Abstract
Huntington’s Disease (HD) is an inherited neurodegenerative condition caused by an expansion of CAG repeats in the Huntingtin (HTT) gene, leading to a toxic form of the HTT protein. Despite advances in understanding the disease and developing symptomatic treatments, effective therapies for modifying its progression remain limited. Among emerging and novel treatments for central nervous system (CNS) disorders, gene therapy (GT), particularly using adeno-associated virus (AAV)-mediated gene delivery, holds great promise. Numerous preclinical and clinical trials are exploring the benefits of AAVs for treating neurodegenerative and genetic diseases. However, while widely used and investigated in rare and genetic disease treatment, AAVs’ potential for HD treatment remains underexplored. The absence of a comprehensive collection of previous reports, advancements, and methodologies regarding exclusively AAV-mediated GT for HD is notable and prompted us to address this gap. The current review compiles the available and emerging information regarding the application of AAVs in HD therapy, outlines the promise of this approach, and highlights the necessity of conducting further studies to achieve efficient HD treatment. The authors hope that the current review will guide further research to unlock the full potential of AAVs in treating HD.
1. Introduction to Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative pathology with complete penetrance, caused by a CAG trinucleotide repeat expansion in the Huntingtin (HTT) gene, located on chromosome 4 [1]. This genetic alteration leads to the production of a mutated form of the HTT protein (known as mHTT), which is neurotoxic [1].
In 1842, Charles Oscar Waters reported the first case of Huntington’s chorea (HC) which was later termed as HD after George Huntington provided a powerful description in 1872 [2]. Over the years, our understanding of this disease has significantly improved, especially after the identification of the HTT gene and the CAG repeat mutation in 1993 [3]. These discoveries have been crucial for the development of diagnostic tools and have driven ongoing research into effective therapies [4].
HTT is a widely expressed, multifunctional protein that is essential for maintaining cellular homeostasis, comprising vesicular trafficking, endocytosis, axonal transport, transcriptional regulation, and autophagy [5,6]. HTT also associates with other protein complexes, enabling intracellular transport processes and sustaining synaptic activity [7]. In addition, HTT controls the transcription of neuronal survival-related genes by interacting with some transcription factors and chromatin-modifying proteins [8].
In HD patients, the mHTT protein is present at abnormally elevated levels [9], with the associated mechanisms outlined in Section 1.1. mHTT disrupts normal interactions between proteins, which causes misfolded protein aggregates to build up in both nuclear and cytoplasmatic compartments [10]. These aggregates destabilize the ubiquitin-proteasome system and autophagic pathways, impairing the clearance of damaged proteins and organelles [11]. Additionally, by interfering with several transcription factors (e.g., CBP) and disrupting histone acetylation, mHTT disrupts transcriptional regulation, causing broad alterations in neuronal transcriptional profiles [12,13]. mHT progressively impairs mitochondrial bioenergetics and perturbs Ca2+ signaling, thereby amplifying oxidative stress and excitotoxicity and promoting excitotoxic neuronal death [14]. Finally, mHTT strongly impacts synaptic integrity and function. Abnormal protein interactions prevent the proper trafficking and recycling of synaptic vesicles, regulate inhibitory signaling, causing altered release of neurotransmitter, changes in postsynaptic receptor composition, and impairments in synaptic plasticity [15,16,17].
1.1. Genetics and Epigenetics
The HTT gene, located on chromosome 4 (4p16.3), encompasses 67 exons and extends over a physical distance of 180 kb of genomic DNA (Figure 1), with transcription occurring from the telomere to the centromere [18]. Differential polyadenylation of the 3′ untranslated region (3’UTR), encoded in the final exon, results in the production of two transcripts (10.4 kb and 13.7 kb), which are found in some cell types, including neurons [19]. HD is caused by a dominant mutation with complete penetrance, leading to an increase in CAG trinucleotide repeats in the HTT gene, specifically within the first exon (Figure 1). This mutation results in the biosynthesis of an HTT protein with an abnormally elongated polyQ sequence [20]. The wild-type HTT protein (23 CAG repeats) is composed of 3144 amino acids (348 kDa), and is conserved across mammalian species, indicating its role in survival [21]. The HTT protein comprises an N-terminal region that includes various elements [22]: (i) the HTTNT, a 17 amino acid sequence, fundamental for nuclear export and endoplasmic reticulum targeting; (ii) the polyQ repeat, linked to pathology initiation; (iii) a proline-rich domain (PRD) that evokes protein aggregation. Proteolytic cleavage of N-terminal fragments (HTTNT, polyQ, and PRD) is more frequent in HD, and these fragments play a crucial role in HD pathogenesis [23]. The rest of the HTT protein is structured into clusters of anti-parallel alpha-helical HEAT repeats, which serve as scaffold motifs for other protein macromolecules [22]. The mHTT protein, primarily its N-terminal fragments, can form aggregates and interfere with cell signaling, axonal transport, transcription, translation, and synaptic function, while promoting apoptosis. These anomalies contribute to neuronal dysfunction and the progression of HD [24,25].
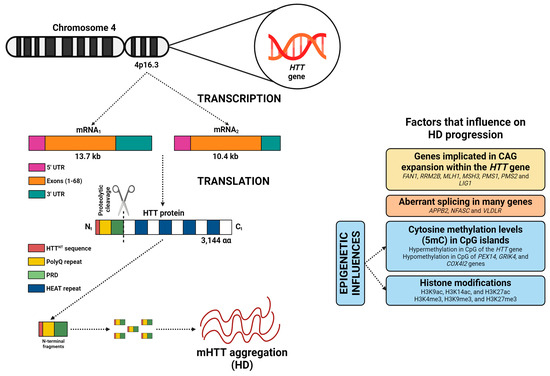
Figure 1.
The HTT gene contains 67 exons and produces two mRNAs (10.4 and 13.7 kb), differing by a 3′ UTR sequence. The resulting HTT protein includes an N-terminal region with a nuclear export signal (HTTNT), a polyglutamine repeat (polyQ), and a proline-rich domain (PRD), followed by clusters of anti-parallel alpha-helical HEAT repeats. In HD, the proteolytic cleavage of N-terminal fragments by peptidases is more frequent, and these fragments are believed to play a role in disease pathogenesis (by formation of mHTT protein). However, numerous factors significantly influence disease progression, including the involvement of genes associated with DNA repair, aberrant splicing, methylation levels of cytosines (5mC) in CpG islands across some genes, and the histone methylation and acetylation levels. Image adapted from BioRender (https://www.biorender.com/).
In unaffected individuals, CAG repeat lengths usually range from 17 to 20 [26]. However, once the CAG repeats reach 27 or more, the risk of developing HD increases exponentially [27,28,29,30]. Moreover, Handsaker et al. (2025) [31], using single-cell HTT-CAG and RNA sequencing, demonstrated that striatal projection neurons undergo somatic HTT CAG expansion from 40–45 to 100–500+ repeats. Expansions up to ~150 CAGs were largely tolerated, whereas further increases led to loss of neuronal identity, induction of senescence and apoptotic pathways, and neuronal death [31]. Finally, the largest genome-wide association study identified numerous genes participating in DNA repair which may promote the expansion of CAG repeats, including FAN1, RRM2B, MLH1, MSH3, PMS1, PMS2, and LIG1 (Figure 1) [32].
The age at symptom onset is partly predicted by the inherited number of CAG repeats, along with other factors such as aberrant splicing and epigenetic influences (Figure 1). mRNA splicing is pivotal in HD pathogenesis [33]. Numerous studies have shown that alternative splicing, a mechanism that allows the production of multiple mRNA variants and protein isoforms from a single gene, is altered in HD [34,35,36,37]. In HD, aberrant splicing affects genes related to neuronal function, including APPB2, NFASC, and VLDLR [38].
On the other hand, substantial research has been developed to identify the essential epigenetic factors that contribute to the pathogenesis of HD [39]. A newly published investigation has demonstrated that hypermethylation at CpG sites in the HTT gene, such as cg22982173, is linked to minor progression of HD (Figure 1). Conversely, hypomethylation at some CpG sites near genes including PEX14, GRIK4, and COX4I2, is linked to increased motor progression in HD patients [40]. Alterations in histone acetylation (e.g., H3K9ac, H3K14ac, and H3K27ac) and histone methylation (e.g., H3K4me3, H3K9me3, and H3K27me3) indicate that mHTT protein may impact chromatin structure by promoting epigenetic alterations (Figure 1) [41].
1.2. Signs and Symptoms
The clinical course of HD is divided into four stages of progression [42]: (i) stage 0 (symptomless period); (ii) stage 1 (onset of neurodegeneration); (iii) stage 2 (manifestation of a recognizable clinical signs, including cognitive and/or motor manifestations of HD); (iv) stage 3 (loss of functional ability and difficulties with daily activities).
HD signs and symptoms, which generally start between the ages of 35 and 40 [43], are classified into a triad of progressive motor, cognitive, and behavioral disruptions [44]. Motor dysfunction includes involuntary movements (e.g., chorea and dystonia) and complications with voluntary movements (e.g., dysphagia and akinesia) [45]. Cognitive impairments involve a decline in executive functions, along with memory loss [46]. Neuropsychiatric problems usually include anxiety, depression, aggression, and psychosis [47]. On the other hand, motor, cognitive, and psychiatric deficits may be detected prior to the onset of HD [35]. HD typically progresses over 15–20 years following the onset of motor dysfunction, with this duration not affected by the length of CAG repeats [43].
HD has traditionally been considered to affect only the central nervous system (CNS) [30,36], but recent evidence indicates that other organs are also affected, such as the heart, bones, skeletal muscle, liver, and digestive tract [48,49,50,51]. Further issues include circadian rhythm disruptions, leading to lower sleep efficiency [52].
1.3. Diagnosis
HD is diagnosed through either a confirmed family history or a positive genetic test. The gold standard for this detection is the DNA analysis, which identifies a CAG repeat count in the HTT gene [53]. In addition to the genetic confirmation, the diagnosis requires evidence of motor disorders, as assessed by the Total Motor Score (TMS) of the Unified Huntington’s Disease Rating Scale (UHDRS). The UHDRS-TMS, with a maximum score of 124 points, includes 15 items that evaluate chorea, dystonia, parkinsonism, motor performance, oculomotor function, and balance [54].
Numerous studies are now focusing on functional changes and alterations in brain imaging before the onset of HD symptoms. Evidence suggests that changes in brain volume and neural connections can be detected several years before clinical manifestations emerge [55]. Moreover, many biomarkers (associated with oxidative stress or immune alterations) are being investigated for the early detection of HD through blood analyses [56].
1.4. Epidemiology
HD is found in all human populations worldwide, although its prevalence varies across regions. A relatively high prevalence can be observed in several European countries: United Kingdom (12.3 cases per 100,000) [57], France (8.9 cases per 100,000) [58], Germany (9.26 cases per 100,000) [59], and Italy (10.85 cases per 100,000) [60]. Canada also displays a high prevalence (9.33 cases per 100,000) [61]. In contrast, HD prevalence is low in Asian countries (0.65 cases per 100,000 in Japan [62] and 0.42 cases per 100,000 in China [63]). These differences in prevalence are primarily related to ethnic variation in CAG repeats and generally prevalence is higher in populations with longer average CAG repeats [64].
In addition to the HD prevalence, its socioeconomic impact profoundly increases the overall burden on the health system and influences patients and families’ quality of life [65]. This underlines the urgent need for novel and effective therapies, making research crucial for achieving better outcomes in HD patients.
Aforementioned characteristic of HD as a genetic disorder, highlights the need for a robust support system to tackle the complexity of its treatment [66]. Following section will outline most current treatment options available for HD patients.
2. Current Treatment Strategies
Although a cure is not currently available for HD, a range of pharmacological interventions have been suggested that provide symptom relief [67,68]. Pharmacological treatments, outlined in Table 1, should be combined with other non-pharmacological strategies to enhance disease management, such as surgical therapies and various non-invasive approaches, which collectively aim to improve functional outcomes and quality of life [69].

Table 1.
List of commonly used drugs for treating symptoms associated with HD.
2.1. Pharmacological Therapies
This section explores the pharmacological strategies used in the management of HD, such as drugs to manage motor symptoms, psychiatric issues, and cognitive impairments.
2.1.1. Dopamine-Depleting Agents
Tetrabenazine (TBZ) and deutetrabenazine (DBZ) are dopamine-depleting pharmacologic agents that inhibit the VMAT2 transporter, reducing presynaptic dopamine levels and decreasing chorea [70]. TBZ was the first agent authorized for the treatment of HD-associated chorea [71]. DBZ is an isotopic isomer of TBZ in which six hydrogen atoms in TBZ are replaced with deuterium atoms. While DBZ retains the same function as TBZ, the presence of deuterium atoms extends its half-life, allowing for less frequent administration [74].
The most typical side effects of this compound are parkinsonism, drowsiness, weakness, depression, and acute akathisia, all of which can be mitigated by reducing the dose [72]. Other side effects include insomnia, anxiety, tremors, memory issues, confusion, dizziness, and nausea [73].
2.1.2. Antipsychotics
Although these drugs are primarily indicated for the treatment of psychosis, they also alleviate chorea symptoms through their interaction with the D2 receptor [75]. The following medications are utilized: (i) haloperidol (this drug selectively inhibits dopamine D2 receptors and is highly effective for treating severe chorea; clinicians continue to recommend haloperidol as the first-line antipsychotic due to its great cost-effectiveness) [76,77]; (ii) tiapride (tiapride, which blocks dopamine D2 and D3 receptors, is another cost-effective option for treating chorea in HD, with a mild and well-tolerated side effect profile) [78]; (iii) olanzapine (blocks dopamine -D1, D2, and D4-, serotonin -5-HT-; -5-HT2A and 5-HT2C-, histamine -H1-, α1-adrenergic, and muscarinic receptors; it is employed as an antipsychotic drug for managing symptoms associated with schizophrenia; recently, olanzapine has been used to treat behavioral symptoms; however, the use of this medication has led to significant side effects like dyslipidemia and weight gain) [79,80,81]; (iv) quetiapine (inhibits dopamine -D2- and 5-HT -5-HT2A- receptors; treatment with quetiapine was linked to reduction in choreiform movements and improvement in behavioral symptoms, like psychosis, agitation, irritability, and insomnia) [82,83,84]; (v) risperidone (blocks dopamine -D2- and 5-HT -5-HT2A- receptors, and is used in the management of schizophrenia; this medication improves behavioral and psychiatric assessment scores, as well as motor symptoms associated with HD; however, this drug promotes numerous side effects, like hyperprolactinemia, extrapyramidal symptoms -akathisia, parkinsonian symptoms, and acute dystonia-, weight gain, sedation, and alterations in electrical conductivity of the myocardium) [85,86,87]; (vi) apriprazole (apriprazole acts as a partial agonist at dopaminergic D2 and serotonergic 5-HT1A receptors, and as a selective antagonist at 5-HT2A receptors; it is well tolerated and has shown significant improvements in some motor and behavioral symptoms in HD patients) [88].
2.1.3. Antidepressants
Antidepressants are commonly prescribed to HD patients with depression [89]. Selective 5-HT reuptake inhibitors (SSRIs) are the main antidepressants used, as they inhibit the reuptake of 5-HT into the presynaptic nerve terminal. This situation increases extracellular 5-HT levels, thus facilitating receptor interaction and producing therapeutic outcomes [77]. The following drugs are used for the treatment of HD symptoms associated with depression: (i) citalopram (the use of this medication results in an improvement in depression symptoms) [91]; (ii) escitalopram (escitalopram, like citalopram, markedly reduces depressive symptoms associated with HD) [94]; (iii) fluoxetine (alleviates depression in the short-term, but there are limited clinical trials assessing its effectiveness in HD patients, emphasizing the necessity for additional research) [95,96]; (iv) sertraline (not only alleviates depression but has also been documented to completely solve aggression and obsessive–compulsive disorder symptoms related to HD) [97].
SSRIs are associated with some side effects, such as sexual dysfunction, and weight gain [92]. Some studies have also indicated an increased risk of suicide in adolescents taking SSRIs [93]. This risk factor has not been observed in adults using SSRIs.
2.1.4. Antiglutamatergics
Some studies have examined how glutamate excitotoxicity affects chorea in the context of HD [98]. Two main antiglutamatergic agents have proven to be effective: (i) amantadine (this drug is a non-competitive N-methyl-D-aspartate -NMDA- receptor antagonist; some studies have shown that amantadine provides modest benefits for HD chorea, as indicated by improvements in the UHDRS; high doses, needed for symptom relief, often lead to significant adverse effects, including hallucinations, forgetfulness, agitation, and sleepiness) [99,100,101]; (ii) riluzole (this antiglutamatergic agent has been tested for treating HD-associated chorea; it was found to reduce chorea symptoms in a dose-dependent manner, with higher doses displaying substantial effectiveness after 8 weeks of treatment; adverse effects included increased plasma levels of liver enzymes and a higher risk of suicide) [102,103].
2.1.5. Anticonvulsants
To stabilize mood disorders, HD patients are prescribed anticonvulsants, including lamotrigine and carbamazepine. Multiple studies have indicated that these drugs are effective in reducing the severity of symptoms associated with HD [104,105,106]. Traditionally, anticonvulsants were used to treat seizures caused by the excessive firing of synchronized neuronal populations [107]. Anticonvulsants reduce this synchrony through three mechanisms [108]: (i) modulation of voltage-gated ion channels; (ii) upregulation of inhibitory neurotransmitters; (iii) downregulation of excitatory neurotransmitters.
The following pharmacologic agents were prescribed: (i) valproate (is an anticonvulsant that increases GABA levels in the brain; when administered in combination with olanzapine, it has been observed to help manage agitation and aggression in patients with HD; this combination allows for a reduced dosage of the antipsychotic medication, which in turn decreases the possibility of side effects) [109,110]; (ii) carbamazepine (blocks voltage-gated sodium channels -VGSCs-; although carbamazepine is prescribed as a mood stabilizer for HD, no clinical research has shown specific benefits for HD patients) [112]; (iii) lamotrigine (this medication inhibits VGSCs and blocks the release of excitatory neurotransmitters glutamate and aspartate; while lamotrigine is primarily used a mood stabilizer in HD, certain investigations indicate that its ability to reduce excitatory neurotransmitter activity and excitotoxicity, a pathological feature of HD) [115,116]; (iv) levetiracetam (binds to the synaptic vesicle protein SV2A, which disrupts the release of excitatory neurotransmitters, such as glutamate, thus blocking neuronal firing and hypersynchronization; this medication effectively reduces involuntary movements associated with HD) [117,118].
Anticonvulsants can generate numerous side effects, including hypersensitivity reactions, blood dyscrasias, gastrointestinal problems, and depression [111]. Moreover, carbamazepine may lead to severe skin conditions including Stevens-Johnson syndrome or toxic epidermal necrolysis [113,114].
2.2. Surgical Therapies and Non-Invasive Approaches
Surgical interventions, such as deep brain stimulation (DBS), have shown considerable promise in alleviating the symptoms of chorea in HD patients [119]. However, the invasiveness of DBS, along with the associated risks and highly specialized care required, make it a treatment option that may not be suitable for everyone [120].
A comprehensive care approach involving multiple disciplines is necessary given the wide range of symptoms associated with HD. This group of experts would include physiotherapists, occupational therapists, speech-language pathologists (SLPs), and dietitians. Physiotherapists focus on improving mobility and balance with the aim of helping to prevent falls and maintain physical function [121]. Occupational therapists collaborate with to adapt their home and work environments to satisfy their needs [122]. SLPs aim to protect both communication and swallowing capabilities [123]. Dietitians provide guidance on nutritional strategies to combat weight loss and prevent malnutrition, common features in HD [124]. Psychological sessions can be used in conjunction with pharmacological treatments to alleviate the psychological and cognitive symptoms of HD. This approach improves significantly the mental health of patients with HD [125].
Current therapies for HD are notably restricted, mainly due to their inability to alter the disease’s progression. The complex pathophysiology of HD, along with the variability in clinical manifestations and the absence of reliable biomarkers for diagnosis and monitoring, presents major challenges in developing effective treatments. Commercial treatments in HD target symptoms without affecting the cause of the disease [67,68]. Additionally, due to its gradual progression, it is difficult to observe the effects of interventions in clinical trials [126]. These limitations highlight the need for experimental therapies that target the HD etiology, including gene silencing methods which would effectively prevent neurodegenerative processes. Developing new therapies is vital for addressing the unmet medical needs of HD patients and enhancing their quality of life [67].
3. AAV-Delivered Genetic Targeting in Huntington’s Disease
Gene therapy (GT) is a revolutionary medical method that utilizes genetic material as a therapeutic agent to treat genetic diseases. It normally involves the introduction of functional genes to the patients’ cells or the correction of defective genes by deletion or modification of genetic codes [127]. In the GT approach, genetic materials, including DNA and RNA molecules, are introduced to the cells through delivery vehicles such as viral and non-viral vectors [128,129].
Among available viral vectors, AAVs are the most extensively employed and clinically validated platform for in vivo GT [130]. Wild-type AAVs are small, nonenveloped, and non-pathogenic viruses from the Parvoviridae family. Their viral genome, which is composed of a 4.7 kb single-stranded linear DNA genome, is replaced by the transgene expression cassette to generate recombinant AAVs (rAAVs) useful for GT. The expression cassette in rAAVs is flanked by two inverted terminal repeats (ITRs) and packaged in various naturally occurring AAV serotypes with specific capsid features and tropisms [131,132]. rAAVs usually remain mostly episomal in transduced cells and rarely integrate into the host genome [133]. Despite having integration reports in animals [134], the direct relationship between AAVs and genotoxicity has not been reported yet. Although rAAVs can stimulate the immune system, produce minimal serious inflammatory responses [135,136], and provide high levels of sustainable expression [137,138]. They are being used as a vector of choice in numerous clinical trials and offer successful treatments to patients suffering from different rare disorders like spinal muscular atrophy, Duchenne muscular dystrophy, and hemophilia A and B disorders [139,140,141,142,143].
AAV’s ability to transduce non-dividing cells such as neurons has made them a valuable therapeutic option for genetic CNS disorders [144,145,146]. Regardless of this, genetic material transfer to the CNS has always been a challenge [147], and administration into the cerebrospinal fluid and intraparenchymal injection are suggested to ease the burden to some extent [148,149]. Nonetheless, the problem still stands with AAV’s ability to easily cross barriers into the CNS, thus a great deal of attempts is made to improve the precision and efficacy of AAV-mediated gene delivery to different neuronal populations [150,151]. For instance, most recently, even a focused ultrasound was used to temporarily disrupt the blood–brain barrier (BBB) to improve AAV delivery into the brain of an HD mouse model [152]. Up to date, several AAV serotypes are being utilized in GT programs, with some exhibiting high brain transduction efficacy. For instance, AAV9 penetrates the BBB and efficiently transduces CNS cells [153,154]. Moreover, it has been shown that the variant AAV-PHP.B achieves successful gene transfer throughout the CNS, up to 40 times more efficiently than AAV9 [155]. Common AAV serotypes that are currently used in preclinical and clinical studies for CNS disease are discussed elsewhere [156,157].
Although rAAV-mediated GT has been widely used and investigated for treating genetic diseases in both preclinical and clinical phases, its potential for HD treatment remains largely unexplored. Generally, GT for HD treatment could be feasible by reducing the production of the toxic HTT protein in affected cells. Among various GT approaches, vector expressed editing tools have emerged as a particularly promising strategy that have been able to slow neuronal degeneration in the brain of patients and experimental animals [158]. Although promising, relatively few studies, compared with other CNS diseases, have addressed the application of rAAV-mediated GT for HD in the last two decades, highlighting a significant opportunity for further research and improvement in this area. In this section, we chronologically present significant studies, with a summary outlined in Table 2. The studies summarized in Table 2 have mainly employed various targeting strategies, including RNA interference (RNAi), short hairpin RNA (shRNA), microRNA (miRNA), zinc-finger proteins, antibodies, and CRISPR/Cas9 systems. Each approach provides unique advantages and limitations. For instance, RNAi- and miRNA-based methods usually offer robust HTT silencing but might raise concerns about off-target effects. Zinc-finger and CRISPR/Cas9 systems offer higher specificity but face challenges of editing efficiency and long-term safety. Antibody-based strategies could reduce aggregates but do not resolve transcriptional dysregulations.
Table 2.
Chronological overview of rAAV-mediated GT studies for HD.
At the preclinical level, it has been shown that the rAAV5 delivered GT tool for RNAi can suppress the expression of mutant HTT in the R6/1 HD transgenic mouse and ameliorate the HD phenotype [159]. Additionally, an rAAV1-delivered RNAi mechanism has also been successful in suppressing mHTT at both the mRNA and protein levels in cell culture and in HD mouse brain. This treatment was able to improve the behavioral and neuropathological abnormalities associated with HD [160].
Similarly, rAAV5-mediated delivery of RNAi into the HD model mouse striatum after the onset of disease successfully ameliorated neuropathological abnormalities by inhibiting mutant gene expression [161]. The benefits of HTT silencing were also observed in a transgenic mouse model of HD, where siRNA delivered by rAAV1/8 attenuated neuronal pathology and delayed the abnormal behavioral phenotype [162]. Furthermore, the neuroprotective efficacy of shRNA-mediated knockdown of HTT expression was shown in a study where the GT tool was delivered to rats using rAAV-HD70 [163].
The therapeutic efficacy of rAAV1 delivered nonallele-specific RNAi therapeutics for HD was reported by a significant reduction in both wild-type and mutant HTT levels in HD mice, leading to improved motor coordination and survival [164]. Supporting the use of rAAV-mediated RNAi as a therapy for HD, another study demonstrated that partial suppression of wild-type HTT expression using a rAAV2/1 delivery system was well tolerated in the non-human primate putamen, without neuronal degeneration or immune response. A 45% decrease in HTT protein levels in the mid- and caudal putamen of rhesus monkeys did not lead to motor deficits or neuronal degeneration [165]. In HD patient iPSC-derived neuronal cultures, the efficacy of rAAV5-miHTT was demonstrated by a reduction in HTT mRNA and protein levels, without off-target effects in gene expression and regulation in neuronal cells and astrocytes [166].
Furthermore, the potential of rAAV2/1 as a delivery tool for synthetic zinc finger that suppresses mHTT expression at both mRNA and protein levels in the brain of the R6/2 HD mouse model was also addressed [167]. Bilateral delivery of an rAAV2 vector encoding an anti-HTT short hairpin RNA to the striatum of rhesus monkeys resulted in a reduction in HTT mRNA and protein levels without side effects up to 6 months post administration [168]. Intrajugular vein administration of rAAV9 expressing a mutant HTT-specific RNAi construct was proven effective in reducing mHTT expression in multiple brain regions and peripheral tissues affected in mice [169].
Another study reported the benefits of rAAV2/1-mediated RNAi in reducing both wild-type and mutant HTT in striatum cells of the YAC128 mouse model of HD. This treatment also led to improvements in behavioral deficits, and a reduction in HTT aggregation, without significant neurotoxicity following intracranial injections [170]. Similar findings were reported in several HD mouse models following one-time striatal rAAV-ZFP application, which led to HTT-lowering and improvement in histopathological, electrophysiological and biomarker deficits [171,172]. Another group of scientists highlighted the high potential of rAAV1 and rAAV2 to transduce the cortico-striatal tissues predominantly affected in HD in the non-human primate brain, suggesting RNAi-based therapy for targeting neurons that degenerate in HD [173].
The design of artificial microRNA-expression constructs, their incorporation into rAAV5, their total silencing of both wild-type and mtHTT, and their allele-specific silencing have been investigated and reported in vitro and in the humanized transgenic Hu128/21 HD mouse model [174]. Moreover, AAVs have also been found beneficial in developing antibody-based therapies for HD. For instance, rAAV6-INT41 was shown to reduce mHTT in the R6/2 mouse model [175,176]. The practicability of total and allele-selective HTT silencing induced by therapeutic microRNAs delivered in rAAVs to the brain without safety concerns has also been demonstrated. Researchers reported the successful suppression of mutant HTT aggregate formation in HD rats by intracerebral administration of rAAV5-miHTT-155. While this construct showed high efficiency, rAAV5-miSNP50T was proven to be more precise in mutant HTT allele selectivity [177].
Meanwhile, the rAAV-delivered CRISPR/Cas9 based edition of the mutant HTT allele was also reported to reduce human mutant expression in the treated hemispheres of BacHD mice [178]. Furthermore, it was shown that an rAAV-delivered CRISPR/Cas9-mediated system can suppress endogenous mHTT expression in the striatum of mHTT-expressing mice and mitigate neuropathology in the adult brain [179]. The therapeutic value of a single intracranial administration of rAAV5-miHTT for decreasing HTT levels of mRNA and protein was validated in a large animal model, a transgenic HD (tgHD) minipig model, in which both mRNA and protein levels of HTT were reduced in the transduced regions of the brain [180].
Disruption in the expression of the mHTT gene in the striatum of the R6/2 mouse model of HD was reported using the rAAV-delivered CRISPR-Cas9 system, resulting in decreased neuronal inclusions and improvements in lifespan and certain motor deficits [181]. It is worth mentioning that rAAV5-miHTT can reduce HTT protein in the striatum and cortex of Q175 HD mice in a dose-dependent manner, emphasizing the benefits of rAAV-mediated HTT-lowering GT for HD [182]. Non-targeted HTT downregulation with rAAV5-miHTT in the humanized Hu128/21 mouse model of HD was sustained for seven months, as addressed by Caron et al. (2020) [183].
A significant reduction in HTT gene expression in deeper tissues and outer layers of the brain of non-human primates by a gene therapy candidate, VY-HTT01, encapsulated in an rAAV1 delivering RNAi mechanism was presented at the 2018 edition of the ESGCT congress [184,185]. The safety and tolerability of rAAV5-miHTT in the non-human primate Macaca fascicularis and Sprague-Dawley rats were demonstrated by intrastriatal administration, with widespread vector DNA and miHTT transgene distribution in the brain areas associated with HD pathology [186]. The widespread biodistribution and durable efficiency of rAAV5-miHTT were also shown in HD-relevant regions of the minipigs brain [187]. There is also a report on the application of rAAV1 and rAAV2 in vitro and in vivo for HTT gene targeting using primary artificial miRNA (pri-amiRNA) delivered to numerous species, such as mice and non-human primates [188]. rAAV5-miHTT-mediated HTT suppression was reported to benefit brain health in an HD mouse model [189]. The neuroprotective effect of hepatoma-derived growth factor (HDGF) in HD treatment was described by employing rAAV8-mediated delivery of HDGF to the striatum of an HD mouse model, which could reduce mHTT; however, there were no significant changes in neurological phenotypes [190]. Finally, most recently, rAAV5 delivered engineered microRNA targeting the HTT exon 1 sequence (rAAV5-miHTT), could significantly reduce HTT mRNA and protein levels in the brain of HD mouse models, heterozygous zQ175 knock-in mice and humanized Hu128/21 mice [191]. Additional studies reporting the benefits of various rAAV serotypes to the HD treatment, along with insights into the disease’s mechanisms and pathogenesis are outlined elsewhere [194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211].
At the clinical level, the Phase I/II clinical study (clinical trial No. NCT04120493), a total HTT-lowering therapy, investigates the safety, tolerability, and efficacy of rAAV5-miHTT in adults with early manifest HD [192], which is widely described elsewhere [212]. The safety and efficacy of AMT-130 in European adults is addressed in the clinical trial No. NCT05243017 [213]. Additionally, the phase I/II clinical trial No. NCT05541627, evaluates the safety, tolerability, and preliminary efficacy of one-time intracerebral bilateral injections of AB-1001 (AAVrh10.CAG.hCYP46A1) in adults with early manifest HD. This rAAV-mediated GT aims to express the human cholesterol 24-hydroxylase gene within the striatum of individuals [193].
The GT studies mentioned here use different mechanisms to lower mutant HTT or reduce its toxic effects. Several approaches, including those using RNAi, shRNA, and miRNA, silence HTT mRNA, leading to lower protein levels and fewer aggregates. Zinc-finger proteins and CRISPR/Cas9 act directly at the DNA level, and this offers higher specificity but raises long-term safety questions. Antibody-based methods aim to neutralize toxic protein fragments rather than preventing their production. Altogether, these approaches reduce mutant HTT burden and improve motor, cognitive, or survival outcomes in models, showing both promise and the challenges that remain for translation to patients.
4. Future Directions
AAVs have shown significant promise in the treatment of genetic and rare diseases, successfully bringing several therapeutic options from the lab to the patient’s bedside. Currently, many preclinical and clinical trials are being conducted for the treatment of genetic diseases by AAVs, with neurodegenerative diseases attracting growing interest for their therapeutic possibilities. Among therapeutic options, AAVs ability to transduce various CNS cells has made them valuable therapeutic and research tools for CNS disorders such as HD. AAV-delivered therapeutic transgenes remain episomal in target cells, which ensures their stability in non-dividing cells and long-term expression of therapeutic tools. This characteristic is particularly advantageous in the treatment of HD, in which the primary concern is with non-dividing cells, specifically neurons. Consequently, a single dose administration may provide a lifelong therapy for HD patients [158]. It is worth mentioning that recent clinical trials with antisense oligonucleotides (ASOs), such as those targeting HTT mRNA, have not shown sustained efficacy in HD, mainly due to limited CNS penetration and the need for repeated dosing [214]. In contrast, AAV-based GT may overcome these obstacles by enabling widespread CNS transduction and long-term expression of therapeutic material after a single administration.
As noted in the literature review, AAVs are not only useful in HD modeling [163,215,216,217], but they also hold a great deal of promise for treating HD. Generally, AAV-mediated therapeutic strategies for treating HD generally focus on the goal of reducing toxic HTT protein production. Reducing mutant HTT protein alleviates toxic effects, but its complete elimination is not recommended due to the critical functions of wild-type HTT, which include its key role in transcription regulation, normal mitochondrial activity, and protecting brain cells from apoptosis. Furthermore, its absence may lead to embryonic lethality and neurodegeneration. Meanwhile, the long-term consequences of HTT reduction in various organs of the human body, including the CNS, remain poorly understood [9,158,218,219,220,221]. In general, HTT reduction appears to have a safe and favorable profile, particularly when partially reduced [222].
On the other hand, despite its potential, particular challenges are also present in the developmnt of AAV-mediated HD treatment, originating from HD characteristics themselves and AAVs’ limitations. Efficient transduction of CNS cells as previously mentioned, correct tissue targeting, vector production challenges and limited cloning capacity of AAV (approximately 4.5–4.7 kb), immune response and safety precautions are among the important challenges. Advantages and disadvantages of common routes of AAV delivery to the CNS, including intraparenchymal delivery, intravenous delivery, and delivery to cerebrospinal fluid, are widely outlined elsewhere [149]. Moreover, the immune system’s response to AAV capsids and transgene products is well discussed before [136]. Another significant barrier in HD treatment by AAVs is the absence of an adequate animal model that accurately mimics the human condition by expressing both normal and mutant HTT at physiological levels [158]. It seems that a combination of comprehensive in vitro and in vivo studies is still needed to shed light on the practical aspects of HD treatment using AAVs as a GT delivery system. The translation of current knowledge into meaningful clinical improvements in affected patients is still lacking. The authors hope that the current review will help further unlock the full potential of AAVs in treating HD.
Author Contributions
Introduction to Huntington’s Disease, P.M.G. and M.G.-D.; Current Treatment Strategies, M.G.-D.; Current Treatment Strategies, P.M.G.; Future Directions, P.M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable. No new data were generated.
Acknowledgments
We gratefully acknowledge BioRender for providing a professional and scientifically rigorous platform that enabled the creation of high-quality graphical illustrations presented in this review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-HT | Serotonin |
| 5mC | 5-methylcytosine |
| AAV | Adeno-associated virus |
| APPB2 | Amyloid beta precursor protein binding protein 2 |
| ASO | Antisense oligonucleotide |
| BBB | Blood–brain barrier |
| CBP | CREB-binding protein |
| CNS | Central nervous system |
| COX4I2 | Cytochrome c oxidase subunit 4I2 |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| DBS | Deep brain stimulation |
| DBZ | Deutetrabenazine |
| ESGCT | European Society of Gene and Cell Therapy Annual Congress |
| FAN1 | FANCD2/FANCI-associated nuclease 1 |
| GABA | γ-aminobutyric acid |
| GRIK4 | Glutamate ionotropic receptor kainate type subunit 4 |
| GT | Gene therapy |
| HC | Huntington’s chorea |
| HD | Huntington’s disease |
| HDGF | Hepatoma binding growth factor |
| HEAT | Huntingtin, elongation factor 3, protein phosphatase 2A, and the yeast kinase TOR1 |
| HTT | Huntingtin |
| HTTNT | Huntingtin nuclear transport |
| iPSC | Induced pluripotent stem cell |
| ITR | Inverted terminal repeat |
| LIG1 | DNA ligase 1 |
| mHTT | Mutated huntingtin protein |
| miHTT | MicroRNA targeting huntingtin |
| miRNA | MicroRNA |
| MLH1 | MutL homolog 1 |
| MSH3 | MutS Homolog 3 |
| NFASC: | Neurofascin |
| NMDA | N-methyl-D-aspartate |
| PEX14 | Peroxisomal biogenesis factor 14 |
| PMS1 | PMS1 protein homolog 1 |
| PMS2 | PMS1 protein homolog 2 |
| polyQ | Polyglutamine |
| PRD | Proline-rich sequence recognition domains |
| pri-amiRNA | Primary artificial microRNA |
| RNAi | RNA interference |
| RRM2B | Ribonucleotide reductase regulatory TP53 inducible subunit M2B |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| SLP | Speech-language pathologist |
| SSRI | Selective serotonin reuptake inhibitor |
| SV2A | Synaptic vesicle glycoprotein 2A |
| TBZ | Tetrabenazine |
| TMS | Total motor score |
| UHDRS | Unified Huntington’s Disease Rating Scale |
| VGSC | Voltage-gated sodium channel |
| VLDLR | Very low-density lipoprotein receptor |
| VMAT2 | Vesicular monoamine transporter 2 |
| ZFP | Zinc finger protein |
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Vale, T.C.; Cardoso, F. Chorea: A Journey through History. Tremor Other Hyperkinet. Mov. 2015, 5, tre-5-296. [Google Scholar] [CrossRef]
- Wexler, N.S. Huntington’s disease: Advocacy driving science. Annu. Rev. Med. 2012, 63, 1–22. [Google Scholar] [CrossRef]
- La Spada, A.R. A Novel Therapy for Huntington’s Disease. Cerebrum 2018, 2018, cer-11-18. [Google Scholar]
- Tong, H.; Yang, T.; Xu, S.; Li, X.; Liu, L.; Zhou, G.; Yang, S.; Yin, S.; Li, X.-J.; Li, S. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 3845. [Google Scholar] [CrossRef]
- Schultz, J.L.; Neema, M.; Nopoulos, P.C. Unravelling the role of huntingtin: From neurodevelopment to neurodegeneration. Brain 2023, 146, 4408–4410. [Google Scholar] [CrossRef]
- Greco, T.M.; Secker, C.; Ramos, E.S.; Federspiel, J.D.; Liu, J.P.; Perez, A.M.; Al-Ramahi, I.; Cantle, J.P.; Carroll, J.B.; Botas, J.; et al. Dynamics of huntingtin protein interactions in the striatum identifies candidate modifiers of Huntington disease. Cell Syst. 2022, 13, 304–320.e5. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Zeitlin, S.; Goldowitz, D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol. Neurobiol. 2003, 28, 259–276. [Google Scholar] [CrossRef]
- Li, X.; Tong, H.; Xu, S.; Zhou, G.; Yang, T.; Yin, S.; Yang, S.; Li, X.; Li, S. Neuroinflammatory Proteins in Huntington’s Disease: Insights into Mechanisms, Diagnosis, and Therapeutic Implications. Int. J. Mol. Sci. 2024, 25, 11787. [Google Scholar] [CrossRef]
- Podvin, S.; Rosenthal, S.B.; Poon, W.; Wei, E.; Fisch, K.M.; Hook, V. Mutant Huntingtin Protein Interaction Map Implicates Dysregulation of Multiple Cellular Pathways in Neurodegeneration of Huntington’s Disease. J. Huntington’s Dis. 2022, 11, 243–267. [Google Scholar] [CrossRef]
- Sap, K.A.; Geijtenbeek, K.W.; Schipper-Krom, S.; Guler, A.T.; Reits, E.A. Ubiquitin-modifying enzymes in Huntington’s disease. Front. Mol. Biosci. 2023, 10, 1107323. [Google Scholar] [CrossRef] [PubMed]
- Cisbani, G.; Cicchetti, F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 2012, 3, e382. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Reid, S.; Scotter, E.L.; McGregor, A.L.; Mehrabi, N.F.; Singh-Bains, M.K.; Glass, M.; Faull, R.L.M.; Snell, R.G.; Dragunow, M. Inconsistencies in histone acetylation patterns among different HD model systems and HD post-mortem brains. Neurobiol. Dis. 2020, 146, 105092. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.C.; Chavan, M.B.; Gurow, K.; Gupta, M.; Dhaliwal, J.S.; Ming, L.C. The role of mitochondrial dysfunction in Huntington’s disease: Implications for therapeutic targeting. Biomed. Pharmacother. 2025, 183, 117827. [Google Scholar] [CrossRef]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef]
- Paraskevopoulou, F.; Parvizi, P.; Senger, G.; Tuncbag, N.; Rosenmund, C.; Yildirim, F. Impaired inhibitory GABAergic synaptic transmission and transcription studied in single neurons by Patch-seq in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2020293118. [Google Scholar] [CrossRef]
- Xu, C.; Chen, S.; Chen, X.; Ho, K.H.; Park, C.; Yoo, H.; Lee, S.H.; Park, H. Altered exocytosis of inhibitory synaptic vesicles at single presynaptic terminals of cultured striatal neurons in a knock-in mouse model of Huntington’s disease. Front. Mol. Neurosci. 2023, 16, 1175522. [Google Scholar] [CrossRef]
- Ambrose, C.M.; Duyao, M.P.; Barnes, G.; Bates, G.P.; Lin, C.S.; Srinidhi, J.; Baxendale, S.; Hummerich, H.; Lehrach, H.; Altherr, M.; et al. Structure and expression of the Huntington’s disease gene: Evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet. 1994, 20, 27–38. [Google Scholar] [CrossRef]
- Lin, B.; Rommens, J.M.; Graham, R.K.; Kalchman, M.; MacDonald, H.; Nasir, J.; Delaney, A.; Goldberg, Y.P.; Hayden, M.R. Differential 3′ polyadenylation of the Huntington disease gene results in two mRNA species with variable tissue expression. Hum. Mol. Genet. 1993, 2, 1541–1545. [Google Scholar] [CrossRef]
- Liu, L.; Tong, H.; Sun, Y.; Chen, X.; Yang, T.; Zhou, G.; Li, X.J.; Li, S. Huntingtin Interacting Proteins and Pathological Implications. Int. J. Mol. Sci. 2023, 24, 13060. [Google Scholar] [CrossRef]
- Iennaco, R.; Formenti, G.; Trovesi, C.; Rossi, R.L.; Zuccato, C.; Lischetti, T.; Bocchi, V.D.; Scolz, A.; Martínez-Labarga, C.; Rickards, O.; et al. The evolutionary history of the polyQ tract in huntingtin sheds light on its functional pro-neural activities. Cell Death Differ. 2022, 29, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein-protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A worldwide study of the Huntington’s disease mutation: The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef]
- Semaka, A.; Collins, J.A.; Hayden, M.R. Unstable familial transmissions of Huntington disease alleles with 27-35 CAG repeats (intermediate alleles). Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010, 153B, 314–320. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.J.; Chotai, K.; Connarty, M.; Crauford, D.; Curtis, A.; et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Genet. 1996, 59, 16–22. [Google Scholar]
- Ashizawa, T.; Wong, L.J.; Richards, C.S.; Caskey, C.T.; Jankovic, J. CAG repeat size and clinical presentation in Huntington’s disease. Neurology 1994, 44, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Capiluppi, E.; Romano, L.; Rebora, P.; Nanetti, L.; Castaldo, A.; Gellera, C.; Mariotti, C.; Macerollo, A.; Cislaghi, M.G. Late-onset Huntington’s disease with 40-42 CAG expansion. Neurol. Sci. 2020, 41, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Handsaker, R.E.; Kashin, S.; Reed, N.M.; Tan, S.; Lee, W.S.; McDonald, T.M.; Morris, K.; Kamitaki, N.; Mullally, C.D.; Morakabati, N.R.; et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell 2025, 188, 623–639.e19. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Barba Bazan, C.A.; Truant, R. DNA Damage Repair in Huntington’s Disease and Other Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef]
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 2023, 24, 242–254. [Google Scholar] [CrossRef]
- Schilling, J.; Broemer, M.; Atanassov, I.; Duernberger, Y.; Vorberg, I.; Dieterich, C.; Dagane, A.; Dittmar, G.; Wanker, E.; van Roon-Mom, W.; et al. Deregulated Splicing Is a Major Mechanism of RNA-Induced Toxicity in Huntington’s Disease. J. Mol. Biol. 2019, 431, 1869–1877. [Google Scholar] [CrossRef]
- Zsindely, N.; Nagy, G.; Siági, F.; Farkas, A.; Bodai, L. Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model. Int. J. Mol. Sci. 2023, 24, 11942. [Google Scholar] [CrossRef]
- Elorza, A.; Márquez, Y.; Cabrera, J.R.; Sánchez-Trincado, J.L.; Santos-Galindo, M.; Hernández, I.H.; Picó, S.; Díaz-Hernández, J.I.; García-Escudero, R.; Irimia, M.; et al. Huntington’s disease-specific mis-splicing unveils key effector genes and altered splicing factors. Brain 2021, 144, 2009–2023. [Google Scholar] [CrossRef]
- Tano, V.; Utami, K.H.; Yusof, N.A.B.M.; Bégin, J.; Tan, W.W.L.; Pouladi, M.A.; Langley, S.R. Widespread dysregulation of mRNA splicing implicates RNA processing in the development and progression of Huntington’s disease. eBioMedicine 2023, 94, 104720. [Google Scholar] [CrossRef]
- Hyeon, J.W.; Kim, A.H.; Yano, H. Epigenetic regulation in Huntington’s disease. Neurochem. Int. 2021, 148, 105074. [Google Scholar] [CrossRef]
- Lu, A.T.; Narayan, P.; Grant, M.J.; Langfelder, P.; Wang, N.; Kwak, S.; Wilkinson, H.; Chen, R.Z.; Chen, J.; Simon Bawden, C.; et al. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun. 2020, 11, 4529. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Vakili, G.; Cha, J.H. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; Mansbach, A.; Borowsky, B.; Konstantinova, P.; Mestre, T.A.; Panagoulias, J.; Ross, C.A.; Zauderer, M.; et al. A biological classification of Huntington’s disease: The Integrated Staging System. Lancet Neurol. 2022, 21, 632–644. [Google Scholar] [CrossRef]
- Kwa, L.; Larson, D.; Yeh, C.; Bega, D. Influence of Age of Onset on Huntington’s Disease Phenotype. Tremor. Other Hyperkinet. Mov. 2020, 10, 21. [Google Scholar] [CrossRef]
- Andhale, R.; Shrivastava, D. Huntington’s Disease: A Clinical Review. Cureus 2022, 14, e28484. [Google Scholar] [CrossRef]
- Yanagisawa, N. The spectrum of motor disorders in Huntington’s disease. Clin. Neurol. Neurosurg. 1992, 94, S182–S184. [Google Scholar] [CrossRef] [PubMed]
- Puigdellívol, M.; Saavedra, A.; Pérez-Navarro, E. Cognitive dysfunction in Huntington’s disease: Mechanisms and therapeutic strategies beyond BDNF. Brain Pathol. 2016, 26, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, A. Neuropsychiatry of Huntington’s disease. Dialogues Clin. Neurosci. 2007, 9, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.; Middelkoop, H.; Jurgens, C.K.; Witjes-Ané, M.N.; Roos, R.A. Seven-year clinical follow-up of premanifest carriers of Huntington’s disease. PLoS Curr. 2011, 3, RRN1288. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar]
- Chuang, C.L.; Demontis, F. Systemic manifestation and contribution of peripheral tissues to Huntington’s disease pathogenesis. Ageing Res. Rev. 2021, 69, 101358. [Google Scholar] [CrossRef]
- Gómez-Jaramillo, L.; Cano-Cano, F.; González-Montelongo, M.D.C.; Campos-Caro, A.; Aguilar-Diosdado, M.; Arroba, A.I. A New Perspective on Huntington’s Disease: How a Neurological Disorder Influences the Peripheral Tissues. Int. J. Mol. Sci. 2022, 23, 6089. [Google Scholar] [CrossRef]
- Voysey, Z.; Fazal, S.V.; Lazar, A.S.; Barker, R.A. The sleep and circadian problems of Huntington’s disease: When, why and their importance. J. Neurol. 2021, 268, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Craufurd, D.; MacLeod, R.; Frontali, M.; Quarrell, O.; Bijlsma, E.K.; Davis, M.; Hjermind, L.E.; Lahiri, N.; Mandich, P.; Martinez, A.; et al. Diagnostic genetic testing for Huntington’s disease. Pract. Neurol. 2015, 15, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Unified Huntington’s Disease Rating Scale: Reliability and consistency. Huntington Study Group. Mov. Disord. 1996, 11, 136–142.
- van Eimeren, T.; Giehl, K.; Reetz, K.; Sampaio, C.; Mestre, T.A. Neuroimaging biomarkers in Huntington’s disease: Preparing for a new era of therapeutic development. Park. Relat. Disord. 2023, 114, 105488. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Y.; Shang, H. The updated development of blood-based biomarkers for Huntington’s disease. J. Neurol. 2023, 270, 2483–2503. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef]
- Crowell, V.; Houghton, R.; Tomar, A.; Fernandes, T.; Squitieri, F. Modeling Manifest Huntington’s Disease Prevalence Using Diagnosed Incidence and Survival Time. Neuroepidemiology 2021, 55, 361–368. [Google Scholar] [CrossRef]
- Ohlmeier, C.; Saum, K.U.; Galetzka, W.; Beier, D.; Gothe, H. Epidemiology and health care utilization of patients suffering from Huntington’s disease in Germany: Real world evidence based on German claims data. BMC Neurol. 2019, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Griguoli, A.; Capelli, G.; Porcellini, A.; D’Alessio, B. Epidemiology of Huntington disease: First post-HTT gene analysis of prevalence in Italy. Clin. Genet. 2016, 89, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Mayer, M.; Ekwaru, P.; McMullen, S.; Graves, E.; Wu, J.W.; Budd, N.; Maturi, B.; Cowling, T.; Mestre, T.A. Epidemiology and economic burden of Huntington’s disease: A Canadian provincial public health system perspective. J. Med. Econ. 2022, 25, 212–219. [Google Scholar] [CrossRef]
- Adachi, Y.; Nakashima, K. Population genetic study of Huntington’s disease—prevalence and founder’s effect in the San-in area, western Japan. Nihon Rinsho. Jpn. J. Clin. Med. 1999, 57, 900–904. [Google Scholar]
- Chang, C.M.; Yu, Y.L.; Fong, K.Y.; Wong, M.T.; Chan, Y.W.; Ng, T.H.; Leung, C.M.; Chan, V. Huntington’s disease in Hong Kong Chinese: Epidemiology and clinical picture. Clin. Exp. Neurol. 1994, 31, 43–51. [Google Scholar]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef]
- Eddy, C.M.; Rickards, H. Social cognition and quality of life in Huntington’s disease. Front. Psychiatry 2022, 13, 963457. [Google Scholar] [CrossRef] [PubMed]
- van Walsem, M.R.; Howe, E.I.; Iversen, K.; Frich, J.C.; Andelic, N. Unmet needs for healthcare and social support services in patients with Huntington’s disease: A cross-sectional population-based study. Orphanet J. Rare Dis. 2015, 10, 124. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517. [Google Scholar] [CrossRef] [PubMed]
- Burgunder, J.M.; Guttman, M.; Perlman, S.; Goodman, N.; van Kammen, D.P.; Goodman, L. An International Survey-based Algorithm for the Pharmacologic Treatment of Chorea in Huntington’s Disease. PLoS Curr. 2011, 3, RRN1260. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Duarte, G.S.; Costa, J.; Ferreira, J.J.; Wild, E.J. Tetrabenazine Versus Deutetrabenazine for Huntington’s Disease: Twins or Distant Cousins? Mov. Disord. Clin. Pract. 2017, 4, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Frank, S. Tetrabenazine: The first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatr. Dis. Treat. 2010, 6, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Paleacu, D.; Giladi, N.; Moore, O.; Stern, A.; Honigman, S.; Badarny, S. Tetrabenazine treatment in movement disorders. Clin. Neuropharmacol. 2004, 27, 230–233. [Google Scholar] [CrossRef]
- Jankovic, J.; Beach, J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology 1997, 48, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.; Frank, S. Deutetrabenazine in the treatment of Huntington’s disease. Neurodegener. Dis. Manag. 2019, 9, 31–37. [Google Scholar] [CrossRef]
- Cepeda, C.; Murphy, K.P.; Parent, M.; Levine, M.S. The role of dopamine in Huntington’s disease. Prog. Brain Res. 2014, 211, 235–254. [Google Scholar]
- Lee, S.C.; Jayasinghe, I.; Lie, Y.J. Subcutaneous haloperidol for chorea in patients with Huntington’s disease and dysphagia. J. Neurol. Neurosurg. Psychiatry 2022, 93, A99–A100. [Google Scholar]
- Zaporowska-Stachowiak, I.; Stachowiak-Szymczak, K.; Oduah, M.T.; Sopata, M. Haloperidol in palliative care: Indications and risks. Biomed. Pharmacother. 2020, 132, 110772. [Google Scholar] [CrossRef]
- Feleus, S.; van Schaijk, M.; Roos, R.A.C.; de Bot, S.T. The Many Faces of Huntington’s Chorea Treatment: The Impact of Sudden Withdrawal of Tiapride after 40 Years of Use and a Systematic Review. J. Pers. Med. 2022, 12, 589. [Google Scholar] [CrossRef]
- Fulton, B.; Goa, K.L. Olanzapine. A review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs 1997, 53, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Paleacu, D.; Anca, M.; Giladi, N. Olanzapine in Huntington’s disease. Acta Neurol. Scand. 2002, 105, 441–444. [Google Scholar] [CrossRef]
- Eder, U.; Mangweth, B.; Ebenbichler, C.; Weiss, E.; Hofer, A.; Hummer, M.; Kemmler, G.; Lechleitner, M.; Fleischhacker, W.W. Association of olanzapine-induced weight gain with an increase in body fat. Am. J. Psychiatry 2001, 158, 1719–1722. [Google Scholar] [CrossRef]
- Goldstein, J.M. Quetiapine fumarate (Seroquel): A new atypical antipsychotic. Drugs Today 1999, 35, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Niederwieser, G. Quetiapine in Huntington’s disease: A first case report. J. Neurol. 2002, 249, 1114–1115. [Google Scholar] [CrossRef] [PubMed]
- Alpay, M.; Koroshetz, W.J. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics 2006, 47, 70–72. [Google Scholar] [CrossRef]
- Möller, H.J. Risperidone: A review. Expert Opin. Pharmacother. 2005, 6, 803–818. [Google Scholar] [CrossRef]
- Schultz, J.L.; Kamholz, J.A.; Nopoulos, P.C.; Killoran, A. Comparing Risperidone and Olanzapine to Tetrabenazine for the Management of Chorea in Huntington Disease: An Analysis from the Enroll-HD Database. Mov. Disord. Clin. Pract. 2018, 6, 132–138. [Google Scholar] [CrossRef]
- Conley, R.R. Risperidone side effects. J. Clin. Psychiatry 2000, 61 (Suppl. S8), 20–23. [Google Scholar] [PubMed]
- Ciammola, A.; Sassone, J.; Colciago, C.; Mencacci, N.E.; Poletti, B.; Ciarmiello, A.; Squitieri, F.; Silani, V. Aripiprazole in the treatment of Huntington’s disease: A case series. Neuropsychiatr. Dis. Treat. 2009, 5, 1–4. [Google Scholar]
- Ogilvie, A.C.; Carnahan, R.M.; Chrischilles, E.A.; Schultz, J.L. The effects of antidepressants on depressive symptoms in manifest Huntington’s disease. J. Psychosom. Res. 2022, 162, 111023. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef]
- Beglinger, L.J.; Adams, W.H.; Langbehn, D.; Fiedorowicz, J.G.; Jorge, R.; Biglan, K.; Caviness, J.; Olson, B.; Robinson, R.G.; Kieburtz, K.; et al. Results of the citalopram to enhance cognition in Huntington disease trial. Mov. Disord. 2014, 29, 401–405. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. Long-term side effects of SSRIs: Sexual dysfunction and weight gain. J. Clin. Psychiatry 2003, 64 (Suppl. S18), 20–24. [Google Scholar] [PubMed]
- Gibbons, R.D.; Brown, C.H.; Hur, K.; Marcus, S.M.; Bhaumik, D.K.; Erkens, J.A.; Herings, R.M.; Mann, J.J. Early evidence on the effects of regulators’ suicidality warnings on SSRI prescriptions and suicide in children and adolescents. Am. J. Psychiatry 2007, 164, 1356–1363. [Google Scholar] [CrossRef]
- Shetty, S.; Hariharan, A.; Shirole, T.; Jagtap, A.G. Neuroprotective potential of escitalopram against behavioral, mitochondrial and oxidative dysfunction induced by 3-nitropropionic acid. Ann. Neurosci. 2015, 22, 11–18. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, N.; Daniele, F.; Ragone, M.A. Fluoxetine in the treatment of Huntington’s disease. Psychopharmacology 2001, 153, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Benfield, P.; Heel, R.C.; Lewis, S.P. Fluoxetine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in depressive illness. Drugs 1986, 32, 481–508. [Google Scholar] [CrossRef]
- Ranen, N.G.; Lipsey, J.R.; Treisman, G.; Ross, C.A. Sertraline in the treatment of severe aggressiveness in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 1996, 8, 338–340. [Google Scholar]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002, 59, 694–699. [Google Scholar] [CrossRef]
- Lucetti, C.; Del Dotto, P.; Gambaccini, G.; Dell’ Agnello, G.; Bernardini, S.; Rossi, G.; Murri, L.; Bonuccelli, U. IV amantadine improves chorea in Huntington’s disease: An acute randomized, controlled study. Neurology 2003, 60, 1995–1997. [Google Scholar] [CrossRef]
- Harandi, A.A.; Pakdaman, H.; Medghalchi, A.; Kimia, N.; Kazemian, A.; Siavoshi, F.; Barough, S.S.; Esfandani, A.; Hosseini, M.H.; Sobhanian, S.A. A randomized open-label clinical trial on the effect of Amantadine on post Covid-19 fatigue. Sci. Rep. 2024, 14, 1343. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. Dosage effects of riluzole in Huntington’s disease: A multicenter placebo-controlled study. Neurology 2003, 61, 1551–1556. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Dubois, B.; de Yébenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef]
- Stahl, S.M. Anticonvulsants as mood stabilizers and adjuncts to antipsychotics: Valproate, lamotrigine, carbamazepine, and oxcarbazepine and actions at voltage-gated sodium channels. J. Clin. Psychiatry 2004, 65, 738–739. [Google Scholar] [CrossRef]
- Amann, B.; Pantel, J.; Grunze, H.; Vieta, E.; Colom, F.; Gonzalez-Pinto, A.; Naber, D.; Hampel, H. Anticonvulsants in the treatment of aggression in the demented elderly: An update. Clin. Pract. Epidemiol. Ment. Health 2009, 5, 14. [Google Scholar] [CrossRef]
- Wang, H.R.; Woo, Y.S.; Bahk, W.M. Potential role of anticonvulsants in the treatment of obsessive-compulsive and related disorders. Psychiatry Clin. Neurosci. 2014, 68, 723–732. [Google Scholar] [CrossRef]
- Campos, M.S.A.; Ayres, L.R.; Morelo, M.R.S.; Carizio, F.A.M.; Pereira, L.R.L. Comparative efficacy of antiepileptic drugs for patients with generalized epileptic seizures: Systematic review and network meta-analyses. Int. J. Clin. Pharm. 2018, 40, 589–598. [Google Scholar] [CrossRef]
- White, H.S.; Smith, M.D.; Wilcox, K.S. Mechanisms of action of antiepileptic drugs. Int. Rev. Neurobiol. 2007, 81, 85–110. [Google Scholar] [PubMed]
- Winterer, G. Valproate and GABAergic system effects. Neuropsychopharmacology 2003, 28, 2050–2051. [Google Scholar] [CrossRef] [PubMed]
- Grove, V.E., Jr.; Quintanilla, J.; DeVaney, G.T. Improvement of Huntington’s disease with olanzapine and valproate. N. Engl. J. Med. 2000, 343, 973–974. [Google Scholar] [CrossRef]
- Mutanana, N.; Tsvere, M.; Chiweshe, M.K. General side effects and challenges associated with anti-epilepsy medication: A review of related literature. Afr. J. Prim. Health Care Fam. Med. 2020, 12, e1–e5. [Google Scholar] [CrossRef]
- Ambrósio, A.F.; Soares-Da-Silva, P.; Carvalho, C.M.; Carvalho, A.P. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem. Res. 2002, 27, 121–130. [Google Scholar] [CrossRef]
- Zaalouk, T.M.; Bitar, Z.I.; Maadarani, O.S.; Elhabibi, M.E. Carbamazepine-induced Stevens-Johnson syndrome in a patient with history of methotrexate-induced mast cell activation syndrome. Clin. Case Rep. 2020, 9, 256–259. [Google Scholar] [CrossRef]
- Chowta, N.K.; Chowta, M.N.; Ramapuram, J.; Kumar, P.; Fazil, A. Carbamzepine-induced toxic epidermal necrolysis. Indian J. Crit. Care Med. 2011, 15, 123–125. [Google Scholar] [CrossRef]
- Lees, G.; Leach, M.J. Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neurological cultures from rat cortex. Brain Res. 1993, 612, 190–199. [Google Scholar] [CrossRef]
- Wu, J.; Tang, T.; Bezprozvanny, I. Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington’s disease. Neurosci. Lett. 2006, 407, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.; Remi, J.; Remi, C.; Charlesworth, S.; Whalley, H.; Bhatia, R.; Hitchens, M.; Mihalyo, M.; Wilcock, A. Levetiracetam. J. Pain Symptom Manag. 2018, 56, 645–649. [Google Scholar] [CrossRef]
- de Tommaso, M.; Di Fruscolo, O.; Sciruicchio, V.; Specchio, N.; Cormio, C.; De Caro, M.F.; Livrea, P. Efficacy of levetiracetam in Huntington disease. Clin. Neuropharmacol. 2005, 28, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Bai, Y.; Zhang, H.; Liu, H.; Hu, W.; Meng, F.; Yang, A.; Zhang, J. An individual patient analysis of the efficacy of using GPi-DBS to treat Huntington’s disease. Brain Stimul. 2020, 13, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.C.; Shill, H.; Ponce, F.; Aslam, S. Deep brain stimulation in PD: Risk of complications, morbidity, and hospitalizations: A systematic review. Front. Aging Neurosci. 2023, 15, 1258190. [Google Scholar] [CrossRef]
- Busse, M.E.; Khalil, H.; Quinn, L.; Rosser, A.E. Physical therapy intervention for people with Huntington disease. Phys. Ther. 2008, 88, 820–831. [Google Scholar] [CrossRef]
- Kim, K.H.; Song, M.K. Update of Rehabilitation in Huntington’s Disease: Narrative Review. Brain Neurorehabil. 2023, 16, e28. [Google Scholar] [CrossRef]
- Giddens, C.L.; Coleman, A.E.; Adams, C.M. A home program of speech therapy in Huntington’s disease. J. Med. Speech Lang. Pathol. 2010, 18, 1–9. [Google Scholar]
- Ogilvie, A.C.; Nopoulos, P.C.; Schultz, J.L. Quantifying the Onset of Unintended Weight Loss in Huntington’s Disease: A Retrospective Analysis of Enroll-HD. J. Huntington’s Dis. 2021, 10, 485–492. [Google Scholar] [CrossRef]
- Zarotti, N.; Dale, M.; Eccles, F.; Simpson, J. Psychological Interventions for People with Huntington’s Disease: A Call to Arms. J. Huntington’s Dis. 2020, 9, 231–243. [Google Scholar] [CrossRef]
- Andrew, K.M.; Fox, L.M. Supporting Huntington’s Disease Families Through the Ups and Downs of Clinical Trials. J. Huntington’s Dis. 2023, 12, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Krebsbach, P.H. Gene therapy: Design and prospects for craniofacial regeneration. J. Dent. Res. 2009, 88, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Le, Y.; Zhang, Z.; Nian, X.; Liu, B.; Yang, X. Viral Vector-Based Gene Therapy. Int. J. Mol. Sci. 2023, 24, 7736. [Google Scholar] [CrossRef]
- Wang, C.; Pan, C.; Yong, H.; Wang, F.; Bo, T.; Zhao, Y.; Ma, B.; He, W.; Li, M. Emerging non-viral vectors for gene delivery. J. Nanobiotechnology 2023, 21, 272. [Google Scholar] [CrossRef]
- Chen, S.H.; Haam, J.; Walker, M.; Scappini, E.; Naughton, J.; Martin, N.P. Recombinant Viral Vectors as Neuroscience Tools. Curr. Protoc. Neurosci. 2019, 87, e67. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef]
- Dalwadi, D.A.; Calabria, A.; Tiyaboonchai, A.; Posey, J.; Naugler, W.E.; Montini, E.; Grompe, M. AAV integration in human hepatocytes. Mol. Ther. 2021, 29, 2898–2909. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Penaud-Budloo, M.; Kaeppel, C.; Appelt, U.; Le Guiner, C.; Moullier, P.; von Kalle, C.; Snyder, R.O.; Schmidt, M. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol. Ther. 2012, 20, 1177–1186. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Costa-Verdera, H.; Unzu, C.; Valeri, E.; Adriouch, S.; González Aseguinolaza, G.; Mingozzi, F.; Kajaste-Rudnitski, A. Understanding and Tackling Immune Responses to Adeno-Associated Viral Vectors. Hum. Gene Ther. 2023, 34, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Haberman, R.P.; McCown, T.J.; Samulski, R.J. Inducible long-term gene expression in brain with adeno-associated virus gene transfer. Gene Ther. 1998, 5, 1604–1611. [Google Scholar] [CrossRef]
- Muhuri, M.; Levy, D.I.; Schulz, M.; McCarty, D.; Gao, G. Durability of transgene expression after rAAV gene therapy. Mol. Ther. 2022, 30, 1364–1380. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019, 9, 127–128. [Google Scholar] [CrossRef]
- George, L.A.; Ragni, M.V.; Rasko, J.E.J.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K.; et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Leebeek, F.W.G.; Miesbach, W. Gene therapy for hemophilia: A review on clinical benefit, limitations, and remaining issues. Blood 2021, 138, 923–931. [Google Scholar] [CrossRef]
- Sidonio, R.F., Jr.; Pipe, S.W.; Callaghan, M.U.; Valentino, L.A.; Monahan, P.E.; Croteau, S.E. Discussing investigational AAV gene therapy with hemophilia patients: A guide. Blood Rev. 2021, 47, 100759. [Google Scholar] [CrossRef]
- Hoy, S.M. Delandistrogene Moxeparvovec: First Approval. Drugs 2023, 83, 1323–1329. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Hudry, E.; Maguire, C.A.; Sena-Esteves, M.; Breakefield, X.O.; Grandi, P. Viral vectors for therapy of neurologic diseases. Neuropharmacology 2017, 120, 63–80. [Google Scholar] [CrossRef]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, S.; D’Souza, A.; Parayath, N.N.; Bleier, B.S.; Amiji, M.M. Nucleic acid therapies for CNS diseases: Pathophysiology, targets, barriers, and delivery strategies. J. Control. Release 2022, 352, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Chandran, J.; Chowdhury, E.A.; Perkinton, M.; Jamier, T.; Sutton, D.; Wu, S.; Dobson, C.; Shah, D.K.; Chessell, I.; Meno-Tetang, G.M.L. Assessment of AAV9 distribution and transduction in rats after administration through Intrastriatal, Intracisterna magna and Lumbar Intrathecal routes. Gene Ther. 2023, 30, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Daci, R.; Flotte, T.R. Delivery of Adeno-Associated Virus Vectors to the Central Nervous System for Correction of Single Gene Disorders. Int. J. Mol. Sci. 2024, 25, 1050. [Google Scholar] [CrossRef]
- Challis, R.C.; Ravindra Kumar, S.; Chen, X.; Goertsen, D.; Coughlin, G.M.; Hori, A.M.; Chuapoco, M.R.; Otis, T.S.; Miles, T.F.; Gradinaru, V. Adeno-Associated Virus Toolkit to Target Diverse Brain Cells. Annu. Rev. Neurosci. 2022, 45, 447–469. [Google Scholar] [CrossRef]
- Duarte, F.; Ramosaj, M.; Hasanovic, E.; Regio, S.; Sipion, M.; Rey, M.; Déglon, N. Semi-automated workflows to quantify AAV transduction in various brain areas and predict gene editing outcome for neurological disorders. Mol. Ther. Methods Clin. Dev. 2023, 29, 254–270. [Google Scholar] [CrossRef]
- Owusu-Yaw, B.S.; Zhang, Y.; Garrett, L.; Yao, A.; Shing, K.; Batista, A.R.; Sena-Esteves, M.; Upadhyay, J.; Kegel-Gleason, K.; Todd, N. Focused Ultrasound-Mediated Disruption of the Blood-Brain Barrier for AAV9 Delivery in a Mouse Model of Huntington’s Disease. Pharmaceutics 2024, 16, 710. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef]
- Dayton, R.D.; Wang, D.B.; Klein, R.L. The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert Opin. Biol. Ther. 2012, 12, 757–766. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Kang, L.; Jin, S.; Wang, J.; Lv, Z.; Xin, C.; Tan, C.; Zhao, M.; Wang, L.; Liu, J. AAV vectors applied to the treatment of CNS disorders: Clinical status and challenges. J. Control. Release 2023, 355, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Ling, Q.; Herstine, J.A.; Bradbury, A.; Gray, S.J. AAV-based in vivo gene therapy for neurological disorders. Nat. Rev. Drug Discov. 2023, 22, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Konstantinova, P. AAV5-miHTT gene therapy for Huntington disease: Lowering both huntingtins. Expert Opin. Biol. Ther. 2020, 20, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lebron, E.; Denovan-Wright, E.M.; Nash, K.; Lewin, A.S.; Mandel, R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. 2005, 12, 618–633. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef]
- Machida, Y.; Okada, T.; Kurosawa, M.; Oyama, F.; Ozawa, K.; Nukina, N. rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem. Biophys. Res. Commun. 2006, 343, 190–197. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef]
- McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B.; Hobbs, T.; Ojeda, S.R.; Davidson, B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2152–2162. [Google Scholar] [CrossRef]
- Keskin, S.; Brouwers, C.C.; Sogorb-Gonzalez, M.; Martier, R.; Depla, J.A.; Vallès, A.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Lowers Huntingtin mRNA and Protein without Off-Target Effects in Patient-Derived Neuronal Cultures and Astrocytes. Mol. Ther. Methods Clin. Dev. 2019, 15, 275–284. [Google Scholar] [CrossRef]
- Garriga-Canut, M.; Agustín-Pavón, C.; Herrmann, F.; Sánchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef]
- Grondin, R.; Kaytor, M.D.; Ai, Y.; Nelson, P.T.; Thakker, D.R.; Heisel, J.; Weatherspoon, M.R.; Blum, J.L.; Burright, E.N.; Zhang, Z.; et al. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 2012, 135, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Dufour, B.D.; Smith, C.A.; Clark, R.L.; Walker, T.R.; McBride, J.L. Intrajugular vein delivery of AAV9-RNAi prevents neuropathological changes and weight loss in Huntington’s disease mice. Mol. Ther. 2014, 22, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Agustín-Pavón, C.; Mielcarek, M.; Garriga-Canut, M.; Isalan, M. Deimmunization for gene therapy: Host matching of synthetic zinc finger constructs enables long-term mutant Huntingtin repression in mice. Mol. Neurodegener. 2016, 11, 64. [Google Scholar] [CrossRef]
- Hadaczek, P.; Stanek, L.; Ciesielska, A.; Sudhakar, V.; Samaranch, L.; Pivirotto, P.; Bringas, J.; O’Riordan, C.; Mastis, B.; San Sebastian, W.; et al. Widespread AAV1- and AAV2-mediated transgene expression in the nonhuman primate brain: Implications for Huntington’s disease. Mol. Ther. Methods Clin. Dev. 2016, 3, 16037. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Mol. Ther. Nucleic Acids 2016, 5, e297. [Google Scholar] [CrossRef]
- Amaro, I.A.; Henderson, L.A. An Intrabody Drug (rAAV6-INT41) Reduces the Binding of N-Terminal Huntingtin Fragment(s) to DNA to Basal Levels in PC12 Cells and Delays Cognitive Loss in the R6/2 Animal Model. J. Neurodegener. Dis. 2016, 2016, 7120753. [Google Scholar] [CrossRef]
- Denis, H.L.; David, L.S.; Cicchetti, F. Antibody-based therapies for Huntington’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104569. [Google Scholar] [CrossRef] [PubMed]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; van Deventer, S.J.; et al. AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-miHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef]
- Ekman, F.K.; Ojala, D.S.; Adil, M.M.; Lopez, P.A.; Schaffer, D.V.; Gaj, T. CRISPR-Cas9-Mediated Genome Editing Increases Lifespan and Improves Motor Deficits in a Huntington’s Disease Mouse Model. Mol. Ther. Nucleic Acids 2019, 17, 829–839. [Google Scholar] [CrossRef]
- Spronck, E.A.; Brouwers, C.C.; Vallès, A.; de Haan, M.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Gene Therapy Demonstrates Sustained Huntingtin Lowering and Functional Improvement in Huntington Disease Mouse Models. Mol. Ther. Methods Clin. Dev. 2019, 13, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2020, 48, 36–54. [Google Scholar] [CrossRef]
- Voyager Therapeutics Announces Preclinical Data for Huntington’s Disease and Amyotrophic Lateral Sclerosis Programs at the Congress of the European Society of Gene and Cell Therapy. Available online: https://ir.voyagertherapeutics.com/news-releases/news-release-details/voyager-therapeutics-announces-preclinical-data-huntingtons/ (accessed on 28 August 2024).
- Voyager Therapeutics Provides Regulatory Update on VY-HTT01 Program. Voyager Therapeutics Inc. 2020. Available online: https://ir.voyagertherapeutics.com/news-releases/news-release-details/voyager-therapeutics-provides-regulatory-update-vy-htt01-program/ (accessed on 2 September 2024).
- Spronck, E.A.; Vallès, A.; Lampen, M.H.; Montenegro-Miranda, P.S.; Keskin, S.; Heijink, L.; Evers, M.M.; Petry, H.; Deventer, S.J.V.; Konstantinova, P.; et al. Intrastriatal Administration of AAV5-miHTT in Non-Human Primates and Rats Is Well Tolerated and Results in miHTT Transgene Expression in Key Areas of Huntington Disease Pathology. Brain Sci. 2021, 11, 129. [Google Scholar] [CrossRef]
- Vallès, A.; Evers, M.M.; Stam, A.; Sogorb-Gonzalez, M.; Brouwers, C.; Vendrell-Tornero, C.; Acar-Broekmans, S.; Paerels, L.; Klima, J.; Bohuslavova, B.; et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci. Transl. Med. 2021, 13, eabb8920. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, P.; Wang, X.; Chen, F.; Christensen, E.; Thompson, J.; Ren, X.; Kells, A.; Stanek, L.; Carter, T.; et al. Efficient and Precise Processing of the Optimized Primary Artificial MicroRNA in a Huntingtin-Lowering Adeno-Associated Viral Gene Therapy In Vitro and in Mice and Nonhuman Primates. Hum. Gene Ther. 2022, 33, 37–60. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.B.; Stam, A.; Brouwers, C.; Fodale, V.; Bresciani, A.; Vermeulen, M.; Mostafavi, S.; Petkau, T.L.; Hill, A.; Yung, A.; et al. AAV5-miHTT-mediated huntingtin lowering improves brain health in a Huntington’s disease mouse model. Brain 2023, 146, 2298–2315. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, K.; Gutiérrez-Ángel, S.; Keeling, S.; Koyuncu, S.; da Silva Padilha, M.; Feigenbutz, D.; Arzberger, T.; Vilchez, D.; Klein, R.; Dudanova, I. Neuroprotective effects of hepatoma-derived growth factor in models of Huntington’s disease. Life Sci. Alliance 2023, 6, e202302018. [Google Scholar] [CrossRef]
- Sogorb-Gonzalez, M.; Landles, C.; Caron, N.S.; Stam, A.; Osborne, G.; Hayden, M.R.; Howland, D.; van Deventer, S.; Bates, G.P.; Vallès, A.; et al. Exon 1-targeting miRNA reduces the pathogenic exon 1 HTT protein in Huntington disease models. Brain 2024, 147, 4043–4055. [Google Scholar] [CrossRef] [PubMed]
- UCSF Huntington’s Disease Trial: (POC) Study with AMT-130 in Adults with Early Manifest Huntington’s Disease. Available online: https://clinicaltrials.ucsf.edu/trial/NCT04120493 (accessed on 2 September 2024).
- Estevez-Fraga, C.; Tabrizi, S.J.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: November 2022. J. Huntington’s Dis. 2022, 11, 351–367. [Google Scholar] [CrossRef]
- McBride, J.L.; During, M.J.; Wuu, J.; Chen, E.Y.; Leurgans, S.E.; Kordower, J.H. Structural and functional neuroprotection in a rat model of Huntington’s disease by viral gene transfer of GDNF. Exp. Neurol. 2003, 181, 213–223. [Google Scholar] [CrossRef]
- Kells, A.P.; Fong, D.M.; Dragunow, M.; During, M.J.; Young, D.; Connor, B. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol. Ther. 2004, 9, 682–688. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef]
- Ramaswamy, S.; McBride, J.L.; Han, I.; Berry-Kravis, E.M.; Zhou, L.; Herzog, C.D.; Gasmi, M.; Bartus, R.T.; Kordower, J.H. Intrastriatal CERE-120 (AAV-Neurturin) protects striatal and cortical neurons and delays motor deficits in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 2009, 34, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [PubMed]
- Hult, S.; Soylu, R.; Björklund, T.; Belgardt, B.F.; Mauer, J.; Brüning, J.C.; Kirik, D.; Petersén, Å. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. 2011, 13, 428–439. [Google Scholar] [CrossRef]
- Gabery, S.; Sajjad, M.U.; Hult, S.; Soylu, R.; Kirik, D.; Petersén, Å. Characterization of a rat model of Huntington’s disease based on targeted expression of mutant huntingtin in the forebrain using adeno-associated viral vectors. Eur. J. Neurosci. 2012, 36, 2789–2800. [Google Scholar] [CrossRef]
- Zuleta, A.; Vidal, R.L.; Armentano, D.; Parsons, G.; Hetz, C. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2012, 420, 558–563. [Google Scholar] [CrossRef]
- Benraiss, A.; Toner, M.J.; Xu, Q.; Bruel-Jungerman, E.; Rogers, E.H.; Wang, F.; Economides, A.N.; Davidson, B.L.; Kageyama, R.; Nedergaard, M.; et al. Sustained mobilization of endogenous neural progenitors delays disease progression in a transgenic model of Huntington’s disease. Cell Stem Cell 2013, 12, 787–799. [Google Scholar] [CrossRef]
- Monteys, A.M.; Wilson, M.J.; Boudreau, R.L.; Spengler, R.M.; Davidson, B.L. Artificial miRNAs Targeting Mutant Huntingtin Show Preferential Silencing In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2015, 4, e234. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Harris, A.F.; Cabral, D.J.; Keeler, A.M.; Sapp, E.; Ferreira, J.S.; Gray-Edwards, H.L.; Johnson, J.A.; Johnson, A.K.; Su, Q.; et al. Widespread Central Nervous System Gene Transfer and Silencing After Systemic Delivery of Novel AAV-AS Vector. Mol. Ther. 2016, 24, 726–735. [Google Scholar] [CrossRef]
- Connor, B.; Sun, Y.; von Hieber, D.; Tang, S.K.; Jones, K.S.; Maucksch, C. AAV1/2-mediated BDNF gene therapy in a transgenic rat model of Huntington’s disease. Gene Ther. 2016, 23, 283–295. [Google Scholar] [CrossRef]
- Keeler, A.M.; Sapp, E.; Chase, K.; Sottosanti, E.; Danielson, E.; Pfister, E.; Stoica, L.; DiFiglia, M.; Aronin, N.; Sena-Esteves, M. Cellular Analysis of Silencing the Huntington’s Disease Gene Using AAV9 Mediated Delivery of Artificial Micro RNA into the Striatum of Q140/Q140 Mice. J. Huntington’s Dis. 2016, 5, 239–248. [Google Scholar] [CrossRef]
- Pfister, E.L.; DiNardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial miRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington’s Disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, S.; Yang, H.; Zhu, L.; Pan, Y.; Jing, L.; Tang, B.; Li, S.; Li, X.J. Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2020, 117, 20265–20273. [Google Scholar] [CrossRef]
- Maxan, A.; Sciacca, G.; Alpaugh, M.; Tao, Z.; Breger, L.; Dehay, B.; Ling, Z.; Chuan, Q.; Cisbani, G.; Masnata, M.; et al. Use of adeno-associated virus-mediated delivery of mutant huntingtin to study the spreading capacity of the protein in mice and non-human primates. Neurobiol. Dis. 2020, 141, 104951. [Google Scholar] [CrossRef] [PubMed]
- Birolini, G.; Verlengia, G.; Talpo, F.; Maniezzi, C.; Zentilin, L.; Giacca, M.; Conforti, P.; Cordiglieri, C.; Caccia, C.; Leoni, V.; et al. SREBP2 gene therapy targeting striatal astrocytes ameliorates Huntington’s disease phenotypes. Brain 2021, 144, 3175–3190. [Google Scholar] [CrossRef]
- Hyeon, S.J.; Park, J.; Yoo, J.; Kim, S.H.; Hwang, Y.J.; Kim, S.C.; Liu, T.; Shim, H.S.; Kim, Y.; Cho, Y.; et al. Dysfunction of X-linked inhibitor of apoptosis protein (XIAP) triggers neuropathological processes via altered p53 activity in Huntington’s disease. Prog. Neurobiol. 2021, 204, 102110. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: April 2020. J. Huntington’s Dis. 2020, 9, 185–197. [Google Scholar] [CrossRef] [PubMed]
- uniQure Announces Completion of Enrollment in First Cohort of Phase I/II Clinical Trial of AMT-130 for the Treatment of Huntington’s Disease. Available online: https://www.uniqure.com/programs-pipeline/phase-1-2-clinical-trial-of-amt-130 (accessed on 2 September 2024).
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs 2022, 36, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, I.; Fiengo, P.; Remelli, R.; Miragliotta, V.; Rossini, L.; Biotti, I.; Cappelli, A.; Petricca, L.; La Rosa, S.; Caricasole, A.; et al. Recombinant Adeno Associated Viral (AAV) vector type 9 delivery of Ex1-Q138-mutant huntingtin in the rat striatum as a short-time model for in vivo studies in drug discovery. Neurobiol. Dis. 2016, 86, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Lee, S.E.; Cho, I.H. Adeno-Associated Viral Vector Serotype DJ-Mediated Overexpression of N171-82Q-Mutant Huntingtin in the Striatum of Juvenile Mice Is a New Model for Huntington’s Disease. Front. Cell Neurosci. 2018, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- So, K.H.; Choi, J.H.; Islam, J.; Kc, E.; Moon, H.C.; Won, S.Y.; Kim, H.K.; Kim, S.; Hyun, S.H.; Park, Y.S. An Optimization of AAV-82Q-Delivered Rat Model of Huntington’s Disease. J. Korean Neurosurg. Soc. 2020, 63, 579–589. [Google Scholar] [CrossRef]
- Brattås, P.L.; Hersbach, B.A.; Madsen, S.; Petri, R.; Jakobsson, J.; Pircs, K. Impact of differential and time-dependent autophagy activation on therapeutic efficacy in a model of Huntington disease. Autophagy 2021, 17, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef]
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000, 26, 300–306. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).