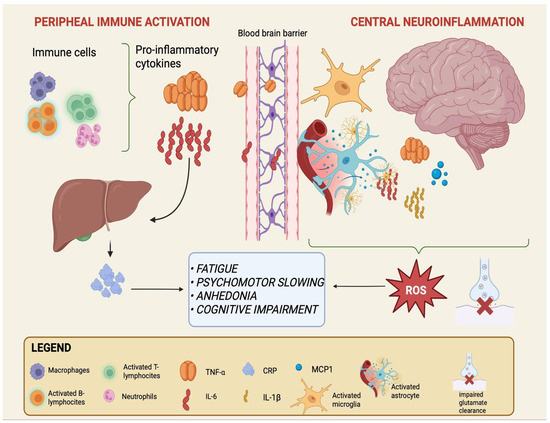
Figure 1.
From peripheral immune activation to central neuroinflammation in mood disorders. Activated immune cells in the periphery (macrophages, neutrophils, and T- and B-lymphocytes) release pro-inflammatory cytokines, primarily IL-6 and TNF-α. IL-6 also stimulates hepatic production of CRP, a systemic inflammation marker. Increased cytokine load can compromise BBB integrity, facilitating their entry into the CNS. Within the brain, astrocytes and microglia become activated and release additional mediators, including IL-6, TNF-α, IL-1β, and MCP-1, while astrocytes show impaired glutamate clearance. This peripheral-to-central immune cascade sustains neuroinflammation and oxidative stress and is associated with core clinical symptoms of mood disorders such as fatigue, psychomotor slowing, anhedonia, and cognitive impairment. Created with BioRender (web-based version). Pinzi, M. (2025) https://BioRender.com/t7otmbn (accessed on 14 September 2025).
3.1.1. Gut–Brain Axis and Microbial Dysbiosis
Alterations in the gut microbiota are among the best-studied contributors to peripheral immune–metabolic activation. Patients with MDD and BD frequently display reduced microbial diversity, characterized by enrichment of pro-inflammatory taxa such as Alistipes and Oscillibacter, and depletion of butyrate-producing genera including Faecalibacterium and Roseburia [45,46]. These compositional changes are associated with functional shifts: reduced SCFA production and increased translocation of lipopolysaccharides (LPSs) and indole derivatives into the circulation [14]. Loss of butyrate (a SCFA) weakens intestinal barrier integrity, while microbial products such as lipopolysaccharide (LPS), peptidoglycan (PGN), flagellin, and trimethylamine-N-oxide (TMAO) translocate into the circulation. Once in peripheral blood—following translocation across a compromised intestinal barrier—these molecules can interact with innate immune receptors, fostering the release of pro-inflammatory cytokines (IL-6, TNF-α, and IL-1β) and amplifying systemic immune activity. This peripheral immune activation may, over time, contribute to BBB dysfunction and facilitate neuroinflammatory processes [13,15,17]. Clinical studies confirm this link: elevated serum LPS and reduced SCFA correlate with higher CRP and IL-6 in MDD, while altered plasma indole-3-propionic acid associates with depressive severity in BD [47]. Multi-omics approaches show that microbial–metabolite signatures not only discriminate MDD from controls but also correlate with symptom severity [38]. Fecal microbiota transfer from depressed patients to germ-free mice induces depressive-like behaviors, supporting causality. Interventional trials with probiotics and prebiotics suggest modest mood improvements, often accompanied by CRP and IL-6 reductions, although with heterogeneous results [44,48]. Together, these data point to a gut-driven immune–metabolic depression subtype. Mechanistic interactions between gut dysbiosis, barrier dysfunction, and neuroinflammation are shown in Figure 2.
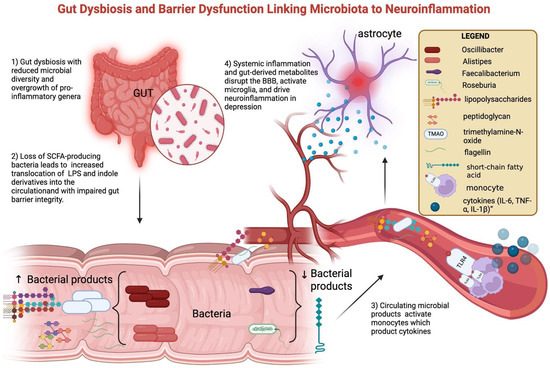
Figure 2.
Gut dysbiosis and barrier dysfunction linking microbiota to neuroinflammation. Gut dysbiosis in mood disorders is characterized by reduced microbial diversity, with an overrepresentation of pro-inflammatory genera (Oscillibacter, Alistipes) and a reduction in SCFA-producing bacteria (Faecalibacterium, Roseburia). Loss of butyrate (a short-chain fatty acid, SCFA) weakens intestinal barrier integrity, while microbial products such as lipopolysaccharide (LPS), peptidoglycan (PGN), flagellin, and trimethylamine-N-oxide (TMAO) translocate into the circulation. These pathogen-associated molecular patterns activate circulating monocytes through pattern-recognition receptors, inducing the release of pro-inflammatory cytokines (IL-6, TNF-α, and IL-1β). Systemic inflammation and gut-derived metabolites promote BBB dysfunction and microglial activation, ultimately driving neuroinflammation and contributing to depressive symptomatology. Created with BioRender.com (web-based version). Pinzi, M. (2025) https://BioRender.com/blocq6n (accessed on 14 September 2025). Original figure created by the authors.
3.1.2. Blood–Brain Barrier Dysfunction
Peripheral immune activation—whether originating from the gut or from other systemic sources—can in turn influence the BBB. Pro-inflammatory cytokines such as TNF-α and IL-6, together with ROS, have been shown to disrupt tight junction proteins, including occludin and claudin-5, leading to increased permeability [12,13]. This condition may permit cytokines, LPS, or other circulating inflammatory mediators to reach the CNS, where they can activate glial cells and foster neuroinflammation [14]. Several biomarkers support the relevance of BBB dysfunction in mood disorders. Elevated serum S100B—a glial-derived protein that enters circulation when barrier permeability is impaired—has been repeatedly reported in both MDD and BD, where higher concentrations correlate with illness severity and even predict poor antidepressant response, underscoring its potential as both a state and prognostic marker [49,50,51,52]. Increased levels of MMP-9, an enzyme that degrades extracellular matrix components, produced in response to inflammatory stimuli, have been associated with acute depressive episodes, while an elevated CSF/serum albumin ratio has been found in subsets of depressed patients, suggesting impaired barrier function [53,54]. Overall, these findings suggest that systemic inflammation and BBB alterations form a bidirectional loop, where peripheral immune signals compromise barrier integrity and, conversely, barrier dysfunction facilitates central immune activation and amplification of neuroinflammation through continuous peripheral-to-central signaling. Although the precise sequence remains unresolved, this model provides a plausible bridge linking systemic immune changes to neuroinflammation in mood disorders [38].
3.2. Intracellular Signaling Pathways Linking Peripheral Inflammation to the CNS
3.2.1. Toll-like Receptors and NF-κB Activation
A central mechanism through which peripheral inflammatory signals influence the CNS involves the activation of pattern-recognition receptors (PRRs) located on the cell surface. Toll-like receptor 4 (TLR4), expressed on the plasma membrane, is responsive to lipopolysaccharides (LPS) and related microbial products that enter systemic blood following increased intestinal permeability [14,55]. In addition, damage-associated molecular patterns (DAMPs) such as HMGB1 or extracellular ATP, released during cellular stress, act as endogenous ligands that further activate TLR4 signaling [56].
Engagement of TLR4 initiates intracellular cascades involving NF-κB, JAK/STAT, and MAPK pathways. These hubs are also downstream of cytokine receptors activated by IL-6, TNF-α, and IL-1β. There is growing evidence implicating TLR4-related intracellular signaling pathways—particularly NF-κB, JAK/STAT, and MAPK cascades—as key mediators in the pathophysiology of mood disorders [57].
In rodent models, NF-κB activation in the hippocampus mediates stress-induced suppression of neurogenesis and depressive-like behavior [18]. In patients with MDD, increased NF-κB activity in peripheral blood mononuclear cells correlates with symptom severity and treatment resistance [18]. Persistent IL-6/STAT3 signaling has been linked to impaired neuroplasticity, while MAPK activation (p38, ERK, JNK) in microglia enhances glutamate release and ROS generation, promoting excitotoxicity [58]. Dysregulated MAPK signaling has been confirmed in postmortem corticolimbic tissue of depressed patients [59]. Activation of these intracellular pathways ultimately results in the production and release of pro-inflammatory cytokines, a phenomenon that has been consistently documented in clinical studies. Indeed, meta-analyses show elevations of CRP, IL-6, IL-12/IL-18, sIL-2R, and TNF-α in MDD. Low-grade inflammation (CRP > 3 mg/L) is present in ~27% of patients, with >1 mg/L in nearly 60%, delineating an “inflamed depression” subgroup [60]. In BD, CRP and TNF-α increase during acute episodes but normalize in euthymia, while IL-6 remains persistently elevated [7]. Transdiagnostically, IL-6, TNF-α, and CRP rise in acute phases of depression, mania, and psychosis, with IL-6 often persisting outside of acute states [61]. Additional hematologic indices (NLR, PLR, and to a lesser extent MLR) also emerge as accessible markers, while immune phenotyping identifies subgroups with neutrophilia, expanded monocytes, CD4+ T cells, and high CRP/IL-6, linked to severity and treatment resistance [62,63]. Key intracellular signaling pathways linking cytokine activity to astrocytic dysfunction are illustrated in Figure 3.
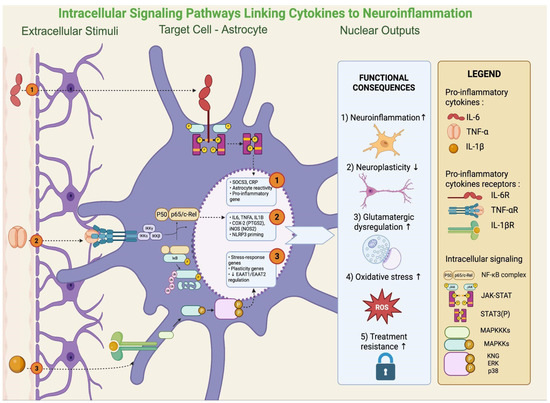
Figure 3.
Intracellular cytokine signaling in astrocytes: pathways sustaining neuroinflammation and plasticity loss. Once bound to their receptors on astrocytes (IL-6R, TNFR, and IL-1β), cytokines activate three main signaling modules: (i) JAK/STAT3, promoting astrocyte reactivity and pro-inflammatory gene programs (e.g., SOCS3, CRP); (ii) NF-κB, inducing transcription of IL6, TNFα, IL1B, PTGS2 (COX-2), NOS2 (iNOS), and NLRP3 priming; and (iii) MAPK pathways (p38/ERK/JNK), regulating stress-response and plasticity-related genes, including EAAT1/2 transporters. Collectively, these cascades amplify neuroinflammation, impair neuroplasticity, disrupt glutamatergic homeostasis, increase oxidative/NO stress, and contribute to treatment resistance. Created with BioRender (web-based version). Pinzi, M. (2025) https://BioRender.com/jnzhwjy (accessed on 14 September 2025).
3.2.2. NOD Receptors, NLRP3 Inflammasome, and Pyroptosis
In parallel to surface receptors, additional PRRs are located within the cytosol, where they sense intracellular danger signals. NOD1/2 receptors detect bacterial peptidoglycans that can translocate into systemic blood under conditions of gut barrier disruption [64]. Their engagement provides a priming signal for transcriptional upregulation of inflammasome components through NF-κB activation [19]. A second level of intracellular sensing involves the NLRP3 inflammasome, a multiprotein complex that responds to cellular stress signals such as mitochondrial dysfunction, ROS accumulation, or extracellular ATP. Full activation of NLRP3 requires this dual input: PRR-mediated priming plus an intracellular danger signal. Once assembled, the inflammasome promotes cleavage of pro–IL-1β and pro–IL-18 into their active forms, driving pyroptotic cell death and amplifying inflammatory cascades [19]. Preclinical studies show that chronic stress enhances NLRP3 activation in the hippocampus and prefrontal cortex, leading to increased IL-1β release, inhibition of neurogenesis, disruption of synaptic plasticity, and the emergence of depressive-like behavior [65]. Conversely, both genetic deletion of the NLRP3 gene and pharmacological inhibition of the NLRP3 inflammasome with MCC950 have been shown to reverse these effects, restoring neuroplasticity and behavioral resilience in animal models [66]. Furthermore, conventional antidepressants reduce NLRP3 activation and IL-1β release in experimental models, suggesting that part of their therapeutic effects may be mediated through modulation of innate immune–inflammatory pathways [67]. Conventional monoaminergic antidepressants (SSRIs/SNRIs/TCAs/MAOIs) enhance synaptic serotonin and norepinephrine and, with chronic administration, promote downstream plasticity adaptations; however, clinical improvement typically unfolds over weeks rather than days, reflecting an indirect mechanism of action [68]. Despite proven efficacy versus placebo and broadly comparable effectiveness across agents, outcome heterogeneity is high [69]. In routine care, only about one-third of patients remit at the first adequate trial, with diminishing returns across subsequent steps [70]. Importantly, baseline inflammatory burden is associated with poorer antidepressant outcomes, underscoring a biology not fully addressed by monoaminergic modulation [71]. Emerging stratification data indicate that CRP can inform drug choice: CRP < 1 mg/L predicts better response to the SSRI escitalopram, whereas higher CRP favors noradrenergic/dopaminergic options (e.g., nortriptyline or bupropion-containing regimens) [72]. Collectively, these findings frame conventional agents as necessary but insufficient for biomarker-enriched subgroups, supporting precision approaches that integrate immune–metabolic targets alongside monoamines. Thus, intracellular sensing through NOD receptors and the NLRP3 inflammasome represents a complementary route to TLR4, linking microbial translocation and cellular stress to neuroinflammatory processes in mood disorders [19,20].
3.3. Final Common Pathways: Mitochondria, Glia, Plasticity, and Kynurenine Metabolism
The downstream consequences of peripheral triggers (Section 3.1) and intracellular signaling pathways (Section 3.2) converge on a set of final common biological hubs that define the so-called “inflammatory depression” phenotype [25]. These hubs include mitochondrial dysfunction and oxidative stress, glial activation and impaired neuroimmune homeostasis, neurotrophic and synaptic alterations, and dysregulation of the kynurenine pathway. Collectively, they translate molecular immune activation into functional and structural brain deficits observed in MDD and BD [24,73]. Their interplay explains clinical dimensions such as fatigue, cognitive impairment, anhedonia, and reduced responsiveness to conventional treatments.
3.3.1. Mitochondrial Dysfunction and Oxidative Stress
Mitochondria are critical integrators of immune and metabolic stress. Pro-inflammatory cytokines such as TNF-α and IL-1β impair the electron transport chain, reducing ATP synthesis and enhancing ROS production, which further activate the NLRP3 inflammasome and amplify IL-1β/IL-18 release [21,43]. This creates a feed-forward loop between oxidative stress and immune activation. Beyond energy production, mitochondrial dysfunction alters calcium buffering and apoptotic signaling, thereby increasing neuronal vulnerability.
Clinical studies consistently show elevated peripheral markers of oxidative stress in mood disorders, including malondialdehyde (MDA), 8-hydroxy-2′-deoxyguanosine (8-OHdG), and isoprostanes, alongside reduced antioxidant defenses such as total antioxidant capacity, uric acid, and zinc [24,25]. A meta-analysis by Liu et al. (2015) demonstrated increased oxidative damage markers and decreased antioxidants across 115 studies, with partial normalization after antidepressant treatment [24]. Translational interventions support the therapeutic relevance of targeting redox balance [24]. Berk et al. (2008) showed that NAC supplementation replenishes glutathione and improves symptoms, findings subsequently extended to mood disorders in smaller trials [17,74]. Complementary evidence supports mitochondrial-targeted compounds such as omega-3 fatty acids, which exert both antioxidant and anti-inflammatory effects, and creatine, which enhances bioenergetics. Altogether, mitochondrial dysfunction links systemic inflammation to fatigue, psychomotor slowing, and poor treatment response, highlighting a tractable therapeutic axis.
3.3.2. Glial Activation and Neuroinflammation
Microglia and astrocytes form the primary immune regulators of the CNS22,23. Under chronic inflammatory stimulation, microglia polarize toward an M1-like pro-inflammatory phenotype, releasing IL-1β, TNF-α, glutamate, and ROS, thereby contributing to excitotoxicity and synaptic pruning [75,76,77]. At the same time, the reparative M2-like phenotype, normally associated with neurogenesis and tissue repair, is blunted in mood disorders. Yirmiya et al. (2015) reviewed how such dysregulation contributes to depressive phenotypes [23]. Astrocytic dysfunction is equally important: reduced expression of glutamate transporters (EAAT1/2) impairs clearance of excitatory neurotransmitters, while postmortem studies show reduced GFAP+ astrocytic density in corticolimbic regions of depressed patients [22]. These changes reduce trophic support, impair synaptic homeostasis, and amplify excitotoxic cascades. In vivo evidence from TSPO-PET imaging corroborates these findings. Setiawan et al. (2015) showed elevated TSPO binding in the anterior cingulate and other limbic regions [78]. A meta-analysis by Eggerstorfer et al. (2022) confirmed increased TSPO binding in ACC, hippocampus, and insula [36]. However, not all interventions targeting glial activation have proven effective. For example, Attwells et al. (2021) found no reduction in TSPO binding after minocycline, highlighting the complexity of central immune modulation [79]. By contrast, Raison et al. (2013) demonstrated antidepressant effects in patients with baseline CRP > 5 mg/L [80]. Together, these data indicate that glial alterations are central to inflammatory depression but require biomarker-driven therapeutic strategies.
3.3.3. Neurotrophic and Synaptic Alterations
Inflammatory cytokines such as IL-1β, TNF-α, and IL-6 impair neuroplasticity by reducing BDNF transcription and TrkB activation [27]. This leads to dendritic spine loss, reduced synaptic density, and impaired long-term potentiation. Postmortem studies confirm downregulation of BDNF and TrkB expression in corticolimbic areas of depressed and suicidal patients [26]. Peripheral BDNF levels are likewise decreased in mood disorders. A meta-analysis by Fernandes et al. (2015) found reduced serum BDNF across illness phases of BD, with stronger reductions during acute episodes [27]. From a therapeutic standpoint, the restoration of BDNF signaling is essential. Duman et al. (2016) highlighted how ketamine enhances BDNF release and TrkB signaling, underlying its rapid antidepressant and pro-cognitive effects [28]. Emerging small-molecule TrkB agonists (e.g., 7,8-dihydroxyflavone) have shown antidepressant-like activity in preclinical models [81]. Beyond BDNF, other neurotrophins such as vascular endothelial growth factor (VEGF) and nerve growth factor (NGF) may also contribute, although with less consistent clinical data. Overall, impaired neurotrophic signaling represents a convergent hub linking immune stress, cognitive impairment, and treatment resistance.
3.3.4. Kynurenine Pathway Dysregulation
The kynurenine pathway (KP) represents a biochemical bridge between inflammation, neurotransmission, and energy metabolism [29]. Pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-6) induce IDO and TDO, diverting tryptophan from serotonin synthesis toward kynurenine and downstream metabolites [29,31]. This shift increases neurotoxic quinolinic acid (QA), a potent NMDA agonist, while reducing kynurenic acid (KYNA), which normally exerts neuroprotective effects as an NMDA antagonist and α7 nicotinic receptor modulator [32,33].
Meta-analyses consistently support KP dysregulation. Marx et al. (2021) found elevated KYN/TRP ratios, increased QA, and reduced KYNA across mood disorders [30]. Inam et al. (2023) confirmed QA elevations in CSF of MDD patients. Importantly, specific metabolites such as 3-hydroxykynurenine (3-HK) and anthranilic acid contribute to oxidative stress and neuronal toxicity [34].
Clinically, KP alterations are associated with suicidality. Sublette et al. (2011) showed increased plasma kynurenine in suicide attempters versus controls [82]. Additionally, immunometabolic factors exacerbate KP shifts: Agudelo et al. (2014) demonstrated that obesity and insulin resistance enhance IDO activity, linking systemic metabolism to central neurotransmission [83]. Altogether, KP dysregulation provides mechanistic and prognostic insight, suggesting potential for biomarker-guided and precision-targeted interventions.
3.4. Region-Specific Neuroinflammatory Changes in Mood Disorders
Neuroinflammation in mood disorders does not occur diffusely but follows regionally selective patterns, affecting circuits that regulate affect, cognition, and motivation. Evidence from postmortem studies, neuroimaging, and recent animal models consistently implicates the prefrontal cortex, hippocampus, amygdala, striatum, and insula. In the prefrontal cortex (PFC), reductions in glial cells—particularly astrocytes and oligodendrocytes—are a robust finding in major depression and bipolar disorder [84]. Recent PET imaging studies show increased TSPO ligand binding in the PFC and anterior cingulate in depressed patients, indicating elevated glial activation in these regions [85].
Chronic stress models also show dendritic spine loss and apical dendrite retraction in medial PFC pyramidal neurons, findings that align with fMRI evidence of impaired prefrontal–amygdala connectivity in inflamed depression [86]. The hippocampus shows strong evidence of neuroinflammatory damage. Microglial activation, astrocyte dysfunction, and downregulation of neurogenesis in the dentate gyrus are reported in both animal models and human studies of major depressive disorder [87,88]. Moreover, recent work shows that hippocampal neuronal survival is decreased, and that treatments targeting microglial polarization (e.g., Urolithin B) can restore neuronal function and reduce depressive behavior [89]. In the amygdala, studies continue to find immune-related modulation. PET and preclinical evidence point to amygdala involvement in mood disorders via increased microglial activation and inflammatory cytokine expression, especially under chronic stress [90]. Reward circuitry, including the nucleus accumbens/ventral striatum, is less well studied in PET recently, but fMRI studies confirm that inflammation correlates with blunted reward response and reduced connectivity [91].
The insula also shows elevated TSPO binding in PET studies of depression [90,92]. Functional imaging suggests altered insular activity during interoceptive and affective tasks in patients with high inflammatory states, consistent with abnormal salience processing.
3.5. Individual Differences in Immune Reactivity
Sex, age, and hormonal milieu are key modulators of neuroimmune signaling and contribute to the heterogeneity observed in the inflammation–depression link. Women generally mount stronger innate and adaptive immune responses than men, which has been associated with greater vulnerability to inflammation-related depressive phenotypes [93]. Sex steroids shape these processes in context-dependent ways. Under stable conditions, estrogens and progesterone can reduce microglial activation and dampen inflammatory signaling [94]. However, hormonal fluctuations during reproductive transitions (such as the peripartum and perimenopause) may destabilize neuroimmune balance and increase susceptibility to mood dysregulation. Perimenopause, in particular, is linked to higher rates of depression compared to premenopause, and estradiol withdrawal can trigger symptoms in vulnerable women [95,96,97]. Testosterone is generally associated with anti-inflammatory effects on myeloid cells, including reduced production of pro-inflammatory cytokines, although findings are context-dependent and not fully consistent across immune cell types and experimental settings [98]. Aging represents an additional and robust source of variability. Immunosenescence (decline in adaptive immune competence) and “inflammaging” (persistent, low-grade increases in IL-6, TNF-α, and CRP) create a biological context that enhances vulnerability to late-life depression [99]. Aging-related immune changes have been associated with higher depressive burden and stronger reactivity to inflammatory stressors in older adults [100,101].
Taken together, sex, age, and hormonal status introduce systematic variability in immune reactivity. Considering these biological axes is essential to improve the interpretation of biomarker findings and to refine phenotyping in mood–immune research.
3.6. Therapeutic Implications and Stratification Approaches
Translational research strongly suggests that immune–inflammatory mechanisms contribute to mood disorders and may be targeted therapeutically, but effectiveness appears contingent upon biological enrichment [102,103]. Adjunctive celecoxib has demonstrated antidepressant efficacy in MDD and antimanic effects in BD, while infliximab improved symptoms only in patients with elevated baseline inflammation [80]. These findings underscore that anti-inflammatory strategies are not universally effective but may be beneficial in specific subgroups defined by immune activation [60]. Other experimental approaches expand this rationale. Antioxidants and mitochondrial modulators such as N-acetylcysteine aim to counteract the oxidative stress and bioenergetic dysfunction outlined in Section 3.3.1 [17,74].
Alterations in the kynurenine pathway, described in Section 3.3.4, justify growing scientific interest given their link with suicidality and treatment resistance [30,82]. Likewise, the gut microbiome, discussed in Section 3.1.1, has emerged as another axis of translational relevance, with early probiotic and prebiotic interventions showing modest but inconsistent benefits [104].
Together, these strategies highlight that inflammation intersects with metabolic, mitochondrial, and neurotrophic domains, reinforcing the importance of integrated treatment concepts [105]. Clinical stratification efforts mirror this complexity. Biomarkers such as CRP and IL-6, the most robustly replicated findings across studies, have shown prognostic and predictive utility, including differential response to SSRIs versus noradrenergic/dopaminergic antidepressants [72].
Cellular immune phenotyping, neuroimaging evidence of regional neuroinflammation, BBB dysfunction, and multi-omics signatures integrating immune, metabolic, and genetic data further support the operationalization of an “inflammatory biotype” [38,78].
Such stratification is critical to improve reproducibility and translational impact, paving the way for biomarker-enriched and adaptive trial designs. Finally, rapid-acting antidepressants exemplify how mechanistic insights can transform clinical care. Ketamine/esketamine, through NMDA receptor antagonism, glutamate surge, and downstream restoration of BDNF/TrkB signaling, represents the first agent to demonstrate robust, rapid effects beyond monoaminergic modulation [106,107].
Collectively, these developments signal a paradigm shift: monoamine dysfunction alone is insufficient to explain treatment response, and future strategies must incorporate immune, metabolic, and neuroplastic dimensions [105]. Potential therapeutic targets emerging from neuroinflammation research are summarized in Figure 4.
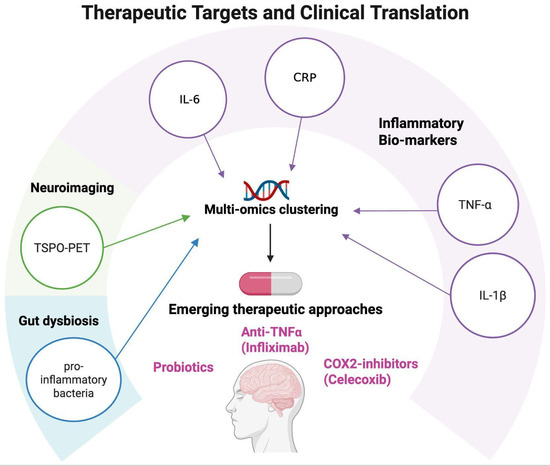
Figure 4.
Therapeutic targets and clinical translation. Schematic representation of potential therapeutic targets emerging from neuroinflammation research in mood disorders. Peripheral biomarkers (CRP, IL-6, TNF-α, and IL-1β), neuroimaging markers (TSPO-PET), and multi-omics clustering approaches can help identify patient subgroups with distinct immune–inflammatory profiles. These insights open the way to personalized interventions, including the use of anti-inflammatory agents such as COX-2 inhibitors (e.g., celecoxib), anti-TNF-α treatments (e.g., infliximab), and probiotic interventions, supporting a translational framework for precision psychiatry. Created with BioRender (web-based version). Pinzi, M. (2025) https://BioRender.com/3akca2h (accessed on 14 September 2025).
3.7. Conflicting Clinical Findings and Study Heterogeneity
Despite encouraging signals, clinical translation of immunomodulatory strategies remains inconsistent, and results often diverge across trials [102,108]. For instance, while infliximab produced benefits in patients with CRP > 5 mg/L, it failed to outperform placebo at the group level in treatment-resistant depression [80].
Similarly, meta-analyses of COX-2 inhibitors report beneficial effects in MDD but less consistent findings in BD, with substantial heterogeneity across populations and study designs [109]. These discrepancies indicate that immune-based interventions cannot be considered universally effective, but rather context-dependent [60]. Neuroimaging further illustrates this variability. Some TSPO-PET studies have demonstrated robust increases in microglial activation in corticolimbic regions, whereas others found more modest or null results, likely due to differences in tracers, patient characteristics, and illness stage [36,78].
At the biomarker level, CRP and IL-6 elevations remain the most consistent findings but results for TNF-α and IL-1β are highly heterogeneous, influenced by comorbidities, medication exposure, and methodological variability [105].
Microbiome interventions show inconsistent clinical benefits despite a strong preclinical rationale [104]. These conflicting results highlight two key lessons. First, they underscore that immune dysregulation is not a ubiquitous feature of mood disorders but likely marks a biologically distinct subgroup, consistent with the concept of an “inflammatory biotype” already emphasized in Section 3.6 [38,63]. Second, they emphasize the need for rigorous stratification to separate true therapeutic signals from noise introduced by sample heterogeneity, methodological diversity, and illness-stage effects.
By explicitly recognizing these inconsistencies, the field can avoid premature generalization and instead refine biomarker-guided approaches that integrate immune, metabolic, and neuroplastic domains.
3.8. Causality and Directionality of the Inflammation–Mood Link
A central question in immunopsychiatry is whether inflammation causes mood disorders or, conversely, arises as their consequence. Current evidence indicates that the relationship is bidirectional. On the one hand, immune activation can precipitate depressive symptoms. Up to 40–50% of patients treated with interferon-α for hepatitis C or melanoma develop depression, an effect mitigated by prophylactic antidepressants [110,111]. Experimental endotoxin challenge in healthy volunteers similarly induces transient increases in IL-6 and TNF-α, accompanied by anhedonia and dysphoria [112].
Conversely, depression itself promotes inflammatory activity via HPA-axis dysregulation, autonomic imbalance, sleep disturbance, and metabolic changes. Prospective cohorts demonstrate this reverse pathway, with depressive symptoms predicting later increases in CRP and IL-6 [113].
Overall, the most reliable conclusion is that the relationship is cyclical: immune activation can trigger depressive symptoms in experimental and clinical models, while depressive states can amplify inflammatory activity in turn. These reciprocal dynamics, moderated by metabolic and demographic factors, underscore the need for biomarker-guided stratification in future clinical trials. An integrated overview of the evidence discussed in this section is provided in Table 1 (MDD) and Table 2 (bipolar and transdiagnostic studies).

Table 1.
Evidence on immune–inflammatory dysregulation in major depressive disorder (MDD). This table summarizes 12 studies investigating the role of neuroinflammation in major depressive disorder. The works are grouped by thematic domains (peripheral biomarkers, kynurenine pathway, neuroimaging, oxidative stress, gut–brain axis, and interventional studies). For each study, the experimental design, sample characteristics, key findings, and clinical implications are reported. The overall picture highlights how specific patient subgroups exhibit distinct immune–inflammatory profiles that may guide precision medicine strategies.
Table 2.
Evidence on Immune–inflammatory dysregulation in bipolar disorder and transdiagnostic studies. This table integrates 8 studies, including both disorder-specific evidence in bipolar disorder and transdiagnostic works that jointly investigated MDD, BD, and, in some cases, schizophrenia. The studies cover some of the same thematic areas as in Table 1 (biomarkers, kynurenine pathway, therapeutic interventions, and multi-omics approaches). Together, these findings highlight shared and disorder-specific immune–inflammatory signatures, supporting the hypothesis of common inflammatory pathways and the need for integrative approaches across mood disorders.
4. Conclusions
Mounting evidence supports the existence of an inflammatory biotype in mood disorders, characterized by systemic cytokine elevations, intracellular signaling through NF-κB, JAK/STAT, MAPK, and NLRP3 pathways, and downstream effects on mitochondria, glia, synaptic plasticity, and kynurenine metabolism. Interfaces such as the gut–brain axis and blood–brain barrier provide mechanistic bridges between peripheral immune activation and central neurobiology, delineating a subgroup of patients with greater severity, cognitive impairment, and reduced treatment response. From a translational perspective, these findings highlight both challenges and opportunities. While anti-inflammatory and immunomodulatory interventions have shown promise, their efficacy appears contingent on baseline immune status, reinforcing the need for biomarker-based patient selection. Future research should therefore prioritize the development of standardized biomarker panels that integrate inflammatory, metabolic, and neuroimaging measures, enabling reproducible stratification across cohorts. Large-scale, adaptive clinical trials are needed to test immunomodulatory, metabolic, and microbiome-targeted interventions in biologically defined subgroups. Integration of multi-omics approaches with machine learning and longitudinal designs will be crucial to unravel causal pathways, identify predictive signatures, and refine treatment algorithms. In parallel, novel therapeutic targets—including inflammasome inhibition, mitochondrial restoration, microbiome modulation, and enhancement of BDNF/TrkB signaling—should be systematically evaluated as adjunctive strategies to conventional monoaminergic agents. Collectively, these directions delineate a roadmap toward a precision psychiatry framework, in which mechanistic insights translate into personalized prevention and treatment of mood disorders.
Author Contributions
Conceptualization, M.P., A.F. and A.C.; methodology, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; validation, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; formal analysis, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; investigation, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; resources, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; data curation, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; writing—original draft preparation, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; writing—review and editing, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; visualization, M.P., A.F., A.C., D.K., G.G., M.B.R., C.P., S.P. and B.P.; supervision, M.P., A.F. and A.C.; project administration, M.P., A.F. and A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
Andrea Fagiolini has received research grants and/or has been a consultant and/or a speaker for Allergan, Angelini, Apsend, Generici DOC, Lundbeck, Italfar-maco, Janssen, Otsuka, Pfizer, Recordati, Roche, Sanofi Aventis, and Sunovion; Alessandro Cuomo is/has been a consultant and/or a speaker for Angelini, Glaxo Smith Kline, Lundbeck, Janssen, Otsuka, Pfizer, and Recordati.
References
- Pastuszak, M.; Cubała, W.J.; Kwaśny, A.; Mechlińska, A. The Search for Consistency in Residual Symptoms in Major Depressive Disorder: A Narrative Review. J. Pers. Med. 2024, 14, 828. [Google Scholar] [CrossRef]
- Aphisitphinyo, S.; Lapid, M.; Coombes, B.; Frank, J.; Gentry, M.; Frye, M. Residual and Subsyndromal Bipolar Symptoms in Older Versus Younger Adults. Am. J. Geriatr. Psychiatry Open Sci. Educ. Pract. 2024, 5, 76–87. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6. [Google Scholar]
- Blackburn, T.P. Depressive disorders: Treatment failures and poor prognosis over the last 50 years. Pharmacol. Res. Perspect. 2019, 7, e00472. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Aggio, V.; Pratesi, M.L.; Greco, G.; Furlan, R. Neuroinflammation in Bipolar Depression. Front. Psychiatry 2020, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Poletti, S.; Mazza, M.G.; Benedetti, F. Inflammatory mediators in major depression and bipolar disorder. Transl. Psychiatry 2024, 14, 247. [Google Scholar] [CrossRef]
- Solmi, M.; Suresh Sharma, M.; Osimo, E.F.; Fornaro, M.; Bortolato, B.; Croatto, G.; Miola, A.; Vieta, E.; Pariante, C.M.; Smith, L.; et al. Peripheral levels of C-reactive protein, tumor necrosis factor-α, interleukin-6, and interleukin-1β across the mood spectrum in bipolar disorder: A meta-analysis of mean differences and variability. Brain Behav. Immun. 2021, 97, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef]
- Halaris, A.; Prochaska, D.; Stefanski, A.; Filip, M. C-reactive protein in major depressive disorder: Promise and challenge. J. Affect. Disord. Rep. 2022, 10, 100427. [Google Scholar] [CrossRef]
- Felger, J.C.; Haroon, E.; Patel, T.A.; Goldsmith, D.R.; Wommack, E.C.; Woolwine, B.J.; Le, N.A.; Feinberg, R.; Tansey, M.G.; Miller, A.H. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol. Psychiatry 2020, 25, 1301–1311. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, J.; Chen, H.; Li, J.; Chen, M. Association between hs-CRP and depressive symptoms: A cross-sectional study. Front. Psychiatry 2024, 15, 1339208. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflammation 2013, 10, 906. [Google Scholar] [CrossRef]
- Maes, M.; Kubera, M.; Leunis, J.C. The gut-brain barrier in major depression: Intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol. Lett. 2008, 29, 117–124. [Google Scholar]
- Skonieczna-Żydecka, K.; Marlicz, W.; Misera, A.; Koulaouzidis, A.; Łoniewski, I. Microbiome-The Missing Link in the Gut-Brain Axis: Focus on Its Role in Gastrointestinal and Mental Health. J. Clin. Med. 2018, 7, 521. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, P.; Li, Y.; Wu, J.; Tan, X.; Zhou, J.; Sun, Z.; Chen, X.; Zhang, G.; Zhang, H.; et al. Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders. Sci Adv. 2020, 6, eaba8555. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Russo, S.J.; Ferguson, D.; Nestler, E.J.; Duman, R.S. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. USA 2010, 107, 2669–2674. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef]
- Liu, T.; Zhong, S.; Liao, X.; Chen, J.; He, T.; Lai, S.; Jia, Y. A Meta-Analysis of Oxidative Stress Markers in Depression. PLoS ONE 2015, 10, e0138904. [Google Scholar] [CrossRef]
- Rowland, T.; Perry, B.I.; Upthegrove, R.; Barnes, N.; Chatterjee, J.; Gallacher, D.; Marwaha, S. Neurotrophins, cytokines, oxidative stress mediators and mood state in bipolar disorder: Systematic review and meta-analyses. Br. J. Psychiatry 2018, 213, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef]
- Fernandes, B.S.; Molendijk, M.L.; Köhler, C.A.; Soares, J.C.; Leite, C.M.G.S.; Machado-Vieira, R.; Ribeiro, T.L.; Silva, J.C.; Sales, P.M.G.; Quevedo, J.; et al. Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: A meta-analysis of 52 studies. BMC Med. 2015, 13, 289. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Williams, R.O. Tryptophan metabolism as a ‘reflex’ feature of neuroimmune communication: Sensor and effector functions for the indoleamine-2, 3-dioxygenase kynurenine pathway. J. Neurochem. 2024, 168, 3333–3357. [Google Scholar] [CrossRef] [PubMed]
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2021, 26, 4158–4178. [Google Scholar] [CrossRef]
- Christmas, D.M.; Potokar, J.P.; Davies, S.J. A biological pathway linking inflammation and depression: Activation of indoleamine 2,3-dioxygenase. Neuropsychiatr. Dis. Treat. 2011, 7, 431. [Google Scholar] [CrossRef] [PubMed]
- Almulla, A.F.; Thipakorn, Y.; Vasupanrajit, A.; Abo Algon, A.A.; Tunvirachaisakul, C.; Hashim Aljanabi, A.A.; Oxenkrug, G.; Al-Hakeim, H.K.; Maes, M. The tryptophan catabolite or kynurenine pathway in major depressive and bipolar disorder: A systematic review and meta-analysis. Brain Behav. Immun. Health 2022, 26, 100537. [Google Scholar] [CrossRef] [PubMed]
- Paul, E.R.; Schwieler, L.; Erhardt, S.; Boda, S.; Trepci, A.; Kämpe, R.; Asratian, A.; Holm, L.; Yngve, A.; Dantzer, R.; et al. Peripheral and central kynurenine pathway abnormalities in major depression. Brain Behav. Immun. 2022, 101, 136. [Google Scholar] [CrossRef]
- Inam, M.E.; Fernandes, B.S.; Salagre, E.; Grande, I.; Vieta, E.; Quevedo, J.; Zhao, Z. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A systematic review and meta-analysis of cerebrospinal fluid studies. Braz J. Psychiatry. 2023, 45, 343–355. [Google Scholar] [CrossRef]
- Köhler, O.; Krogh, J.; Mors, O.; Benros, M.E. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732. [Google Scholar] [CrossRef]
- Eggerstorfer, B.; Kim, J.H.; Cumming, P.; Lanzenberger, R.; Gryglewski, G. Meta-analysis of molecular imaging of translocator protein in major depression. Front. Mol. Neurosci. 2022, 15, 981442. [Google Scholar] [CrossRef]
- Hagenberg, J.; Brückl, T.M.; Erhart, M.; Kopf-Beck, J.; Ködel, M.; Rehawi, G.; Röh-Karamihalev, S.; Sauer, S.; Yusupov, N.; Rex-Haffner, M.; et al. Dissecting depression symptoms: Multi-omics clustering uncovers immune-related subgroups and cell-type specific dysregulation. Brain Behav. Immun. 2025, 123, 353–369. [Google Scholar] [CrossRef]
- Mokhtari, A.; Porte, B.; Belzeaux, R.; Etain, B.; Ibrahim, E.C.; Marie-Claire, C.; Lutz, P.E.; Delahaye-Duriez, A. The molecular pathophysiology of mood disorders: From the analysis of single molecular layers to multi-omic integration. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 116, 110520. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, R.; Yang, B.; Han, J.; Li, Y. Dissecting the shared genetic landscape of anxiety, depression, and schizophrenia. J. Transl. Med. 2024, 22, 373. [Google Scholar] [CrossRef] [PubMed]
- Gal, Z.; Torok, D.; Gonda, X.; Eszlari, N.; Anderson, I.M.; Deakin, B.; Petschner, P.; Juhasz, G.; Bagdy, G. New Evidence for the Role of the Blood-Brain Barrier and Inflammation in Stress-Associated Depression: A Gene-Environment Analysis Covering 19,296 Genes in 109,360 Humans. Int. J. Mol. Sci. 2024, 25, 11332. [Google Scholar] [CrossRef]
- Dutcher, E.G.; Pama, E.A.C.; Lynall, M.E.; Khan, S.; Clatworthy, M.R.; Robbins, T.W.; Bullmore, E.T.; Dalley, J.W. Early-life stress and inflammation: A systematic review of a key experimental approach in rodents. Brain Neurosci. Adv. 2020, 4, 2398212820978049. [Google Scholar] [CrossRef]
- Paganin, W.; Signorini, S. Inflammatory biomarkers in depression: Scoping review. BJPsych Open 2024, 10, e165. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef]
- Simpson, C.A.; Mu, A.; Haslam, N.; Schwartz, O.S.; Simmons, J.G. Feeling down? A systematic review of the gut microbiota in anxiety/depression and irritable bowel syndrome. J. Affect. Disord. 2020, 266, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Schiweck, C.; Dalile, B.; Balliet, A.; Aichholzer, M.; Reinken, H.; Erhardt, F.; Freiling, J.; Bouzouina, A.; Uckermark, C.; Reif, A.; et al. Circulating short-chain fatty acids are associated with depression severity and predict remission from major depressive disorder. Brain Behav. Immun. Health 2025, 48, 101070. [Google Scholar] [CrossRef]
- Huang, R.; Wang, K.; Hu, J. Effect of Probiotics on Depression: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8, 483. [Google Scholar] [CrossRef]
- Jang, B.S.; Kim, H.; Lim, S.W.; Jang, K.W.; Kim, D.K. Serum S100B Levels and Major Depressive Disorder: Its Characteristics and Role in Antidepressant Response. Psychiatry Investig. 2008, 5, 193–198. [Google Scholar] [CrossRef]
- Ambrée, O.; Bergink, V.; Grosse, L.; Alferink, J.; Drexhage, H.A.; Rothermundt, M.; Arolt, V.; Birkenhäger, T.K. S100B Serum Levels Predict Treatment Response in Patients with Melancholic Depression. Int. J. Neuropsychopharmacol. 2015, 19, pyv103. [Google Scholar] [CrossRef]
- Schroeter, M.L.; Abdul-Khaliq, H.; Diefenbacher, A.; Blasig, I.E. S100B is increased in mood disorders and may be reduced by antidepressive treatment. Neuroreport 2002, 13, 1675–1678. [Google Scholar] [CrossRef]
- Arolt, V.; Peters, M.; Erfurth, A.; Wiesmann, M.; Missler, U.; Rudolf, S.; Kirchner, H.; Rothermundt, M. S100B and response to treatment in major depression: A pilot study. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2003, 13, 235–239. [Google Scholar] [CrossRef]
- Rybakowski, J.K.; Remlinger-Molenda, A.; Czech-Kucharska, A.; Wojcicka, M.; Michalak, M.; Losy, J. Increased serum matrix metalloproteinase-9 (MMP-9) levels in young patients during bipolar depression. J. Affect. Disord. 2013, 146, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodriguez, E.M.; Beurel, E. Blood brain barrier and inflammation in depression. Neurobiol. Dis. 2022, 175, 105926. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Medicine 2015, 21, S6–S12. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox Control of Microglial Function: Molecular Mechanisms and Functional Significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, D.; Caso, J.R.; Javier Meana, J.; Callado, L.F.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Intracellular inflammatory and antioxidant pathways in postmortem frontal cortex of subjects with major depression: Effect of antidepressants. J. Neuroinflamm. 2018, 15, 251. [Google Scholar] [CrossRef]
- Osimo, E.F.; Baxter, L.J.; Lewis, G.; Jones, P.B.; Khandaker, G.M. Prevalence of low-grade inflammation in depression: A systematic review and meta-analysis of CRP levels. Psychol. Medicine 2019, 49, 1958–1970. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, Y.; Wang, X.; Jiang, Z.; Zhu, L.; Fang, S. Neutrophil-to-Lymphocyte Ratio, Platelet-to-Lymphocyte Ratio, and Monocyte-to-Lymphocyte Ratio in Depression: An Updated Systematic Review and Meta-Analysis. Front. Psychiatry 2022, 13, 893097. [Google Scholar] [CrossRef]
- Lynall, M.E.; Turner, L.; Bhatti, J.; Cavanagh, J.; De Boer, P.; Mondelli, V.; Jones, D.; Drevets, W.C.; Cowen, P.; Harrison, N.A.; et al. Peripheral Blood Cell–Stratified Subgroups of Inflamed Depression. Biol. Psychiatry 2020, 88, 185–196. [Google Scholar] [CrossRef]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 2010, 16, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Yue, N.; Huang, H.; Zhu, X.; Han, Q.; Wang, Y.; Li, B.; Liu, Q.; Wu, G.; Zhang, Y.; Yu, J. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J. Neuroinflamm. 2017, 14, 102. [Google Scholar] [CrossRef]
- Placeres-Uray, F.; Gorthy, A.S.; Torres, M.D.; Atkins, C.M. Inhibition of microglia priming by NLRP3 reduces the impact of early life stress and mild TBI. J. Neuroinflamm. 2025, 22, 185. [Google Scholar] [CrossRef]
- Alcocer-Gómez, E.; Casas-Barquero, N.; Williams, M.R.; Romero-Guillena, S.L.; Cañadas-Lozano, D.; Bullón, P.; Sánchez-Alcazar, J.A.; Navarro-Pando, J.M.; Cordero, M.D. Antidepressants induce autophagy dependent-NLRP3-inflammasome inhibition in Major depressive disorder. Pharmacol. Res. 2017, 121, 114–121. [Google Scholar] [CrossRef]
- Taylor, M.J.; Freemantle, N.; Geddes, J.R.; Bhagwagar, Z. Early onset of selective serotonin reuptake inhibitor antidepressant action: Systematic review and meta-analysis. Arch. Gen. Psychiatry 2006, 63, 1217–1223. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet Lond. Engl. 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D.; et al. Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef]
- Uher, R.; Tansey, K.E.; Dew, T.; Maier, W.; Mors, O.; Hauser, J.; Dernovsek, M.Z.; Henigsberg, N.; Souery, D.; Farmer, A.; et al. An Inflammatory Biomarker as a Differential Predictor of Outcome of Depression Treatment With Escitalopram and Nortriptyline. Am. J. Psychiatry 2014, 171, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Sacher, J.; Neumann, J.; Fünfstück, T.; Soliman, A.; Villringer, A.; Schroeter, M.L. Mapping the depressed brain: A meta-analysis of structural and functional alterations in major depressive disorder. J. Affect. Disord. 2012, 140, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-Acetyl Cysteine as a Glutathione Precursor for Schizophrenia—A Double-Blind, Randomized, Placebo-Controlled Trial. Biol. Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef]
- Banasr, M.; Duman, R.S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 2008, 64, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Duman, R.S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. USA 2008, 105, 751–756. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of Translocator Protein Density, a Marker of Neuroinflammation, in the Brain During Major Depressive Episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef]
- Attwells, S.; Setiawan, E.; Rusjan, P.M.; Xu, C.; Kish, S.J.; Vasdev, N.; Houle, S.; Santhirakumar, A.; Meyer, J.H. A double-blind placebo-controlled trial of minocycline on translocator protein distribution volume in treatment-resistant major depressive disorder. Transl. Psychiatry 2021, 11, 334. [Google Scholar] [CrossRef]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression: The Role of Baseline Inflammatory Biomarkers. JAMA Psychiatry 2013, 70, 31. [Google Scholar] [CrossRef]
- Castrén, E.; Antila, H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 2017, 22, 1085–1095. [Google Scholar] [CrossRef]
- Sublette, M.E.; Galfalvy, H.C.; Fuchs, D.; Lapidus, M.; Grunebaum, M.F.; Oquendo, M.A.; Mann, J.J.; Postolache, T.T. Plasma kynurenine levels are elevated in suicide attempters with major depressive disorder. Brain Behav. Immun. 2011, 25, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef]
- Öngür, D.; Drevets, W.C.; Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. USA 1998, 95, 13290. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Chang, Y.H.; Xie, X.T.; Wang, X.Y.; Ma, H.Y.; Liu, M.C.; Zhang, H.M. PET Imaging Unveils Neuroinflammatory Mechanisms in Psychiatric Disorders: From Microglial Activation to Therapeutic Innovation. Mol. Neurobiol. 2025, in press. [Google Scholar] [CrossRef]
- Radley, J.J.; Rocher, A.B.; Miller, M.; Janssen, W.G.M.; Liston, C.; Hof, P.R.; McEwen, B.S.; Morrison, J.H. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex 2006, 16, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Zhang, J. Neuroinflammation, memory, and depression: New approaches to hippocampal neurogenesis. J. Neuroinflamm. 2023, 20, 283. [Google Scholar] [CrossRef]
- Fang, S.; Wu, Z.; Guo, Y.; Zhu, W.; Wan, C.; Yuan, N.; Chen, J.; Hao, W.; Mo, X.; Guo, X.; et al. Roles of microglia in adult hippocampal neurogenesis in depression and their therapeutics. Front. Immunol. 2023, 14, 1193053. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, D.; Yu, G.; Du, H.; Xu, L.; Cao, Y.; Cui, M.; Wang, W.; Wang, D.; Liu, J.; et al. Alleviation of Microglia Mediating Hippocampal Neuron Impairments and Depression-Related Behaviors by Urolithin B via the SIRT1-FOXO1 Pathway. CNS Neurosci. Ther. 2025, 31, e70379. [Google Scholar] [CrossRef]
- Meyer, J.H.; Cervenka, S.; Kim, M.J.; Kreisl, W.C.; Henter, I.D.; Innis, R.B. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry 2020, 7, 1064–1074. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef]
- Setiawan, E.; Attwells, S.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Xu, C.; Sharma, S.; Kish, S.; Houle, S.; et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: A cross-sectional study. Lancet Psychiatry 2018, 5, 339–347. [Google Scholar] [CrossRef]
- Bekhbat, M.; Neigh, G.N. Sex differences in the neuro-immune consequences of stress: Focus on depression and anxiety. Brain Behav. Immun. 2018, 67, 1–12. [Google Scholar] [CrossRef]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen Prevents the Lipopolysaccharide-Induced Inflammatory Response in Microglia. J. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [CrossRef]
- Zhu, J.; Jin, J.; Tang, J. Inflammatory pathophysiological mechanisms implicated in postpartum depression. Front. Pharmacol. 2022, 13, 955672. [Google Scholar] [CrossRef]
- Liang, G.; Kow, A.S.F.; Yusof, R.; Tham, C.L.; Ho, Y.C.; Lee, M.T. Menopause-Associated Depression: Impact of Oxidative Stress and Neuroinflammation on the Central Nervous System—A Review. Biomedicines 2024, 12, 184. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Ben Dor, R.; Martinez, P.E.; Guerrieri, G.M.; Harsh, V.L.; Thompson, K.; Koziol, D.E.; Nieman, L.K.; Rubinow, D.R. Effects of Estradiol Withdrawal on Mood in Women With Past Perimenopausal Depression: A Randomized Clinical Trial. JAMA Psychiatry 2015, 72, 714–726. [Google Scholar] [CrossRef]
- Vancolen, S.; Sébire, G.; Robaire, B. Influence of androgens on the innate immune system. Andrology 2023, 11, 1237–1244. [Google Scholar] [CrossRef]
- Wrona, M.V.; Ghosh, R.; Coll, K.; Chun, C.; Yousefzadeh, M.J. The 3 I’s of immunity and aging: Immunosenescence, inflammaging, and immune resilience. Front. Aging 2024, 5, 1490302. [Google Scholar] [CrossRef] [PubMed]
- Szymkowicz, S.M.; Gerlach, A.R.; Homiack, D.; Taylor, W.D. Biological factors influencing depression in later life: Role of aging processes and treatment implications. Transl. Psychiatry 2023, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.L.; Barbosa, I.G.; Bauer, M.E.; Miranda, A.S. Immunopsychiatry of late life depression: Role of ageing-related immune/inflammatory processes in the development and progression of depression. Acta Neuropsychiatr. 2025, 37, e67. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of Anti-inflammatory Treatment on Depression, Depressive Symptoms, and Adverse Effects: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Na, K.S.; Lee, K.J.; Lee, J.S.; Cho, Y.S.; Jung, H.Y. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: A meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 79–85. [Google Scholar] [CrossRef]
- Wallace, C.J.K.; Milev, R. The effects of probiotics on depressive symptoms in humans: A systematic review. Ann. Gen. Psychiatry 2017, 16, 14. [Google Scholar] [CrossRef]
- Halaris, A. Inflammation, Heart Disease, and Depression. Curr. Psychiatry Rep. 2013, 15, 400. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Ketamine and rapid-acting antidepressants: A new era in the battle against depression and suicide. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Q.; Wang, Q. Effect of celecoxib on improving depression: A systematic review and meta-analysis. World J. Clin. Cases 2022, 10, 7872–7882. [Google Scholar] [CrossRef]
- Gędek, A.; Szular, Z.; Antosik, A.Z.; Mierzejewski, P.; Dominiak, M. Celecoxib for Mood Disorders: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2023, 12, 3497. [Google Scholar] [CrossRef]
- Raison, C.L.; Woolwine, B.J.; Demetrashvili, M.F.; Borisov, A.S.; Weinreib, R.; Staab, J.P.; Zajecka, J.M.; Bruno, C.J.; Henderson, M.A.; Reinus, J.F.; et al. Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment. Pharmacol. Ther. 2007, 25, 1163–1174. [Google Scholar] [CrossRef]
- Schaefer, M.; Sarkar, R.; Knop, V.; Effenberger, S.; Friebe, A.; Heinze, L.; Spengler, U.; Schlaepfer, T.; Reimer, J.; Buggisch, P.; et al. Escitalopram for the Prevention of Peginterferon-α2a–Associated Depression in Hepatitis C Virus–Infected Patients Without Previous Psychiatric Disease. Ann. Intern. Med. 2012, 157, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-Induced Anhedonia: Endotoxin Reduces Ventral Striatum Responses to Reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.C.; Rand, K.L.; Muldoon, M.F.; Kamarck, T.W. A Prospective Evaluation of the Directionality of the Depression-Inflammation Relationship. Brain Behav. Immun. 2009, 23, 936–944. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).